ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2012; 8(2):195-213. doi:10.7150/ijbs.3805 This issue Cite
Review
TGFβ Signaling and Cardiovascular Diseases
Evangelia Pardali1 ![]() , Peter ten Dijke2
, Peter ten Dijke2
1. Department of Cardiology and Angiology, University Hospital Münster, Münster, Germany.
2. Department of Molecular Cell Biology and Centre for Biomedical Genetics, Leiden University Medical Center, Leiden, The Netherlands.
Abstract
Transforming growth factor β (TGFβ) family members are involved in a wide range of diverse functions and play key roles in embryogenesis, development and tissue homeostasis. Perturbation of TGFβ signaling may lead to vascular and other diseases. In vitro studies have provided evidence that TGFβ family members have a wide range of diverse effects on vascular cells, which are highly dependent on cellular context. Consistent with these observations genetic studies in mice and humans showed that TGFβ family members have ambiguous effects on the function of the cardiovascular system. In this review we discuss the recent advances on TGFβ signaling in (cardio)vascular diseases, and describe the value of TGFβ signaling as both a disease marker and therapeutic target for (cardio)vascular diseases.
Keywords: atherosclerosis, BMP, hypertension, Smad, TGFβ, vessel calcification.
Introduction
Transforming growth factor β (TGFβ) is the prototype member of a highly evolutionary conserved secreted family of cytokines. The TGFβ family consists of TGFβ, activins, inhibins, nodals, anti-mullerian hormone (AMH) and bone morphogenetic proteins (BMPs) [1, 2]. TGFβ family members affect various biological processes including cell proliferation, differentiation, migration, adhesion, apoptosis and extracellular matrix (ECM) production. Consequently TGFβ members play crucial role in embryonic development, adult tissue homeostasis and in the pathogenesis of a variety of diseases such as cancer, autoimmune diseases, fibrosis and cardiovascular diseases. They all exert their cellular effects via heteromeric complexes of type I and type II serine/threonine kinase receptors located at the plasmamembrane, and the Smad transcription factors, which have a pivotal role in intracellular signaling [1, 3].
TGFβ family members act in a highly contextual manner and can either stimulate or inhibit angiogenesis. Genetic studies in mice and human have robustly confirmed the notion that TGFβ signaling is essential for regulation of vasculogenesis and angiogenesis [3-6]. Inactivation of different components of TGFβ signaling in mice results in embryonic lethality mainly due to defects in the vascular system. Likewise studies in human further corroborated the importance of TGFβ signaling in vascular function. Mutations in TGFβ family genes or mutations, which lead to missregulated TGFβ signaling result in vascular pathologies, such as arteriovenous malformations (AVMs), aneurysms, hypertension, atherosclerosis and cardiovascular disease [3, 4, 7]. Cardiovascular disease is by far the largest chronic health problem causing significant morbidity, mortality and despite the advances in preventive and therapeutic strategies, it remains the leading cause of death worldwide.
Due to its role in the pathophysiology of cardiovascular disease, TGFβ has been considered as a potent therapeutic target in a wide spectrum of cardiovascular disorders. However, because of its multifunctional and context dependent action interference with TGFβ-dependent processes must be carefully designed and the effects of TGFβ signaling or its inhibition on the different cell types involved must be carefully studied. Several recent reviews have discussed the role of TGFβ signaling in angiogenesis and tumor angiogenesis [4, 5, 8]. In the present review we will focus on the role of TGFβ family signaling in (cardio)vascular diseases.
TGFβ signaling
TGFβ family proteins are generated as dimeric inactive precursor homodimers. The prototypic family member TGFβ1 is secreted as an inactive latent precursor consisting of TGFβ1 and a latency associated peptide (LAP) [1, 2]. This complex is associated with the latent TGFβ binding proteins (LTBP). TGFβ gets activated by proteolytic cleavage of LAP and LTBP by thrombospondin, plasmin, reactive oxygen species, acidic microenvironment, matrix metalloproteinases (MMP2 and 9) and β6 integrin [3]. Interestingly several factors involved in cardiovascular injury regulate TGFβ expression and activation. BMPs appear to be secreted in an active form; they are made in the cytoplasm as dimeric pro-protein complexes, which are cleaved by specific proteases. Once secreted their bioavailability is regulated through reversible interactions with extracellular antagonists such as Noggin, Chordin, Gremlin, cross-veinless 2 [CV2, also referred to as BMP endothelial cell precursor derived regulator (BMPER)] and matrix GLA protein (MGP).
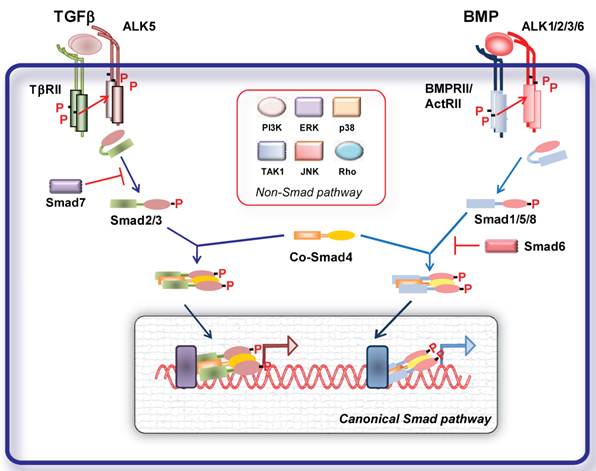
TGFβ family members elicit their cellular effects by inducing heterotetrameric complexes of type I and type II serine/threonine kinase transmembrane receptors [1] (Fig. 1). A number of type I and II receptors have been identified. There are five type II receptors: activin receptor type IIA (ActRIIA), activin receptor type IIB (ActRIIB), BMP type II receptor (BMPRII), TGFβ type II receptor (TβRII) and AMH type II receptor (AMHRII). There are seven type I receptors, termed activin receptor-like kinases (ALKs), ALK1- 7 [1]. Different ligands can bind to different combinations of type I and type II receptors resulting in signal specificity. TGFβ signals in most cells via TGFβ type II receptor (TβRII) and TβRI (ALK5); activins, inhibins and nodal signal via activin receptor type IIA (ActRIIA) and IIB and ALK4; BMPs via BMP type II receptor (BMPRII), ActRIIs and ALK1, 2, 3 and 6 [1] (Fig. 1).
Accessory receptors have been identified that regulate access of TGFβ family members to signaling receptors. For TGFβ, endoglin and betaglycan, and the inhibitory pseudoreceptor BMP and activin membrane-bound inhibitor homolog (BAMBI) have been best characterized. TGFβ accessory receptors have a short intracellular domain that lacks catalytic activity. TGFβ2 can bind to TβRII only in the presence of betaglycan. Endoglin expression enhances BMP signaling, while has inhibitory effects on TGFβ/ALK5 signaling [6]. Soluble forms of betaglycan and endoglin have been shown also to interfere with signaling by sequestering TGFβ family ligands from binding to signaling receptors.
Ligand binding to the constitutively active type II receptor instigates the recruitment and phosphorylation of the type I receptor on specific serine and threonine residues in its intracellular juxtamembrane region. Upon activation type I receptor recruits the receptor regulated (R-)Smad proteins. Smads are intracellular signal transducers for the TGFβ superfamily and regulate transcription of target genes. Smad proteins are classified into three groups the R-Smad1, -2, -3, -5 and -8, common-mediator Smads (Co-Smad4), and inhibitory Smads (I-Smad6 and -7) [1, 2] (Fig. 1). Activated type I receptors recruit and phosphorylate R-Smads at two serine residues in their extreme carboxyl termini. ALK4 and -5 (and -7) mediate R-Smad2 and -3 phosphorylation, while ALK1, -2, -3 and -6 mediate phosphorylation of R-Smad1, -5 and -8. TGFβ, activins, inhibins, nodal signal usually through Smad2 and -3 [1, 2]. Studies on endothelial cells (ECs) revealed that TGFβ can signal through two distinct type receptors, i.e. ALK5 and the ALK1, resulting in activation of both Smad2/3 and Smad1/5/8, respectively [9]. Recently, it was demonstrated that TGFβ can signal via the Smad1 pathway not only on ECs but also in tumor cells. BMPs and AMH signal through Smad1, -5 and -8. In human pulmonary artery endothelial cells (PAECs) BMP9 was shown to induce phosphorylation of Smad1/5 and Smad2 via BMPRII/ActRII/ALK1. While activation of both BMPR-II and ActRII were required for Smad1 phosphorylation, ActRII mediated a greater proportion of phosphorylation on Smad2 [10].
Activated R-Smads interact with Smad4 and the tri- or dimeric formed complexes can translocate to the nucleus, where, together with other transcription factors, co-activators and co-repressors, they regulate target gene expression. Inhibitory (I)- Smads, i.e. Smad6 and -7, antagonize signaling by inhibiting R-Smad activation either by competing with R-Smads for type I receptor interaction and/or by recruiting specific ubiquitin ligases or phosphatases to the activated receptor complex, thereby targeting it for proteosomal degradation or dephosphorylation, respectively [1, 2, 11]. Additionally I-Smads can negatively regulate TGFβ family signaling via degradation of R-Smads and through inhibition of R-Smad-Co-Smad4 complex formation. Smad7 inhibits signaling from all branches of the TGFβ superfamily, whereas Smad6 selectively inhibits BMP signaling [1, 2, 11].
Given the complexity and the variety of the TGFβ family responses, the basic Smad signaling pathway is surprisingly simple. Besides this highly conserved signaling core, TGFβ ligands can regulate the activity of a number of other signaling pathways (non canonical pathways). TGFβ receptor activation results in activation of several other non-Smad signaling pathways in a context dependent manner (Fig. 1). Non-Smad signaling pathways can involve activation of TGFβ activated kinase (TAK)-1, Extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen activated protein (MAP) kinases, Rho GTPases, PI3K-AKT and TRAF6 pathway, which crosstalk with the Smad pathways [12]. In addition TGFβ signaling can be further regulated by different signaling cascades. Classical signaling cascades such as MAP kinases can influence the function and stability of Smad proteins through different types of modifications such as phosphorylation, ubiquitination and sumoylation [2]. Thus, cellular responses to TGFβ signaling result from the dynamic regulation of canonical and non-canonical cascades.
TGFβ signaling and EC function
Genetic studies in mice and humans have provided evidence for the importance of components of the TGFβ signaling pathway in vascular morphogenesis and angiogenesis. Deletion of components of the TGFβ pathway (ligands, receptors and intracellular Smad mediators) in mice leads to embryonic lethality due to vascular abnormalities (Table 1) (reviewed in [3, 4, 7, 8]).
Signal transduction by TGFβ family members. TGFβ and BMP dimers induce heteromeric complex formation between specific type II and type I receptors. The type II receptors then transphosphorylate the type I receptors, leading to their activation. Subsequently, the type I receptor propagates the signal into the cell by phosphorylating receptor-regulated (R)-Smads, which form heteromeric complexes with Smad4 (common (Co)-Smad) and translocate in the nucleus where by interacting with other transcription factors regulate gene transcriptional responses (canonical Smad signaling pathway). Inhibitory (I) Smads 6 and 7 inhibit receptor activation of R-Smads. In addition, the activated type I receptors can activate non-Smad pathways (non-Smad signaling pathway).
