Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(2):249-257. doi:10.7150/ijbs.3967 This issue Cite
Research Paper
Interleukin-22 Ameliorates Cerulein-Induced Pancreatitis in Mice by Inhibiting the Autophagic Pathway
Dechun Feng1#, Ogyi Park1#, Svetlana Radaeva2, Hua Wang1, Shi Yin1, Xiaoni Kong1, Mingquan Zheng3, Sam Zakhari2, Jay K. Kolls3, Bin Gao1 ![]()
1. Laboratory of Liver Diseases, National Institute on Alcohol Abuse and Alcoholism, NIH, Bethesda, MD 20892, USA;
2. Division of Metabolism and Health Effects, National Institute on Alcohol Abuse and Alcoholism, NIH, Bethesda, MD 20892, USA;
3. Department of Genetics, Louisiana State University Health Sciences Center, New Orleans, LA 70112, USA.
# DF and OP contributed equally to this work.
Received 2011-12-16; Accepted 2012-1-1; Published 2012-1-6
Abstract
Pancreatitis occurs when digestive enzymes are activated in the pancreas. Severe pancreatitis has a 10-30% mortality rate. No specific treatments for pancreatitis exist now. Here, we discovered that interleukin-22 (IL-22) may have therapeutic potential in treating acute and chronic pancreatitis. Wild-type and IL-22 knockout mice were equally susceptible to cerulein-induced acute and chronic pancreatitis, whereas liver-specific IL-22 transgenic mice were completely resistant to cerulein-induced elevation of serum digestive enzymes, pancreatic necrosis and apoptosis, and inflammatory cell infiltration. Treatment of wild-type mice with recombinant IL-22 or adenovirus IL-22 markedly attenuated the severity of cerulein-induced acute and chronic pancreatitis. Mechanistically, we show that the protective effect of IL-22 on pancreatitis was mediated via the induction of Bcl-2 and Bcl-XL, which bind to Beclin-1 and subsequently inhibit autophagosome formation to ameliorate pancreatitis. In conclusion, IL-22 ameliorates cerulein-induced pancreatitis by inhibiting the autophagic pathway. IL-22 could be a promising therapeutic drug to treat pancreatitis.
Keywords: IL-22, pancreatitis, autophagy, Bcl-2, Beclin-1.
Introduction
Pancreatitis is inflammation of the pancreas that can occur in acute or chronic forms [1, 2]. Acute pancreatitis is marked by the sudden onset of right upper quadrant or epigastric pain, whereas chronic pancreatitis is characterized by a recurring or persistent abdominal pain with or without steatorrhea or diabetes mellitus [1, 2]. Alcohol consumption is a leading cause for both acute and chronic pancreatitis, followed by gallstones and autoimmune diseases etc [1-3]. Many alcoholic patients develop chronic pancreatitis soon after surviving a second attack of acute pancreatitis [3, 4]. Mild pancreatitis is usually self-limited, with less than 5% mortality, whereas severe acute pancreatitis is associated with 10-30% mortality [5, 6]. The current treatment for pancreatitis is supportive and consists of fluid and salt replacement and pain management. No specific treatments are available at this time [1, 2]. It is generally believed that pancreatitis is caused by the self-digestion of pancreatic acinar cells following the conversion of the inactive trypsinogen to the active trypsin [1, 2, 7]. However, the mechanisms underlying the pathogenesis of pancreatitis are not fully understood.
Interleukin-22 (IL-22), a recently identified type II cytokine that belongs to the IL-10 family [8], can be produced by many types of cells, including Th17, γδT, NK, and NKT cells, but it only targets epithelial cells due to the restricted expression of IL-22R1 on these cells [9-11]. Through binding to IL-22R1 on various types of epithelial cells, including hepatocytes and intestinal epithelial cells, IL-22 has been shown to protect against concanavalin A-induced hepatitis [12-14], inflammatory bowel disease [15], and ulcerative colitis [16]. Similarly, acinar cells, the pancreatic epithelial cells, also express high levels of IL-22R1 [17]. However, the functions of IL-22 in acinar cells remain largely unknown. Because IL-22 is a well-documented survival factor for hepatocytes and liver cancer cells [12, 14, 18], we hypothesize that IL-22 may also protect against acinar cell death and pancreatitis. In this paper, we tested this hypothesis in a model of pancreatitis induced by the injection of cerulein. Our results reveal that IL-22 treatment ameliorates cerulein-induced pancreatitis by inhibiting autophagy, suggesting that IL-22 may have therapeutic potential for the treatment of pancreatitis.
Materials and Methods
Materials
Anti-STAT3, anti-phospho-STAT3, anti-Bcl-2, anti-LC3, anti-USP9x, anti-Bcl-XL, and anti-Beclin-1 antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti-Mcl-1 antibody (Abcam, Cambridge, MA), anti-VMP1 antibody (Enzo Life Sciences, Farmingdale, NY), and anti-MPO antibody (Biocare Medical, Concord, CA) were also purchased. IL-22 ELISA kit was obtained from R&D Systems (Minneapolis, MN).
Mice
Liver-specific IL-22 transgenic mice in a C57BL/6 background were generated as described previously [14]. The IL-22TG heterozygous male mice were bred with female C57BL/6 mice, producing approximately 50% IL-22TG heterozygous mice and 50% WT littermate controls. All animal experiments were approved by the NIAAA animal care and use committee. IL-22 knockout (KO) mice on a C57BL/6 background were kindly provided by Dr. Wenjun Ouyang (Genentech, San Francisco, California).
Histological analysis and immunohistochemistry
Formalin-fixed pancreas samples were processed, and 4-μm thick paraffin sections were stained with hematoxylin and eosin (H&E) for histological analysis. To detect apoptotic cells, the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed using a kit from Milipore (Temecula, CA). To detect myeloperoxidase (MPO) and Microtubule-associated protein 1A/1B-light chain 3 (LC3), pancreas sections were incubated with primary antibodies after antigen retrieval and visualized by DAB.
Administration of mice with IL-22 adenovirus
IL-22 adenovirus and control adenovirus-empty vectors were made as described previously [14]. Mice were injected intravenously (i.v.) with adenovirus-IL-22 (5×108 pfu) or adenovirus-empty vector (5×108 pfu)
Experimental pancreatitis
Acute pancreatitis was induced by hourly intraperitoneal (i.p.) injections of 50 μg/kg cerulein (Sigma) for 7 times as described [19]. Mice were sacrificed 7 hours after the first cerulein injection. For exogenous recombinant IL-22 treatment, recombinant IL-22 (1 μg/g) (Genescript, Piscataway, NJ) was administered (i.p) to mice 2 hours before the first cerulein injection. Chronic pancreatitis was induced by 7 hourly injections (i.p) of 50 μg/kg cerulein once a week for 4 weeks. Mice received adenovirus-IL-22 or adenovirus-empty vector during the third week.
Blood chemistry
Serum amylase and lipase levels were determined using the IDEXX analyzer system (IDEXX Laboratories, Westbrook, ME).
Western blot and immunoprecipitation
Western blots were performed as described previously [20]. For immunoprecipitation, protein was extracted from the pancreas of WT or IL-22TG mice and incubated with Bcl-2 or Bcl-XL antibodies overnight. Protein G beads were then added and incubated for 4 hours. Immunoprecipitates were analyzed by Western blot.
Electron microscope (EM)
One-mm3 pancreatic tissue samples were fixed with 2.5% glutaraldehyde in 0.15 M sodium cacodylate (pH 7.4) overnight, postfixed with 1% OsO4, stained en bloc with 1.5% uranyl acetate, dehydrated in ethanol, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and Reynold's lead citrate and examined with a Hitachi-600 electron microscope.
Statistical analysis
Data are expressed as the mean ± SD. To compare values obtained from three or more groups, one-factor analysis of variance (ANOVA) was used, followed by Tukey's post hoc test. To compare values obtained from two groups, the Student's t-test was performed. Statistical significance was assumed at the P<0.05 level.
Results
IL-22TG mice are resistant to cerulein-induced acute and chronic pancreatitis
To determine the effects of IL-22 on pancreatitis, we compared cerulein-induced acute pancreatitis in WT, IL-22KO, and IL-22TG mice. As illustrated in Figure 1A, without cerulein treatment (phosphate buffered saline [PBS] -treated groups), pancreas appearance and weight and serum amylase and lipase levels were similar between the WT, IL-22KO, and IL-22TG mice. Repeated injection of cerulein, which is a decapeptide analog of cholecystokinin, increased the pancreas and body weight ratio and serum amylase and lipase levels in the WT and IL-22KO mice, but not in the IL-22TG mice. In addition, as shown in Figure 1A, serum IL-22 levels were undetectable in the PBS-treated WT and IL-22KO mice but were detected at high levels in the IL-22TG mice. Repeated injection of cerulein did not elevate serum IL-22 levels in the WT or IL-22KO mice, nor did it affect serum IL-22 levels in the IL-22TG mice.
IL-22TG mice are resistant to cerulein-induced acute pancreatitis. WT, IL-22KO, and IL-22TG mice were treated seven times with cerulein or PBS injections to induce acute pancreatitis. Mice were sacrificed one hour after the last injection. The ratio of the pancreas/body weight, serum levels of IL-22, and amylase and lipase levels were measured (A). Representative H&E staining, TUNEL staining, and MPO staining are shown in the left panel of (B). The pathology score and the number of TUNEL+ and MPO+ cells are summarized in the right panel of (B). *** P < 0.001 compared to cerulein-treated WT mice. Data are representative of three different experiments, 5~7 mice in each group. In panel A, UD: undetectable.