Misexpression of TGFβ signaling components lead to (cardio)vascular abnormalities.
| Gene | Human disease | Animal model | Reference |
|---|---|---|---|
| Ligands | |||
| TGFβ1 | Camurati-Engelmann disease | KO: embryonic lethal with vascular defects or postnatal lethality from autoimmune disease. | [13-16] |
| TGFβ2 | Kawasaki disease | KO: aortic arch defects, cardiac septal defects, perinatal lethality. | [17,18] |
| TGFβ3 | unknown | KO: cleft palate, delayed lung maturation, die shortly after birth. | [19,20] |
| Receptors | |||
| TβRII | MFS2, LDS | KO: embryonic lethal, vascular defects | [21,136] |
| TβRI/ALK5 | LDS | KO: embryonic lethal, angiogenesis defects | [22,145,146] |
| ALK1 | HHT | KO: embryonic lethal, reduced VSMC differentiation, dilated vessels, AVMs | [23,24] |
| BMPRII | PAH | KO: pre-angiogenesis lethality Transgenic BMPR2-mutant allele: pulmonary hypertension | [25-27] |
| Accessory receptors | |||
| ENG (endoglin) | HHT | KO: embryonic lethal due to vascular defects, reduced vSMC differentiation, heart defects. Het: vascular lesions similar to HHT. | [28-30] |
| Soluble Endoglin TGFBR3 (betaglycan) | Pre-eclampsia Familial intracranial aneurysm | KO: poorly formed cardiac septa, incomplete compaction of ventricular walls | [51] [31] |
| Smads | |||
| Smad1 | Unknown | KO: embryonic lethal due to defects in chorion-allantoic circulation | [32] |
| Smad3 | Aortic aneurysm-osteoarthritis | [33-37] | |
| Smad4 | JP and HHT | KO: embryonic lethal | [38-40] |
| Smad5 | Unknown | KO: embryonic lethal due to angiogenesis defects | [41-43] |
| Smad6 | Unknown | KO: heart abnormalities, aortic ossification and elevated blood pressure | [44] |
| Smad7 | Unknown | KO: embryonic lethal due to cardiovascular defects | [45] |
| Smad8 | PAH | Defective pulmonary vascular remodeling | [46] |
Abbreviations: KO, knockout; Het, heterozygous; AVMs, arteriovenous malformations; LDS, Loeys-Dietz syndrome; MFS2, Marfan syndrome type 2; HHT, hereditary haemorrhagic telangiectasia; PAH, pulmonary arterial hypertension; JP, Juvenile polyposis; VSMC, vascular smooth muscle cell.
Numerous examples of divergent and contradictory responses of ECs to TGFβ have been reported. It is well accepted today that the TGFβ paradox is due to a number of factors which significantly alter how ECs respond to TGFβ: cellular density, TGFβ concentration, duration of treatment, presence of serum components, surrounding matrix, micro/macrovessel origin of ECs and also species derivation [3, 4, 47, 48]. The inhibitory effects of TGFβ on EC migration and proliferation are mediated by the TGFβ/ALK5/Smad2/3 signaling pathway. In contrast, TGFβ signaling via the TGFβ/ALK1/endoglin leads to proliferation and migration [9, 49]. It was demonstrated that despite that the two pathways are opposed to each other, ALK5 is required for efficient ALK1 signaling and the ratio of the two receptors as well as endoglin expression determines the relative response to TGFβ [9, 49]. Interestingly endoglin can be shed off the EC membrane and result to a soluble form (solEng). solEng was shown to interfere with angiogenesis and high levels of solEng contribute to EC dysfunction that is observed in preeclampsia. solEng inhibits VEGF-induced angiogenesis, EC proliferation [50-52] as well as tumor growth. It has been suggested that solEng exerts its effects by inhibiting TGFβ signaling [52]. However, recent studies revealed that solEng specifically binds to BMP9 and 10, suggesting that solEng may exerts its effects by interfering with BMP9/10 signaling in vivo [50]. It will be interesting to characterise the exact mechanism by which solEng interferes with vascular endothelial growth factor (VEGF)-induced angiogenesis. It can be hypothesized that solEng by scavenging TGFβ ligands affects the delicate balance required for angiogenesis. In addition to the control of TGFβ signaling by its classical receptors, additional receptor associated proteins have been identified for TGFβ in ECs. For example, vascular endothelial (VE)-cadherin has been shown to recruit TGF-β receptors I and II into an active receptor complex and regulate TGFβ signaling [53]. Moreover, the cytoplasmic domain of ALK1 was demonstrated to bind casein kinase (CK)2β, the regulatory subunit of protein kinase CK2, which results in enhancement of ALK1 signaling [54].
BMPs have also been implicated in EC function and angiogenesis. BMP2, -4, -6 and -7 have been shown to induce angiogenesis, EC proliferation and migration [3, 4, 8]. Interestingly, although BMP4 was shown to induce capillary sprouting in a EC spheroid sprouting assay [55], other reports have suggested that BMP4 inhibits basal and VEGF induced angiogenesis in a human umbilical vein EC (HUVEC)-fibroblast sprouting angiogenesis assay [56]. Other BMPs, such as BMP9, have also inhibitory effects on EC activation, proliferation and angiogenesis [10, 57, 58]. However, BMP9 was also reported to induce angiogenesis in vivo and EC proliferation [59]. Additionally, BMP9 in cooperation with TGFβ can induce tumor angiogenesis by inducing EC proliferation [60]. This BMP9 paradox needs to be further investigated and the factors and signaling molecules which determine the pro- or anti-angiogenic effect of BMP9 need to be characterized. The effects of BMPs on EC function and angiogenesis can be modulated by expression of different BMP antagonists and regulators. BMPER is a differentially expressed protein in embryonic endothelial precursor cells. It was shown initially that BMPER interacts with BMPs and when overexpressed antagonizes their activity [61]. In contrast, in a BMPER-deficient zebrafish model BMPER behaves as a BMP agonist. Moreover, depletion of BMPER from ECs results in reduced sprouting and BMPER is required for the pro-angiogenic effects of BMP4 [61]. These results suggest that BMPER plays a pivotal role in fine-tuning BMP activity in angiogenesis. Another BMP antagonist induced by hypoxia is chordin-like 1 (CHL-1). CHL-1 was shown to antagonize the anti-angiogenic effects of BMP4, and alters the homeostatic balance between BMP4 and VEGF to synergize in driving retinal angiogenesis [56]. Matrix GLA protein (MGP) is expressed in EC and plays crucial role in EC function by affecting BMP, TGFβ and VEGF signaling [62]. MGP inhibits BMP2 and BMP4 activity by direct interaction as determined by co-immunoprecipitation [62, 63], while potentiates TGFβ signaling. It was suggested that MGP induces VEGF expression and angiogenesis by potentiating TGFβ signaling, thereby leading to increased VEGF expression and angiogenesis.
TGFβ signaling does not affect only proliferative and migratory EC responses, it also affects vascular permeability. Inhibition of TGFβ signaling by the of TβRI/ALK5 kinase chemical inhibitor SB431542 results in increased expression of the EC specific component Claudin-5 and inhibits expression of adhesion molecules [64]. One of the best characterized factors which can modulate the EC barrier is VEGF. TGFβ is a known regulator of VEGF in a variety of cell types, including epithelial cells [65], macrophages [66], osteoblasts [67] and perivascular cells [68]. Thus, TGFβ can modulate EC barrier properties by influencing VEGF synthesis in the capillary compartment. TGFβ can have also direct effects on EC permeability. Exogenous TGFβ can increases permeability in EC cultures [69, 70]. However, inhibition of TGFβ signaling by neutralizing TGFβ antibody or by SB431542 in co-cultures of brain ECs and pericytes [71] or inhibition of TGFβ signaling by the SB431542 in retinal ECs and mural cell precursors co-cultures [72] leads to increased EC permeability. In addition, it was shown that TGFβ contributes to the maintenance of the EC barrier properties of the retinal microvasculature in vivo. Systemic inhibition of TGFβ in adult mice led to increased vessel permeability as demonstrated by decreased association between the tight junction proteins ZO-1 and occludin [72]. Moreover, adenoviral infection with constitutively active ALK5 (caALK5), but not caALK1, induces EC retraction by decreasing EC permeability [73]. In addition in the EC-specific Smad4 knockout mice it has been reported that EC permeability defects, due to impaired EC gap junction assembly, lead to impaired vessel maturation and embryonic lethality [74]. Moreover, thrombospondin, an activator of latent TGFβ, could rescue the increased vascular permeability and impaired vascular maturation reported in Akt1 knockout mice [75]. Interestingly, inhibition of TGFβ signaling in vivo by pharmacological inhibition of ALK5 resulted in enhanced delivery of nanoparticles to the tumor tissue extravascular space due to increased vessel leakiness [76]. In addition, solEng disrupts EC function and thus results in enhanced capillary permeability. Taken together, these studies suggest that TGFβ signaling plays a critical role in EC function and as a result in vascular development and vascular function.
TGFβ signaling in mural cells
TGFβ signaling besides operating as a rheostat that controls EC differentiation, viability and function, it plays also important role in smooth muscle cell (SMC) differentiation and function. Several studies provided evidence that SMCs are a major cell type through which TGFβ affects vascular development and disease. TGFβ upregulates differentiation markers and induces contractile function in SMC precursors [77]. TGFβ potentiates vascular SMC (VSMC) differentiation by increasing the expression of α-smooth muscle actin (SMA) and smooth muscle myosin [78, 79]. At low concentrations it promotes proliferation of SMC, whereas, at high concentrations it inhibits SMC proliferation and migration [80-82]. It was shown that TGFβ-induced expression of SMC marker genes involves activation of protein kinase N (PKN) and p38 MAPK pathways resulting in differentiation of SMCs [83]. TGFβ1 regulates phenotype transition in SMC by regulation of specific contractile proteins such as αSMA, SM22 α and calponin 1 in different cells including 10T1/2 cells, neural crest cells and fibroblasts [84-86]. In the neural crest stem cell line Monc-1, RhoA modulates TGFβ1-induced SMC differentiation [87]. In 10T1/2 cells, ERK1/2 activity and PI3K/AKT are involved in TGFβ1-regulated differentiation [86, 88].
Genetic studies in mice revealed that inactivation of critical TGFβ signaling molecules in all cells results in failure of vascular development which correlates with a lack of SMC investment of nascent vessels [3, 4]. In adult arteries, SMCs express TGFβ receptors and appear to respond to TGFβ by synthesizing ECM that contributes to the growth of atherosclerotic and restenotic lesions [89-91]. Lineage-specific ablation of TGFβ signaling in VSMCs resulted in alterations in vascular morphogenesis in mice, including impaired elastogenesis and vessel widening [92]. TGFβ actions on SMC are both direct and indirect, mediated through downstream effects of TGFβ signaling in other cell types. For example, TGFβ acts directly on ECs, with consequent paracrine effects on SMCs [93, 94]. TGFβ could act directly on cells of the immune system, with effects on SMC mediated through alterations in plasma cytokines.
BMPs play also important role in SMC function. BMPs were reported to regulate migration, proliferation, and apoptosis of VSMCs and to modulate the expression of SMC phenotypic markers in vitro. Interestingly, the effects of BMPs on VSMCs vary, depending on the anatomic origin of the cells, their state of differentiation or dedifferentiation, and culture conditions. BMP2, BMP4 and to a lesser extent BMP7 are reported to inhibit VSMC proliferation [95-99]. It was shown that BMP2 and BMP7-induced apoptosis of pulmonary SMCs (PASMCs) [100]. It was also shown that BMP2 inhibits SMC proliferation without stimulating ECM synthesis and that adenovirus-mediated transfer of the BMP2 gene inhibited injury-induced intimal hyperplasia suggesting the possibility of therapeutic application of BMP2 for the prevention of vascular proliferative disorders [97]. BMP7 inhibits TGFβ and platelet-derived growth factor (PDGF)-BB-induced SMC growth and stimulates the expression of SMC-specific markers by inducing p21 and I-Smad expression [101, 102]. In addition, BMP7 exhibited anti-inflammatory activity by downregulating intercellular adhesion molecule-1 (ICAM-1) expression [101]. These results suggest that BMP7 may be used in clinic in order to prevent vascular proliferative disorders and to maintain vascular integrity. In another study, BMP2 was shown to be expressed by human aortic SMCs (HASMC) and to induce migration of HASMC in a concentration- and time-dependent manner. In addition, BMP2 synergistically induced PDGF-induced chemotaxis. In contrast, BMP2 had no significant effect on HASMC proliferation [98]. Both BMP7 and BMP4 promote apoptotic cell death in human primary PASMCs by activating caspase-8 and caspase-9. PASMCs expressing mutant forms of BMPRII, identified in idiopathic pulmonary arterial hypertension (IPAH) patients were resistant to BMP-mediated proapoptotic effects [103]. Interestingly it was shown that pulmonary microvascular ECs (PMVEC) secrete BMP4 in response to hypoxia and promote proliferation and migration of VSMCs in a BMP4-dependent fashion [95]. It was shown that BMP4 inhibited proliferation of PASMCs isolated from proximal pulmonary arteries, but stimulated proliferation of PASMCs from peripheral arteries, and conferred protection from apoptosis. Although these differences were not caused by differential activation of BMP signaling pathways, the pro-proliferative effect of BMP4 on peripheral PASMCs was found to be p38MAPK/ERK-dependent [99]. The role of BMP signaling in PAH will be discussed in more detail in following sessions.
Vascular tone and TGFβ signaling
EC-Mural cell interactions are important not only for the growth and maintenance of intact vessels but also for physiological vessel function. Vessel tone regulation is controlled by the contractile state of SMC. EC regulate the SMCs contractile function by releasing a number of vasoactive soluble factors [104]. These factors stimulate either contraction or relaxation of mural cells and thus modulate capillary flow. Nitric oxide (NO) and prostacyclin (PGI2) exert vasodilatory effects on SMCs [105]. NO has also antiproliferative effects on SMC. EC dysfunction, including decreased endothelial nitric-oxide synthase (eNOS) activity and loss of bioactive NO, plays a prominent role in the development of pulmonary arterial hypertension (PAH) [106]. On the other hand endothelin (ET)-1 is a potent vasoconstrictor which stimulates contraction of mural cells via ET-A and ET-B receptors and release of NO from ECs via endothelial ET-B receptors [107].
Several studies have suggested that TGFβ plays an important role in the regulation of vascular tone and reactivity, since TGFβ signaling is involved in regulating the expression of both vasodilators and vasoconstrictors. It was demonstrated that TGFβ/ALK5/Smad3 pathway induces EC expression of ET-1, resulting in decreased EC migration and proliferation [108, 109]. In addition TGFβ1 induces PGI2 production by ECs [110], and increases EC levels of cycloxygenase-1 [111] and prostacyclin-stimulating factor [112], which induce synthesis of the vasodilatory molecule prostacyclin. In addition, iloprost, a prostacyclin analog induces increased TGFβ1 expression by SMCs [113], suggesting a potential feedback mechanism. TGFβ regulates also eNOS activity, the enzyme responsible for baseline NO signaling in ECs [114, 115]. In contrast, addition of a synthetic NO donor diethylenetriamine nitric oxide adduct (DETA)/NO to cultured human coronary SMCs was shown to induce TGFβ mRNA and protein expression in human coronary smooth muscle cells [116]. Another study has demonstrated that aortas from NO synthase deficient mice displayed enhanced basal TGFβ1 activity; ECs from these animals showed increased Smad phosphorylation and transcriptional activity. In the same study it was shown that NO reduced the half-life of ectopically expressed Smad2 by enhancing its ubiquitination. Thus, NO pathway in ECs interferes with TGFβ/Smad2 signaling by directing the proteasomal degradation of activated Smads and may act as a feedback mechanism to limit activation of NO by TGFβ [117]. These results suggest that, since TGFβ signaling influences both vasodilators and vasoconstrictors, misregulation of TGFβ signaling may result in perturbation of the vessel tone. Consistent with this notion it was shown that intravascular infusion of TGFβ in mice results in decreased vascular resistance and vasolidation. In addition, it was suggested that chronically increased levels of TGFβ1 in the brain of Alzheimer leads to impaired constriction of cerebral vessels due to impaired ET-1 mediated contractions [118]. The role of TGFβ signaling in hypertension will be further discussed in later sections.
Overexpression of solEng in mice resulted in impaired peripheral microvessel blood flow response and collapsed retinal microvessels [72, 119]. Moreover, overexpression of solEng leads to brain AVMs in mice [120]. In addition, solEng was found to inhibit TGFβ-mediated activation of eNOS, thereby affecting vascular tone [52]. Similarly, mice heterozygous for endoglin have a severely vasoactive response as a result of decreased eNOS expression due to eNOS decreased half-life [115]. In addition Eng+/− mice exhibit reduced matrigel-dependent capillary tube formation and delayed reperfusion in a hindlimb ischemia model [121]. Moreover, it was shown that Acetylcholine-induced vasorelaxation it was significantly increased in adult Eng+/- vs. control vessels. These vasodilatory responses were inhibited by the nitric oxide synthase inhibitor N (G)-nitro-L- arginine methyl ester (L-NAME), suggesting eNOS dependence. In addition eNOS uncoupling was observed in lung tissues of adult, but not newborn, heterozygous mice and was associated with increased production of reactive O2 species (ROS) in adult Eng+/- vs. control lungs. These data suggest that eNOS-dependent H2O2 generation in Eng+/- lungs accounts for the heightened pulmonary vasorelaxation [122]. In a recent study thalidomide induced vessel maturation by mural cell recruitment in Eng-heterozygous mice and Hereditary hemorrhagic telangiectasia (HHT) patients, partly by inducing PDGF-B expression in ECs, further supporting the important role of endoglin in EC-SMC interactions [123].
BMPs were also shown to play important role in vessel tone regulation and vascular function. Addition of exogenous BMP2 induces expression and activity of eNOS in intrapulmonary artery and pulmonary EC preparations [124]. In the same study it was shown that BMP2 heterozygous mice develop severe hypoxic pulmonary hypertension while BMP4 heterozygous mice are protected. This was shown to be due to decreased endothelial nitric oxide synthase (eNOS) expression and activity in the pulmonary vasculature of hypoxic Bmp2+/- but not Bmp4+/- mice. It was also shown that activation of BMPRII promotes pulmonary artery EC (PAEC) survival, proliferation, and migration by activating eNOS [125]. BMP2 and BMP4 were shown to stimulate eNOS phosphorylation resulting in increased eNOS activity. Inhibition of eNOS activation by NOS inhibitors hindered BMP2 and BMP4 induced PAEC migration. Interestingly functional deletion of the extracellular BMP modulator BMPER results in reduced eNOS expression [126]. BMP4 infusion stimulated aortic NADPH oxidase activity which led to endothelial dysfunction and subsequent impaired vasorelaxation and increased systolic blood pressure in mice [127]. BMP4 upregulation has been linked with hypertension. Addition of BMP4 in cultured human coronary arterial ECs exerts prooxidant, prohypertensive effects [128]. BMP4 treatment of mouse aortaes led to impaired endothelium-dependent relaxations, exaggerated endothelium-dependent contractions via cyclooxygenase-2 (COX-2) upregulation in a reactive oxygen species (ROS) dependent manner [129]. It has been also reported that increased pulmonary vascular remodelling in Alk1+/− mice leads to signs of PAH and it has been shown that this is associated with eNOS-dependent ROS production and it can be averted by anti-oxidant treatment [130]. Interestingly prostacyclin analogues were shown to enhance BMP induced Smad1/5 phosphorylation by suppressing Smad6 expression [131]. Furthermore, prostacyclin analogues rescued the defects in Smad1/5 phosphorylation in PASMCS from PAH patients and inhibited SMC proliferation and prevented PAH progression in the monocrotaline rat model of PAH [131].
Perturbation of TGFβ signaling in cardiovascular diseases
The critical role of TGFβ signaling in vascular function was further verified by studies in human. Genetic analysis revealed that mutations in genes of the TGFβ family or genes which affect TGFβ signaling function lead to vascular dysfunction and disease (Table 1).
Mutations in the ALK1 and endoglin genes lead to an autosomal dominant vascular dysplasia known as HHT. HHT is characterized by the development of telangiectasias and AVMs [3, 4]. HHT type 1 is caused by mutations in endoglin [28, 132], whereas type 2 is due to mutations in ALK1 [23, 24]. Recently, it was reported that a subset of patients with juvenile polyposis (JP), carrying mutations in the Smad4 gene, can also develop HHT [38]. Interestingly targeted deletions of Acvrl1 (ALK1), TβR1 (ALK5), TβR2 and Eng in mice result in vascular abnormalities highly reminiscent of those described in patients with HHT. The vascular abnormalities and fragile leaky vessels are due to abnormal EC proliferation and SMC recruitment [3]. Thalidomide treatment of HHT patients was shown to enhance blood vessel stabilization and reduce nosebleed frequency [123]. Moreover, it has been shown that VEGF levels are elevated in skin telangiectatic lesions of HHT patients and that anti-angiogenic drugs, such as thalidomide and bevacizumab (anti-VEGF antibody), are effective in treating gastrointestinal bleedings and liver AVMs, respectively [133-135].
Endoglin can be shed from the cell surface by MMP14 and this result in a soluble form of endoglin [51]. Excessive production of solEng by trophoblasts in placenta has been reported in patients affected by pre-eclampsia [52], whether this is caused by excessive MMP14 activity remains to be shown. Pre-eclampsia is characterized by EC dysfunction resulting in vasoconstriction, endo-organ ischemia and increased vascular permeability which leads to a raise in blood pressure, is a major cause of maternal, fetal, and neonatal mortality [52]. It was shown that solEng interferes with EC function [51, 52]. Recent studies have shown that solEng can bind to BMP9 and BMP10 and inhibit their signaling [50]. In addition solEng can inhibit VEGF induced angiogenesis in vitro and in vivo. Although the exact mechanism is not known these results suggest that sol-endoglin may be used in anti-angiogenic therapeutic interventions.
Marfan syndrome (MFS) is a genetic disorder characterized by defects in the cardiovascular system and in aorta and heart valves due to increased TGFβ signaling activity. MFS is caused by mutations in the fibrillin gene [136, 137] and affects the skeletal system, ocular system, cardiovascular system, pulmonary system as well as other organs. Fibrillin interacts with LTBP and control TGFβ bioavailability [138]. MFS type 2 is caused by mutation in the TβRII gene locus. Interestingly it was shown recently that mutations in TβRII associated with MFS2 and LDS have differential effects on TGFβ-induced Smad and ERK signaling. It was suggested that the balance between TGFβ-induced Smad and ERK might be an important determinant of phenotypic severity in disorders related to different mutations in TβRII [139]. In a mouse model of MFS inhibition of TGFβ signaling by a TGFβ neutralizing antibody or the angiotensin II type 1 receptor inhibitor losartan resulted in decreased aortic root dilatation, elastic fiber degeneration, Smad2 activation, and reversal of skeletal myopathy [140-142]. These results further confirmed the contribution of excess TGFβ signaling to the vascular pathology of MFS. Although the mechanism by which losartan protects against aortic aneurysm and reduces Smad2 activation is not fully understood clinical studies have been initiated and losartan is now used in clinical trials with MFS [143]. Recent observations demonstrated an essential role for the TGFβ non-Smad signaling in aneurysm pathogenesis in MFS mice. It was demonstrated that TGFβ-induced ERK1/2 activation contributes to aortic aneurysm progression and that antagonism of this pathway may be therapeutically useful [144].
The Loeys-Dietz (LDS) syndrome is an aortic-aneurysm syndrome characterized by cardiovascular manifestations caused by inactivating mutations in the TGFβ type I or II receptors [145, 146]. Although the molecular mechanism of LDS is still poorly understood, LDS is most likely related to TGFβ signaling defects. Interestingly the aortae of patients with LDS, who carry heterozygous inactivating mutations in either TβRII or TβRI display increased TGFβ-SMAD signaling and fibrosis [146].
Recently, mutations in Smad3 have been linked with a syndrome characterised by aneurysms, dissections and tortuosity throughout the arterial tree in association with mild craniofacial features and skeletal and cutaneous anomalies [35]. In addition the affected individuals displayed early-onset osteoarthritis. These findings implicate the TGFβ signaling pathway as a putative pharmacological target for the development of new treatment strategies for arterial wall anomalies and osteoarthritis.
As discussed earlier TGFβ signaling is critical for vascular tone regulation and its perturbation leads to impaired blood flow. Hypertension is a cardiovascular risk factor which contributes to the development of organ damage such as renal sclerosis, stroke, and coronary heart disease. Recent studies have suggested that there is a link between increased levels of circulating TGFβ and hypertension [147, 148]. It has been shown that emilin, an extracellular matrix protein, regulates TGFβ availability and that it is involved in the regulation of arterial diameter. Genetic studies have shown that disruption of emilin-1 expression results in increased conversion of pro-TGFβ to the mature form and as a result increased TGFβ signaling [149]. This leads to a reduction in arterial lumen diameter with a resultant increase in vascular resistance and hypertension. It has been postulated that this is due to increased TGFβ signaling which leads to cytostasis of VSMCs and decreased arterial wall expansion. On the other hand, reduced arterial diameter and hypertension may be a secondary consequence of vascular remodelling in mice. This phenotype could be rescued genetically by disruption of one the TGFβ alleles and reduction of TGFβ levels [149]. Interestingly, missregulation and reduction of TGFβ signaling in ECs, due to increased levels of solEng, results also in increased blood pressure in pre-eclampsia [52].
IPAH is a disease characterized by constriction of the pre-capillary pulmonary arteries, which results in sustained pulmonary arterial pressure. IPAH patients are prone to heart failure [150, 151]. PAH is characterized by excessive proliferation of ECs and VSMCs [152, 153], increased vascular inflammation, increased vasoconstriction and reduced dilation. Genetic studies in human revealed that loss of function mutations in BMP type II receptor (BMPRII) were linked with heritable pulmonary arterial hypertension (HPAH) [26, 154-157]. BMPRII expression by ECs and VSMCs is significantly reduced not only in HPAH cases with a BMPRII mutation, but also in patients with secondary PAH [158-160]. Mice expressing a BMPR-II mutant lacking C-terminal tail in the pulmonary SMCs develop pulmonary vascular lesions similar to PAH. Interestingly it was shown that disruption of BMPRII in PASMCs leads to reduced BMP2 and BMP4 signaling, while signaling by BMP6 and BMP7 is enhanced [161]. Furthermore, it was shown that BMP4 is upregulated in hypoxia-induced PAH and that heterozygous Bmp4/lacZ mice are protected from the development of hypoxia-induced PAH, VSMC proliferation, and vascular remodelling. Hypoxia induced PMVEC to secrete BMP4 and promoted proliferation and migration of VSMCs in a BMP4-dependent manner [95]. In addition reduced BMP/Smad signaling resulted in activation of the p38 MAPK pathway which led in aberrant PASMC proliferation [99, 162, 163].
Recently mutations in the downstream mediator Smad8, which is highly expressed in the pulmonary vasculature, were identified in patients with HPAH [164]. This is surprising, since loss of Smad8 function in canonical BMP signaling is largely compensated by Smad1/5. In the same study it was shown that Smad8 mutations lead to decreased miR-21 levels and that miR-21 overexpression results in anti-proliferative effects in PAEC and PASMC. Overexpression of Smad8 corrected microRNA processing and reversed the hyperproliferative phenotype [165]. Loss of Smad8 function in mice results in abnormal vascular remodeling, increased vascular inflammation and pulmonary tumors [46]. Interestingly, certain HHT2 patients develop PAH-like syndromes, suggesting that ALK1 mutations can also be involved in PAH [166, 167].
The role of TGFβ in PAH has been also investigated. Abnormal TGF-β signaling has been observed in the pulmonary vasculature of PAH patients [96, 168]. TGFβ induces apoptosis in PMVEC. Although TGFβ induced both Smad2 and Smad1/5 activation, only Smad2 was shown to be important in TGFβ-induced apoptosis of PMVEC. Thus, imbalance of ALK1 and ALK5 may contribute to the development of pulmonary artery hypertension [169]. Other studies provided evidence that increased TGFβ signaling participates in the pathogenesis of experimental PAH. The development of Monocrotaline-induced-PAH was associated with increased vascular cell apoptosis and increased TGFβ signaling as documented by phosphorylated Smad2 expression. Inhibition of TGFβ signaling significantly attenuated the development of the PAH and reduced pulmonary vascular remodeling [170].
Although it is recognized that PAH is characterized by increased number of SMCs, this may not be only due to increased proliferation of SMCs. Evolving experimental evidence suggests alternative sources for SMC cells. It has been suggested that ECs may transdifferentiate into mesenchymal-like cells expressing SMA, resulting thus in increased numbers of SMC-like cells in vascular pathologies. This hypothesis is supported by the demonstration that transdifferentiation of pulmonary arteriolar ECs into SMC-like cells occurs during hypoxia-induced pulmonary vascular remodeling and is regulated by myocardin [171]. This process during which ECs lose endothelial features, and acquire mesenchymal-like characteristics has been termed Endothelial-to-Mesenchymal Transition (EndoMT). Several groups have provided evidence that EndoMT is critical in early vascular development. For example, it was shown that EndoMT plays an important role in aortic and pulmonary artery development [172, 173]. Future studies will shed more light into the role of EndoMT in the pathology of PAH and other vascular diseases.
TGFβ signaling in atherosclerosis and vascular calcification
Atherosclerosis is a chronic inflammatory disease of the arterial wall leading to regional ischemia and ischemic complications such as myocardial infarction and stroke, which greatly contribute to morbidity and mortality. Atherosclerosis is characterised by immunological mechanisms leading to vascular inflammation and plaque formation [174] (Fig. 2). TGFβ ligands, its receptors as well as Smad proteins have been found to be expressed in fibro-fatty lesions and fibrous plaques [175-177]. Consistent with such an expression profile, TGFβ has been found to affect the properties and function of all the cell types that are known to be present in atherosclerotic lesions such as ECs, SMCs, monocytes/macrophages, platelets, T cells and myofibroblasts. It has been suggested originally that TGFβ exerts anti-atherogenic effects [178] thus having a protective role either by regulating vascular cell proliferation, plaque stability, immune system function or by its anti-inflammatory function [179, 180]. Interestingly, it was shown that there is an inverse relationship between serum TGFβ1 levels and the development of atherosclerosis and that concentration of active TGFβ1 is severely depressed in advanced atherosclerosis [181-183]. In addition, mutations of TβRII have been associated with atherosclerotic and restenotic vascular cells [184]. In contrast to the atheroprotective effects of TGFβ several studies have shown that atherogenic stimuli such as shear stress [185], oxidized cholesterol [186], angiotensin II [187] or growth factors such as PDGF result in increased TGFβ expression [188]. At the same time increased TGFβ1 expression was found in atherosclerotic clinical specimens and increased levels of TGFβ1 were correlated with advanced atherosclerosis [189, 190] (Fig. 2).
Endoglin expression was shown to be upregulated in VSMC in human atherosclerotic plaques further supporting a role for endoglin in the maintenance of vascular integrity and in the response of the vessel wall to injury [89, 191]. In addition it was shown that solEng levels are significantly higher in diabetic patients and in patients with hypertension and diabetes [192]. Higher solEng levels were found in patients with diabetes who had retinopathy, in patients with diabetes who had a high probability of 10-year cardiovascular risk, and in patients with diabetes and hypertension who had three or more damaged target organs than in those with no organs affected [192]. Changes in solEng plasma levels have been reported in other pathologies such as atherosclerosis and coronary artery disease [193]. Also, changes of solEng plasma levels after an acute myocardial infarct are accurate predictors of acute mortality in these patients [194].
Several studies suggested that BMPs play also an important role in atherosclerosis by modulating EC inflammation and cell differentiation. It was shown that BMP4 is increased in response to the high fat diet and BMP2 and -4 have been implicated in proinflammatory effects in the endothelium [195-197]. Furthermore, interference with the BMP pathway leads to reduced, while enhanced BMP activity results in increased atherosclerotic lesion formation in Apolipoprotein (Apo)E knockout mice [195-197]. It was suggested that this might be due to the effects of BMP signaling on EC function. BMP2 and BMP4 have been implicated as mediators of endothelial inflammation in response to shear stress, oxidative stress and pro-inflammatory cytokines [198, 199]. Whether these effects are due to effects on mononuclear cell function is not known (Fig. 2).
A histological and clinical hallmark of atherosclerosis is arterial calcification. Although initially it was considered that vascular calcification is a passive process of accumulation of dying cells, later studies suggested that vascular calcification is a highly regulated process. Several studies have provided evidence that vascular calcification is the result of an osteogenic process, which leads to mineralisation of the ECM and cell differentiation towards osteoblast-like lineages. It has recently become appreciated that under certain pathologic contexts and culture conditions, VSMCs can differentiate to acquire an osteogenic phenotype, and express lineage markers consistent with bone-forming cells as well as calcification-regulating proteins commonly found in bone [200-202]. Considerable evidence suggests that TGFβ family members, particularly the BMPs, are important modulators of vascular calcification [203, 204], probably by inducing ECM production that is amenable to mineralization and by favoring differentiation toward osteoblast-like lineages (Fig. 2). After the demonstration that BMP2 is expressed in calcified human atherosclerotic plaques it was shown that BMPs can direct osteogenic programming of vascular mesenchymal progenitors of the pericyte lineage [205]. Later it was shown that BMP signaling as well as a variety of inflammatory stimuli can promote expression of osteoblast lineage markers such as alkaline phosphatase in cultured VSMCs [200, 201, 206-209]. Treatment of SMC with BMP2 enhanced vascular calcification via increased phosphate uptake and induction of osteogenic phenotype modulation in SMC [210]. Moreover, BMPs may induce VSMC to express proteins, such as osteoprotegerin which may confer protection from vascular calcific and atheromatous disease [211]. Recently, it was shown that inhibition of BMP signaling in ApoE−/− mice by transgene expression of MGP resulted in diminished Smad1/5/8 signaling, as well as reduced inflammation, lesion formation, and calcification after fat feeding [197]. On the other hand MGP deficient ApoE−/− mice displayed enhanced Smad1/5/8 signaling and extensive medial calcification as previously reported [197]. In addition, BMPs were shown to be increased in diabetic vasculopathy. BMP2 was shown to be expressed in adventitial myofibroblasts in the aorta of fat-fed diabetic Low-density lipoprotein receptor (LDLR)−/− mice and to induce an osteogenic phenotype in a subset of the myofibroblasts by inducing Msx2 expression, which is associated with bone formation [212]. In diabetic rats both BMP2 and BMP4 were shown to be induced. Interestingly, transgenic expression of MGP resulted in reduced BMP activity and diminished calcification in the entire media [213]. MGP, noggin and chordin are downregulated in dedifferentiated VSMCs, which would make them more susceptible to BMP action [195]. Thus, the balance between endothelial BMPs and their inhibitors may determine the health of the endothelium, and an efficient limitation of endothelial BMP activity may confer protection against atherosclerosis. TGFβ was also shown to play important role in vascular calcification. TGFβ is present in calcified aortic valves and regulates vascular calcification and osteoblastic differentiation of VSMCs [214, 215].
Involvement of TGFβ and BMP signaling in atherosclerosis. Elevated expression of TGFβ and BMP2 and -4 during atherosclerosis leads to lesion formation and vascular calcification. Vascular calcification may be a result of smooth muscle cells (SMC) dedifferentiation to osteogenic SMC or differentiation of ECs in to mesenchymal osteogenic cells via endothelial to mesenchymal transition (EndoMT).
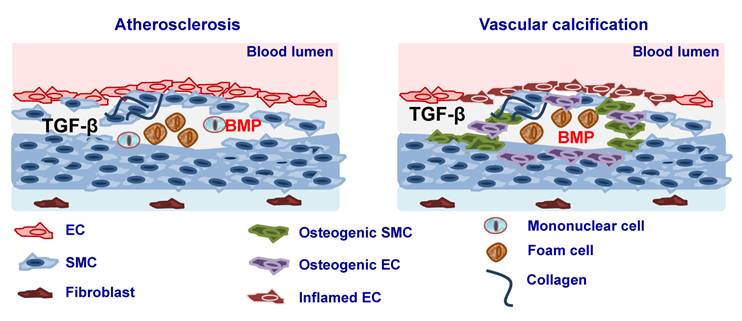
It has been proposed that during lesion formation enhanced BMP signaling may promote developmental conditions and trigger the differentiation of multipotential susceptible cells into the osteogenic lineage. It has been suggested that several cell populations might enter into an osteocondrogenic lineage reprogramming, however, the relative contribution of the respective cell population to local osteogenesis and vascular calcification in different arteriosclerotic disease states remains to be established. These include the calcifying vascular cells isolated from the aortic media [205, 216]. Myofibroblasts present in the adventitia of diabetic and hyperlipidemic LDLR−/− mice [212] may also acquire an osteogenic phenotype. Pericytes have recently emerged as multipotent adult mesenchymal stem cells and it was shown that they have osteogenic potential [217]. BMPs can direct osteogenic programming of vascular mesenchymal progenitors of the pericyte lineage; however, the relative contribution of pericytes to osteogenic vascular calcification has yet to be clarified. It was proposed that VSMC can differentiate and acquire an osteogenic phenotype and this may directly contribute to medial calcific vascular disease. VSMC transdifferentiation was also shown to contribute to the vascular endochondral ossification mechanisms in mice [218]. Circulating stem cells may also contribute to osteogenic changes in the artery wall. It has been suggested recently that vascular ECs might enter into an osteocondrogenic lineage reprogramming [219]. Several studies have suggested that EC plasticity allows EC to transdifferentiate into messenchymal cells during embryonic development [220] or as a response to chronic inflammation [221, 222]. Endothelial differentiation towards a mesenchymal phenotype has recently been linked to ALK2 activity. It was demonstrated that expression of a constitutively active form of the ALK2 type I receptor in mice causes EndoMT; EC cells acquire a stem cell-like phenotype which enables them to undergo osteogenic differentiation [219]. Patients with fibrodysplasia ossificans progressiva are characterized by heterotopic calcification, caused by mutations in the ALK2 gene. It was shown that chondrocytes and osteoblasts of these patients expressed EC markers. In addition, treatment of HUVECs and human cutaneous microvascular ECs (HCMECs) with either TGFβ2 or BMP-resulted in induction of EndoMT, whereas BMP7 and VEGF blocked it [219]. TGFβ has also been reported to induce EndoMT in ECs in vitro [223-226]. In addition TGFβ1 induces EndMT in EC progenitors via the TGFβ/ALK5/Smad2 signaling. TGFβ treatment resulted in decreased expression of endothelial markers and while inducing mesenchymal marker expression as well as cytoskeletal rearrangements, which promoted cells to acquire a contractile phenotype [225].
Concluding remarks
It is well appreciated that members of the TGFβ family play a crucial role in the regulation of both normal and abnormal vascular function. TGFβ ligands regulate angiogenesis through their actions either on ECs and/or on mural cells, affecting e.g., proliferation, migration and most importantly cell differentiation, underlying the important role of TGFβ signals in angiogenesis. TGFβ signaling regulates also EC/SMC interactions and by this way further control vascular function. Missregulation of TGFβ signaling, both in human and mice, leads to vascular dysfunction and vascular pathologies. Recently, it has become evident that TGFβ signaling acts as a rheostat rather than an on-off switch to regulate vascular function. The effects of TGFβ ligands are highly context dependent. Ligand concentration, cell density, cell type, media and culture conditions may determine specific cellular responses to TGFβ family members. In addition the effects of TGFβ signals depend on the interplay between different members of the TGFβ family.
Due to its role in cardiovascular disease pathophysiology, TGFβ signaling components have been considered as potent therapeutic targets in a wide spectrum of cardiovascular disorders. Several classes of inhibitors targeting ligand-receptor interactions have been developed to interfere with the TGFβ pathway. Such inhibitors include TGFβ neutralizing antibodies, anti-receptor antibodies, receptor ectodomain proteins that act as ligand traps. In addition small chemical inhibitors have been developed which interfere with the kinase activity of type I TGFβ receptor. In a mouse model of MFS it was shown that systemic delivery of pan-TGFβ neutralizing antibodies prevents aortic aneurysm, developmental emphysema and skeletal myopathy [138, 141, 227]. Administration of losartan had similar effects by inhibiting TGFβ ligand expression, receptors, and activators such as thrombospondin and MMPs [227]. However, the same anti-TGFβ neutralizing antibody treatment induced lethal cardiovascular pathologies in mouse models of aneurysm and atherosclerosis [228, 229]. In addition, preclinical observations suggested that angiotensin II type I-receptor blockade results in decreased TGFβ signaling. This was translated into clinical studies and the effects of angiotensin II type I inhibitors are being studied in MFS patients [140, 143]. Based on the observations that the pathology of PAH results from a misbalance in BMP and TGFβ signaling pathways, targeting of these pathways has been considered as a potential therapeutic intervention for the treatment of PAH. Inhibition of TGFβ signaling as a potential treatment of PAH is under investigation [230, 231]. Specific BMP agonists may also be useful in treatment PAH. Interestingly, administration of iloprost also restores deficiency in Smad1/Smad5 phosphorylation and Id1 levels in BMPRII deficient PASMCs, while prostacyclin analogues are currently used for treatment of PAH in clinic since they restore Id1 expression in PASMCs [131]. Both TGFβ and BMP signaling have been implicated in atherosclerosis and vascular calcification. Several therapeutic approaches to interfere with BMP signaling such as recombinant ligands, antagonists, soluble receptors and neutralizing antibodies have been exploited [232, 233]. Recently Dorsomorphin, a BMP receptor antagonist [233], has been succesfully used for inhibition of BMP signaling. In addition, identification of novel molecules, such as LDN-193189 with enhanced specificity to antagonize BMP receptor activity [234] offer new therapeutic possibilities.
In conclusion, due to its multifunctional and context dependent function interference with TGFβ-dependent processes must be carefully designed and the effects of TGFβ signaling or its inhibition on the different cell types involved must be carefully studied. Thus, development of new methods through high throughput screening techniques will result in the identification of new generation molecules targeting TGFβ and BMP signaling. In this regard, targeting the downstream regulators Smads might be a more selective diminution of the TGFβ signaling.
Acknowledgements
We thank Gonzalo Sánchez-Duffhues and Marie Jose Goumans for critical reading and suggestions. Studies on the role of TGFβ in cardiovascular diseases are supported by Centre for Biomedical Genetics, Netherlands research council (NWO-MW), Netherlands Institute for Regenerative Medicin (NIRM) the Le Ducq foundation and the 'Innovative Medizinische Forschung' (IMF PA121004).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465-71
2. Heldin CH, Moustakas A. Role of Smads in TGFβ signaling. Cell Tissue Res. 2011 Epub ahead of print
3. ten Dijke P, Arthur HM. Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8(11):857-69
4. Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20(9):556-67
5. Pardali E, ten Dijke P. Transforming growth factor-β signaling and tumor angiogenesis. Front Biosci. 2009;14:4848-61
6. ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11(1):79-89
7. Doetschman T, Barnett JV, Runyan RB. et al. Transforming growth factor β signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2011 Epub ahead of print
8. van Meeteren LA, Goumans MJ, ten Dijke P. TGF-β Receptor Signaling Pathways in Angiogenesis; Emerging Targets for Anti-Angiogenesis Therapy. Curr Pharm Biotechnol. 2011 Epub ahead of print
9. Goumans MJ, Valdimarsdottir G, Itoh S. et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell. 2003;12(4):817-28
10. Upton PD, Davies RJ, Trembath RC. et al. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem. 2009;284(23):15794-804
11. Itoh S, ten Dijke P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19(2):176-84
12. Guo X, Wang XF. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009;19(1):71-88
13. Dickson MC, Martin JS, Cousins FM. et al. Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development. 1995;121(6):1845-54
14. Kulkarni AB, Huh CG, Becker D. et al. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90(21):770-4
15. Letterio JJ, Geiser AG, Kulkarni AB. et al. Autoimmunity associated with TGF-β1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest. 1996;98(9):2109-19
16. Shull MM, Ormsby I, Kier AB. et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359(6397):693-9
17. Sanford LP, Ormsby I, Gittenberger-de Groot AC. et al. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124(13):2659-70
18. Shimizu C, Jain S, Davila S. et al. Transforming growth factor-β signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet. 2011;4(1):16-25
19. Kaartinen V, Voncken JW, Shuler C. et al. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11(4):415-21
20. Proetzel G, Pawlowski SA, Wiles MV. et al. Transforming growth factor-β3 is required for secondary palate fusion. Nat Genet. 1995;11(4):409-14
21. Oshima M, Oshima H, Taketo MM. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179(1):297-302
22. Larsson J, Goumans MJ, Sjostrand LJ. et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001;20(7):1663-73
23. Johnson DW, Berg JN, Baldwin MA. et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13(2):189-95
24. Oh SP, Seki T, Goss KA. et al. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. 2000;97(6):2626-31
25. Beppu H, Ichinose F, Kawai N. et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell MolPhysiol. 2004;287(6):L1241-L7
26. Lane KB, Machado RD, Pauciulo MW. et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81-4
27. West J, Fagan K, Steudel W. et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109-14
28. Arthur HM, Ure J, Smith AJ. et al. Endoglin, an ancillary TGFβ receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217(1):42-53
29. Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104(10):1343-51
30. Li DY, Sorensen LK, Brooke BS. et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284(5419):1534-7
31. Stenvers KL, Tursky ML, Harder KW. et al. Heart and liver defects and reduced transforming growth factor β2 sensitivity in transforming growth factor β type III receptor-deficient embryos. Mol Cell Biol. 2003;23(12):4371-85
32. Lechleider RJ, Ryan JL, Garrett L. et al. Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev Biol. 2001;240(1):157-67
33. Ashcroft GS, Yang X, Glick AB. et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1(5):260-6
34. Datto MB, Frederick JP, Pan L. et al. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol Cell Biol. 1999;19(4):2495-504
35. van de Laar IM, Oldenburg RA, Pals G. et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121-6
36. Yang X, Letterio JJ, Lechleider RJ. et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18(5):1280-91
37. Zhu Y, Richardson JA, Parada LF. et al. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94(6):703-14
38. Gallione CJ, Repetto GM, Legius E. et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363(9412):852-9
39. Sirard C, de la Pompa JL, Elia A. et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12(1):107-19
40. Yang X, Li C, Xu X. et al. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95(7):3667-72
41. Chang H, Huylebroeck D, Verschueren K. et al. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126(8):1631-42
42. Chang H, Zwijsen A, Vogel H. et al. Smad5 is essential for left-right asymmetry in mice. Dev Biol. 2000;219(1):71-8
43. Yang X, Castilla LH, Xu X. et al. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 1999;126(8):1571-80
44. Galvin KM, Donovan MJ, Lynch CA. et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24(2):171-4
45. Chen Q, Chen H, Zheng D. et al. Smad7 is required for the development and function of the heart. J Biol Chem. 2009;284(1):292-300
46. Huang Z, Wang D, Ihida-Stansbury K. et al. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum Mol Genet. 2009;18(15):2791-801
47. Goumans MJ, Valdimarsdottir G, Itoh S. et al. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002;21(7):1743-53
48. Liu Z, Kobayashi K, van Dinther M. et al. VEGF and inhibitors of TGFβ type-I receptor kinase synergistically promote blood-vessel formation by inducing α5-integrin expression. J Cell Sci. 2009;122(Pt18):3294-302
49. Lebrin F, Goumans MJ, Jonker L. et al. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004;23(20):4018-28
50. Castonguay R, Werner ED, Matthews RG. et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem. 2011;286(34):30034-46
51. Hawinkels LJ, Kuiper P, Wiercinska E. et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70(10):4141-50
52. Venkatesha S, Toporsian M, Lam C. et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642-9
53. Rudini N, Felici A, Giampietro C. et al. VE-cadherin is a critical endothelial regulator of TGF-β signalling. EMBO J. 2008;27(7):993-1004
54. Lee NY, Haney JC, Sogani J. et al. Casein kinase 2β as a novel enhancer of activin-like receptor-1 signaling. FASEB J. 2009;23(11):3712-21
55. Zhou Q, Heinke J, Vargas A. et al. ERK signaling is a central regulator for BMP-4 dependent capillary sprouting. Cardiovasc Res. 2007;76(3):390-9
56. Kane R, Godson C, O'Brien C. Chordin-like 1, a bone morphogenetic protein-4 antagonist, is upregulated by hypoxia in human retinal pericytes and plays a role in regulating angiogenesis. Mol Vis. 2008;14:1138-48
57. David L, Mallet C, Mazerbourg S. et al. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953-61
58. Scharpfenecker M, van Dinther M, Liu Z. et al. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci. 2007;120(Pt6):964-72
59. Suzuki Y, Ohga N, Morishita Y. et al. BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J Cell Sci. 2010;123(10):1684-92
60. Cunha SI, Pardali E, Thorikay M. et al. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp Med. 2010;207(1):85-100
61. Heinke J, Wehofsits L, Zhou Q. et al. BMPER is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ Res. 2008;103(8):804-12
62. Bostrom K, Zebboudj AF, Yao Y. et al. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-β1 activity in endothelial cells. J Biol Chem. 2004;279(51):52904-13
63. Yao Y, Zebboudj AF, Shao E. et al. Regulation of bone morphogenetic protein-4 by matrix GLA protein in vascular endothelial cells involves activin-like kinase receptor 1. J Biol Chem. 2006;281(45):33921-30
64. Watabe T, Nishihara A, Mishima K. et al. TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J Cell Biol. 2003;163(6):1303-11
65. Bian ZM, Elner SG, Elner VM. Thrombin-induced VEGF expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2007;48(6):2738-46
66. Jeon SH, Chae BC, Kim HA. et al. Mechanisms underlying TGF-β1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol. 2007;81(2):557-66
67. Tokuda H, Hatakeyama D, Akamatsu S. et al. Involvement of MAP kinases in TGF-β-stimulated vascular endothelial growth factor synthesis in osteoblasts. Arch Biochem Biophys. 2003;415(1):117-25
68. Darland DC, Massingham LJ, Smith SR. et al. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264(1):275-88
69. Birukova AA, Adyshev D, Gorshkov B. et al. ALK5 and Smad4 are involved in TGF-β1-induced pulmonary endothelial permeability. FEBS Lett. 2005;579(18):4031-7
70. Lee YH, Kayyali US, Sousa AM. et al. Transforming growth factor-β1 effects on endothelial monolayer permeability involve focal adhesion kinase/Src. Am J Respir Cell Mol Biol. 2007;37(4):485-93
71. Dohgu S, Takata F, Yamauchi A. et al. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-β production. Brain Res. 2005;1038(2):208-15
72. Walshe TE, Saint-Geniez M, Maharaj AS. et al. TGF-β is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS One. 2009;4(4):e5149
73. Antonov AS, Antonova GN, Fujii M. et al. Regulation of endothelial barrier function by TGF-β type I receptor ALK5: Potential role of contractile mechanisms and heat shock protein 90. J Cell Physiol. 2012Jan;227(2):759-71
74. Lan Y, Liu B, Yao H. et al. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol. 2007;27(21):7683-92
75. Chen J, Somanath PR, Razorenova O. et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11(11):1188-96
76. Kano MR, Komuta Y, Iwata C. et al. Comparison of the effects of the kinase inhibitors imatinib, sorafenib, and transforming growth factor-β receptor inhibitor on extravasation of nanoparticles from neovasculature. Cancer Sci. 2009;100(1):173-80
77. Bjorkerud S. Effects of transforming growth factor-β1 on human arterial smooth muscle cells in vitro. Arterioscler Thromb. 1991;11(4):892-902
78. Hautmann MB, Madsen CS, Owens GK. A transforming growth factor β (TGFβ) control element drives TGFβ-induced stimulation of smooth muscle α-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272(16):10948-56
79. Seay U, Sedding D, Krick S. et al. Transforming growth factor-β-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J Pharmacol Exp Ther. 2005;315(3):1005-12
80. Ding R, Darland DC, Parmacek MS. et al. Endothelial-mesenchymal interactions in vitro reveal molecular mechanisms of smooth muscle/pericyte differentiation. Stem Cells Dev. 2004;13(5):509-20
81. Goumans MJ, Ward-van Oostwaard D, Wianny F. et al. Mouse embryonic stem cells with aberrant transforming growth factor β signalling exhibit impaired differentiation in vitro and in vivo. Differentiation. 1998;63(3):101-13
82. Hirschi KK, Rohovsky SA, D'Amore PA. PDGF, TGF-β, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141(3):805-14
83. Deaton RA, Su C, Valencia TG. et al. Transforming growth factor-β1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280(35):31172-81
84. Chambers RC, Leoni P, Kaminski N. et al. Global expression profiling of fibroblast responses to transforming growth factor-β1 reveals the induction of inhibitor of differentiation-1 and provides evidence of smooth muscle cell phenotypic switching. Am J Pathol. 2003;162(2):533-46
85. Chen S, Lechleider RJ. Transforming growth factor-β-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94(9):1195-202
86. Lien SC, Usami S, Chien S. et al. Phosphatidylinositol 3-kinase/Akt pathway is involved in transforming growth factor-β1-induced phenotypic modulation of 10T1/2 cells to smooth muscle cells. Cell Signal. 2006;18(8):1270-8
87. Chen S, Crawford M, Day RM. et al. RhoA modulates Smad signaling during transforming growth factor-β-induced smooth muscle differentiation. J Biol Chem. 2006;281(3):1765-70
88. Kawai-Kowase K, Sato H, Oyama Y. et al. Basic fibroblast growth factor antagonizes transforming growth factor-β1-induced smooth muscle gene expression through extracellular signal-regulated kinase 1/2 signaling pathway activation. Arterioscler Thromb Vasc Biol. 2004;24(8):1384-90
89. Bot PT, Hoefer IE, Sluijter JP. et al. Increased expression of the transforming growth factor-β signaling pathway, endoglin, and early growth response-1 in stable plaques. Stroke. 2009;40(2):439-47
90. Majesky MW, Lindner V, Twardzik DR. et al. Production of transforming growth factor β 1 during repair of arterial injury. J Clin Invest. 1991;88(3):904-10
91. Schulick AH, Taylor AJ, Zuo W. et al. Overexpression of transforming growth factor β1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci U S A. 1998;95(12):6983-8
92. Choudhary B, Zhou J, Li P. et al. Absence of TGFβ signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47(2):115-21
93. Battegay EJ, Raines EW, Seifert RA. et al. TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. 1990;63(3):515-24
94. Carvalho RL, Jonker L, Goumans MJ. et al. Defective paracrine signalling by TGFβ in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131(24):6237-47
95. Frank DB, Abtahi A, Yamaguchi DJ. et al. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res. 2005;97(5):496-504
96. Morrell NW, Yang X, Upton PD. et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790-5
97. Nakaoka T, Gonda K, Ogita T. et al. Inhibition of rat vascular smooth muscle proliferation in vitro and in vivo by bone morphogenetic protein-2. J Clin Invest. 1997;100(11):2824-32
98. Willette RN, Gu JL, Lysko PG. et al. BMP-2 gene expression and effects on human vascular smooth muscle cells. J Vasc Res. 1999;36(2):120-5
99. Yang X, Long L, Southwood M. et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96(10):1053-63
100. Zhang S, Fantozzi I, Tigno DD. et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L740-54
101. Dorai H, Sampath TK. Bone morphogenetic protein-7 modulates genes that maintain the vascular smooth muscle cell phenotype in culture. J Bone Joint SurgAm. 2001;83A(Suppl 1):S70-S8
102. Dorai H, Vukicevic S, Sampath TK. Bone morphogenetic protein-7 (osteogenic protein-1) inhibits smooth muscle cell proliferation and stimulates the expression of markers that are characteristic of SMC phenotype in vitro. J Cell Physiol. 2000;184(1):37-45
103. Lagna G, Nguyen PH, Ni W. et al. BMP-dependent activation of caspase-9 and caspase-8 mediates apoptosis in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L1059-67
104. Volpe M, Cosentino F. Abnormalities of endothelial function in the pathogenesis of stroke: the importance of endothelin. J Cardiovasc Pharmacol. 2000;35(4S2):S45-8
105. Weber AA, Seul C, Harth V. et al. Inhibition of glycosphingolipid synthesis by threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) and the modulation of IL-1β-stimulated expression of inducible nitric oxide synthase in rat aortic smooth muscle cells. Br J Pharmacol. 1998;123(5):906-10
106. Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev. 2007;15(3):143-9
107. Bourque SL, Davidge ST, Adams MA. The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. Am J Physiol Regul Integr Comp Physiol. 2011;300(6):R1288-95
108. Castanares C, Redondo-Horcajo M, Magan-Marchal N. et al. Transforming growth factor-β receptor requirements for the induction of the endothelin-1 gene. Exp Biol Med (Maywood). 2006;231(6):700-3
109. Castanares C, Redondo-Horcajo M, Magan-Marchal N. et al. Signaling by ALK5 mediates TGF-β-induced ET-1 expression in endothelial cells: a role for migration and proliferation. J Cell Sci. 2007;120(Pt7):1256-66
110. Ristimaki A, Ylikorkala O, Viinikka L. Effect of growth factors on human vascular endothelial cell prostacyclin production. Arteriosclerosis. 1990;10(4):653-7
111. Jackson BA, Goldstein RH, Roy R. et al. Effects of transforming growth factor β and interleukin-1β on expression of cyclooxygenase 1 and 2 and phospholipase A2 mRNA in lung fibroblasts and endothelial cells in culture. Biochem Biophys Res Commun. 1993;197(3):1465-74
112. Hata Y, Clermont A, Yamauchi T. et al. Retinal expression, regulation, and functional bioactivity of prostacyclin-stimulating factor. J Clin Invest. 2000;106(4):541-50
113. Meyer-Kirchrath J, Debey S, Glandorff C. et al. Gene expression profile of the Gs-coupled prostacyclin receptor in human vascular smooth muscle cells. Biochem Pharmacol. 2004;67(4):757-65
114. Schwartz EA, Reaven E, Topper JN. et al. Transforming growth factor-β receptors localize to caveolae and regulate endothelial nitric oxide synthase in normal human endothelial cells. Biochem J. 2005;390(Pt1):199-206
115. Toporsian M, Gros R, Kabir MG. et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96(6):684-92
116. Schmidt A, Geigenmueller S, Voelker W. et al. Exogenous nitric oxide causes overexpression of TGF-β1 and overproduction of extracellular matrix in human coronary smooth muscle cells. Cardiovasc Res. 2003;58(3):671-8
117. Saura M, Zaragoza C, Herranz B. et al. Nitric oxide regulates transforming growth factor-β signaling in endothelial cells. Circ Res. 2005;97(11):1115-23
118. Tong XK, Hamel E. Transforming growth factor-β1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol Pharmacol. 2007;72(6):1476-83
119. Walshe TE, Dole VS, Maharaj AS. et al. Inhibition of VEGF or TGF-β signaling activates endothelium and increases leukocyte rolling. Arterioscler Thromb Vasc Biol. 2009;29(8):1185-92
120. Chen Y, Hao Q, Kim H. et al. Soluble endoglin modulates aberrant cerebral vascular remodeling. Ann Neurol. 2009;66(1):19-27
121. Jerkic M, Rivas-Elena JV, Santibanez JF. et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ Res. 2006;99(3):248-56
122. Belik J, Jerkic M, McIntyre BA. et al. Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1170-8
123. Lebrin F, Srun S, Raymond K. et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16(4):420-8
124. Anderson L, Lowery JW, Frank DB. et al. Bmp2 and Bmp4 exert opposing effects in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298(3):R833-42
125. Gangopahyay A, Oran M, Bauer EM. et al. Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J Biol Chem. 2011;286(38):33134-40
126. Helbing T, Rothweiler R, Ketterer E. et al. BMP activity controlled by BMPER regulates the proinflammatory phenotype of endothelium. Blood. 2011;118(18):5040-9
127. Miriyala S, Gongora Nieto MC, Mingone C. et al. Bone morphogenic protein-4 induces hypertension in mice: role of noggin, vascular NADPH oxidases, and impaired vasorelaxation. Circulation. 2006;113(24):2818-25
128. Csiszar A, Labinskyy N, Jo H. et al. Differential proinflammatory and prooxidant effects of bone morphogenetic protein-4 in coronary and pulmonary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295(2):H569-77
129. Wong WT, Tian XY, Chen Y. et al. Bone morphogenic protein-4 impairs endothelial function through oxidative stress-dependent cyclooxygenase-2 upregulation: implications on hypertension. Circ Res. 2010;107(8):984-91
130. Jerkic M, Kabir MG, Davies A. et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc Res. 2011;92(3):375-84
131. Yang J, Li X, Al-Lamki RS. et al. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res. 2010;107(2):252-62
132. Sorensen LK, Brooke BS, Li DY. et al. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFβ coreceptor. Dev Biol. 2003;261(1):235-50
133. Bauditz J, Lochs H. Angiogenesis and vascular malformations: antiangiogenic drugs for treatment of gastrointestinal bleeding. World J Gastroenterol. 2007;13(45):5979-84
134. Bauditz J, Schachschal G, Wedel S. et al. Thalidomide for treatment of severe intestinal bleeding. Gut. 2004;53(4):609-12
135. Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360(20):2143-4
136. Collod-Beroud G, Le Bourdelles S, Ades L. et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22(3):199-208
137. Mizuguchi T, Matsumoto N. Recent progress in genetics of Marfan syndrome and Marfan-associated disorders. J Hum Genet. 2007;52(1):1-12
138. Neptune ER, Frischmeyer PA, Arking DE. et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407-11
139. Horbelt D, Guo G, Robinson PN. et al. Quantitative analysis of TGFBR2 mutations in Marfan-syndrome-related disorders suggests a correlation between phenotypic severity and Smad signaling activity. J Cell Sci. 2010;123(Pt24):4340-50
140. Brooke BS, Habashi JP, Judge DP. et al. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. 2008;358(26):2787-95
141. Cohn RD, van Erp C, Habashi JP. et al. Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13(2):204-10
142. Matt P, Habashi J, Carrel T. et al. Recent advances in understanding Marfan syndrome: should we now treat surgical patients with losartan?. J Thorac Cardiovasc Surg. 2008;135(2):389-94
143. Lacro RV, Dietz HC, Wruck LM. et al. Rationale and design of a randomized clinical trial of β-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;154(4):624-31
144. Holm TM, Habashi JP, Doyle JJ. et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358-61
145. Loeys BL, Chen J, Neptune ER. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275-81
146. Loeys BL, Schwarze U, Holm T. et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med. 2006;355(8):788-98
147. Laviades C, Varo N, Diez J. Transforming growth factor β in hypertensives with cardiorenal damage. Hypertension. 2000;36(4):517-22
148. Lijnen PJ, Petrov VV, Fagard RH. Association between transforming growth factor-β and hypertension. Am J Hypertens. 2003;16(7):604-11
149. Zacchigna L, Vecchione C, Notte A. et al. Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell. 2006;124(5):929-42
150. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655-65
151. Sastry BK, Narasimhan C, Reddy NK. et al. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover study. J Am Coll Cardiol. 2004;43(7):1149-53
152. Cool CD, Stewart JS, Werahera P. et al. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155(2):411-9
153. Humbert M, Morrell NW, Archer SL. et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12S):13S-24S
154. Cogan JD, Vnencak-Jones CL, Phillips JA. et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med. 2005;7(3):169-74
155. Jiang Y, Nohe A, Bragdon B. et al. Trapping of BMP receptors in distinct membrane domains inhibits their function in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2011;301(2):L218-27
156. Machado RD, Aldred MA, James V. et al. Mutations of the TGF-β type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27(2):121-32
157. Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:251-64
158. Atkinson C, Stewart S, Upton PD. et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672-8
159. Du L, Sullivan CC, Chu D. et al. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med. 2003;348(6):500-9
160. Menon S, Fessel J, West J. Microarray studies in pulmonary arterial hypertension. Int J Clin Pract Suppl. 2011;169:19-28
161. Yu PB, Beppu H, Kawai N. et al. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem. 2005;280(26):24443-50
162. Dewachter L, Adnot S, Guignabert C. et al. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur Respir J. 2009;34(5):1100-10
163. Rudarakanchana N, Flanagan JA, Chen H. et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11(13):1517-25
164. Shintani M, Yagi H, Nakayama T. et al. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46(5):331-7
165. Drake KM, Zygmunt D, Mavrakis L. et al. Altered microRNA Processing in Heritable Pulmonary Arterial Hypertension: an Important Role for Smad-8. Am J Respir Crit Care Med. 2011 Epub ahead of print
166. Harrison RE, Flanagan JA, Sankelo M. et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865-71
167. Trembath RC. Mutations in the TGF-β type 1 receptor, ALK1, in combined primary pulmonary hypertension and hereditary haemorrhagic telangiectasia, implies pathway specificity. J Heart Lung Transplant. 2001;20(2):175
168. Arcot SS, Lipke DW, Gillespie MN, Olson JW. Alterations of growth factor transcripts in rat lungs during development of monocrotaline-induced pulmonary hypertension. Biochem Pharmacol. 1993;46(6):1086-91
169. Lu Q, Patel B, Harrington EO. et al. Transforming growth factor-β1 causes pulmonary microvascular endothelial cell apoptosis via ALK5. Am J Physiol Lung Cell Mol Physiol. 2009;296(5):L825-38
170. Zaiman AL, Podowski M, Medicherla S. et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 2008;177(8):896-905
171. Zhu P, Huang L, Ge X. et al. Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling. Int J Exp Pathol. 2006;87(6):463-74
172. Arciniegas E, Neves CY, Carrillo LM. et al. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium. 2005;12(4):193-200
173. Arciniegas E, Servin M, Arguello C. et al. Development of the aorta in the chick embryo: structural and ultrastructural study. Atherosclerosis. 1989;76(2-3):219-35
174. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317-25
175. Bobik A, Agrotis A, Kanellakis P. et al. Distinct patterns of transforming growth factor-β isoform and receptor expression in human atherosclerotic lesions. Colocalization implicates TGF-β in fibrofatty lesion development. Circulation. 1999;99(22):2883-91
176. Frostegard J, Ulfgren AK, Nyberg P. et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145(1):33-43
177. Kalinina N, Agrotis A, Antropova Y. et al. Smad expression in human atherosclerotic lesions: evidence for impaired TGF-β/Smad signaling in smooth muscle cells of fibrofatty lesions. Arterioscler Thromb Vasc Biol. 2004;24(8):1391-6
178. Grainger DJ. Transforming growth factor β and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler Thromb Vasc Biol. 2004;24(3):399-404
179. Dabek J, Mazurek U, Gasior Z. et al. Transcriptional activity of genes encoding Transforming Growth Factor β and its receptors in peripheral blood mononuclear cells from patients with acute coronary syndromes. Int J Cardiol. 2006;111(2):275-9
180. Mallat Z, Tedgui A. The role of transforming growth factor β in atherosclerosis: novel insights and future perspectives. Curr Opin Lipidol. 2002;13(5):523-9
181. Borkowski P, Robinson MJ, Kusiak JW. et al. Studies on TGF-β 1 gene expression in the intima of the human aorta in regions with high and low probability of developing atherosclerotic lesions. Mod Pathol. 1995;8(5):478-82
182. Grainger DJ, Kemp PR, Metcalfe JC. et al. The serum concentration of active transforming growth factor-β is severely depressed in advanced atherosclerosis. Nat Med. 1995;1(1):74-9
183. Hering S, Jost C, Schulz H. et al. Circulating transforming growth factor β1 (TGFβ1) is elevated by extensive exercise. Eur J Appl Physiol. 2002;86(5):406-10
184. McCaffrey TA. TGF-βs and TGF-β receptors in atherosclerosis. Cytokine Growth Factor Rev. 2000;11(1-2):103-14
185. Negishi M, Lu D, Zhang YQ. et al. Upregulatory expression of furin and transforming growth factor-β by fluid shear stress in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21(5):785-90
186. Leonarduzzi G, Sevanian A, Sottero B. et al. Up-regulation of the fibrogenic cytokine TGF-β1 by oxysterols: a mechanistic link between cholesterol and atherosclerosis. FASEB J. 2001;15(9):1619-21
187. Siegert A, Ritz E, Orth S. et al. Differential regulation of transforming growth factor receptors by angiotensin II and transforming growth factor-β1 in vascular smooth muscle. J Mol Med (Berl). 1999;77(5):437-45
188. Redondo S, Santos-Gallego CG, Ganado P. et al. Acetylsalicylic acid inhibits cell proliferation by involving transforming growth factor-β. Circulation. 2003;107(4):626-9
189. Panutsopulos D, Papalambros E, Sigala F. et al. Protein and mRNA expression levels of VEGF-A and TGF-β1 in different types of human coronary atherosclerotic lesions. Int J Mol Med. 2005;15(4):603-10
190. Wang XL, Liu SX, Wilcken DE. Circulating transforming growth factor β 1 and coronary artery disease. Cardiovasc Res. 1997;34(2):404-10
191. Conley BA, Smith JD, Guerrero-Esteo M. et al. Endoglin, a TGF-β receptor-associated protein, is expressed by smooth muscle cells in human atherosclerotic plaques. Atherosclerosis. 2000;153(2):323-35
192. Blazquez-Medela AM, Garcia-Ortiz L, Gomez-Marcos MA. et al. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010;8:86
193. Li CG, Bethell H, Wilson PB. et al. The significance of CD105, TGFβ and CD105/TGFβ complexes in coronary artery disease. Atherosclerosis. 2000;152(1):249-56
194. Cruz-Gonzalez I, Pabon P, Rodriguez-Barbero A. et al. Identification of serum endoglin as a novel prognostic marker after acute myocardial infarction. J Cell Mol Med. 2008;12(3):955-61
195. Nakagawa Y, Ikeda K, Akakabe Y. et al. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler Thromb Vasc Biol. 2010;30(10):1908-15
196. Pachori AS, Custer L, Hansen D. et al. Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J Mol Cell Cardiol. 2010;48(6):1255-65
197. Yao Y, Bennett BJ, Wang X. et al. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res. 2010;107(4):485-94
198. Csiszar A, Smith KE, Koller A. et al. Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-kappaB activation by tumor necrosis factor-α, H2O2, and high intravascular pressure. Circulation. 2005;111(18):2364-72
199. Sorescu GP, Song H, Tressel SL. et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95(8):773-9
200. Hruska KA, Mathew S, Saab G. Bone morphogenetic proteins in vascular calcification. Circ Res. 2005;97(2):105-14
201. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99(10):1044-59
202. Tintut Y, Alfonso Z, Saini T. et al. Multilineage potential of cells from the artery wall. Circulation. 2003;108(20):2505-10
203. Jeziorska M. Transforming growth factor-βs and CD105 expression in calcification and bone formation in human atherosclerotic lesions. Z Kardiol. 2001;90(Suppl 3):23-6
204. Simionescu A, Philips K, Vyavahare N. Elastin-derived peptides and TGF-β1 induce osteogenic responses in smooth muscle cells. Biochem Biophys Res Commun. 2005;334(2):524-32
205. Bostrom K, Watson KE, Horn S. et al. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91(4):1800-9
206. Hayashi K, Nakamura S, Nishida W. et al. Bone morphogenetic protein-induced MSX1 and MSX2 inhibit myocardin-dependent smooth muscle gene transcription. Mol Cell Biol. 2006;26(24):9456-70
207. Jono S, Shioi A, Ikari Y. et al. Vascular calcification in chronic kidney disease. J Bone Miner Metab. 2006;24(2):176-81
208. Shioi A, Katagi M, Okuno Y. et al. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-α and oncostatin M derived from macrophages. Circ Res. 2002;91(1):9-16
209. Tintut Y, Abedin M, Cho J. et al. Regulation of RANKL-induced osteoclastic differentiation by vascular cells. J Mol Cell Cardiol. 2005;39(2):389-93
210. Li X, Giachelli CM. Sodium-dependent phosphate cotransporters and vascular calcification. Curr Opin Nephrol Hypertens. 2007;16(4):325-8
211. Bennett BJ, Scatena M, Kirk EA. et al. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler Thromb Vasc Biol. 2006;26(9):2117-24
212. Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol. 2006;26(7):1423-30
213. Bostrom KI, Jumabay M, Matveyenko A. et al. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108(4):446-57
214. Clark-Greuel JN, Connolly JM, Sorichillo E. et al. Transforming growth factor-β1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg. 2007;83(3):946-53
215. Jian B, Narula N, Li QY. et al. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75(2):457-65
216. Watson KE, Bostrom K, Ravindranath R. et al. TGF-β 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest. 1994;93(5):2106-13
217. Canfield AE, Doherty MJ, Wood AC. et al. Role of pericytes in vascular calcification: a review. Z Kardiol. 2000;89(Suppl 2):20-7
218. Speer MY, Yang HY, Brabb T. et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104(6):733-41
219. Medici D, Shore EM, Lounev VY. et al. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16(12):1400-6
220. Azhar M, Runyan RB, Gard C. et al. Ligand-specific function of transforming growth factor β in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238(2):431-42
221. Lipton BH, Bensch KG, Karasek MA. Histamine-modulated transdifferentiation of dermal microvascular endothelial cells. Exp Cell Res. 1992;199(2):279-91
222. Romero LI, Zhang DN, Herron GS. et al. Interleukin-1 induces major phenotypic changes in human skin microvascular endothelial cells. J Cell Physiol. 1997;173(1):84-92
223. Egorova AD, Khedoe PP, Goumans MJ. et al. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res. 2011;108(9):1093-101
224. Mihira H, Suzuki HI, Akatsu Y. et al. TGF-{β}-induced mesenchymal transition of MS-1 endothelial cells requires Smad-dependent cooperative activation of Rho signals and MRTF-A. J Biochem. 2011 Epub ahead of print
225. Moonen JR, Krenning G, Brinker MG. et al. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res. 2010;86(3):506-15
226. Kokudo T, Suzuki Y, Yoshimatsu Y. et al. Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121(Pt 20):3317-24
227. Habashi JP, Judge DP, Holm TM. et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117-21
228. Mallat Z, Gojova A, Marchiol-Fournigault C. et al. Inhibition of transforming growth factor-β signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89(10):930-4
229. Wang Y, Ait-Oufella H, Herbin O. et al. TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120(2):422-32
230. Long L, Crosby A, Yang X. et al. Altered bone morphogenetic protein and transforming growth factor-β signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119(4):566-76
231. Megalou AJ, Glava C, Oikonomidis DL. et al. Transforming growth factor-β inhibition attenuates pulmonary arterial hypertension in rats. Int J Clin Exp Med. 2010;3(4):332-40
232. Hao J, Daleo MA, Murphy CK. et al. Dorsomorphin, a selective small molecule inhibitor of BMP signaling, promotes cardiomyogenesis in embryonic stem cells. PLoS One. 2008;3(8):e2904
233. Yu PB, Deng DY, Beppu H. et al. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J Biol Chem. 2008;283(7):3877-88
234. Cuny GD, Yu PB, Laha JK. et al. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett. 2008;18(15):4388-92
Author contact
![]() Corresponding author: Evangelia Pardali: evangelia.pardalide
Corresponding author: Evangelia Pardali: evangelia.pardalide
Received 2011-11-15
Accepted 2011-12-1
Published 2012-1-1