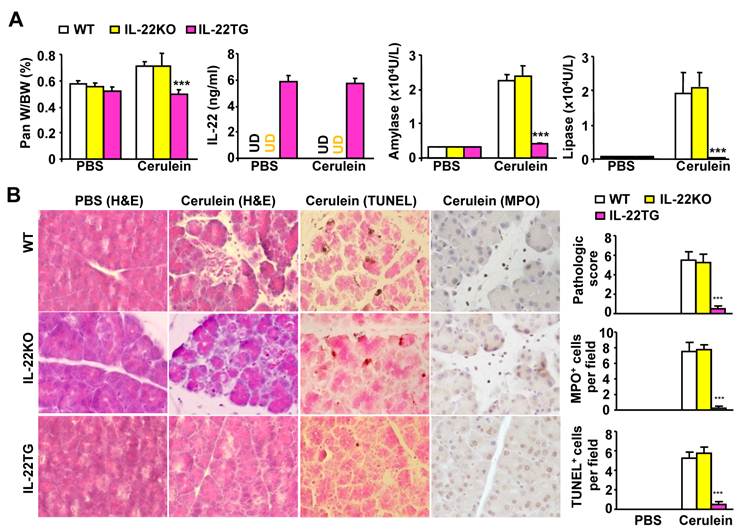
The histological analyses shown in Figure 1B indicate significant tissue necrosis in the cerulein-treated WT and IL-22KO mice, but not in the IL-22TG mice. The numbers of TUNEL+ apoptotic acinar cells and MPO+ neutrophils were elevated in the WT and IL-22KO mice after cerulein treatment, whereas this elevation was barely detectable in the cerulein-treated IL-22TG mice (Fig. 1B).
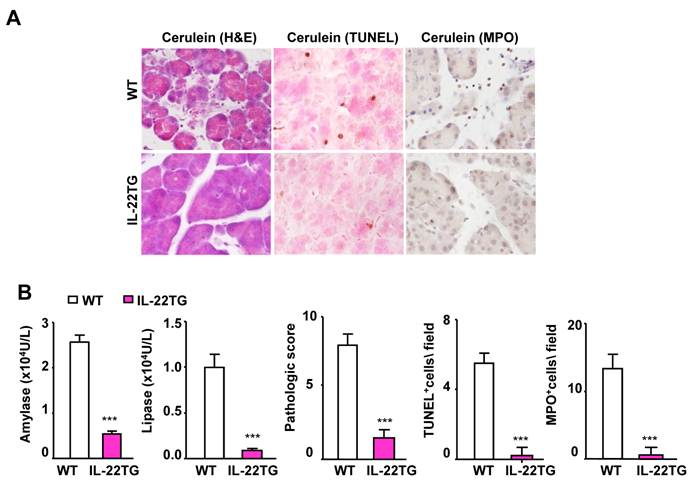
We also tested the severity of cerulein-induced chronic pancreatitis (4-week treatment) in the WT and IL-22TG mice. As illustrated in Figure 2A, 4-week repeated cerulein treatment markedly induced chronic pancreatitis, as evidenced by an elevation in serum amylase and lipase levels and of pancreatic necrosis, TUNEL+ cells, and MPO+ neutrophils in the pancreases of the WT mice. In contrast, such repeated cerulein treatment induced only mild pancreatitis in the IL-22TG mice (Fig. 2A and 2B).
IL-22TG mice are resistant to cerulein-induced chronic pancreatitis. WT and IL-22TG mice received seven cerulein or PBS injections weekly for four weeks to induce chronic pancreatitis. Mice were sacrificed one hour after the last injection. Representative H&E staining, TUNEL staining, and MPO staining are shown (A). Serum levels of amylase and lipase were measured. The pathology score and the number of TUNEL+ and MPO+ cells are summarized (B). ***P<0.001 compared to cerulein-treated WT mice. Data are representative of three different experiments, 5~7 mice in each group.
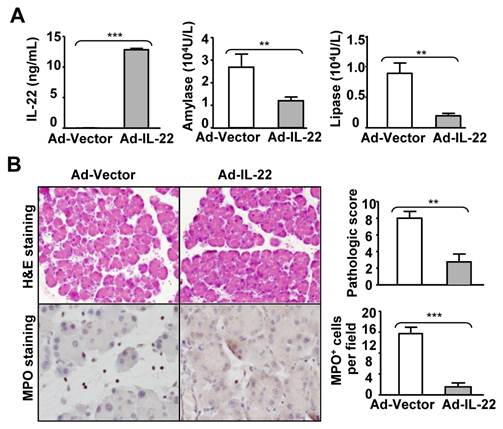
Treatment with adeno-IL-22 ameliorates chronic cerulein-induced pancreatitis in mice. C57BL6 mice received a set of seven injections of cerulein weekly for four weeks. Ad-vector or Ad-IL-22 was injected on the 3rd week. Mice were sacrificed one hour after the last cerulein injection. Serum levels of IL-22, amylase and lipase levels were measured (A). Representative H&E and MPO staining are shown in the left panel of (B). The pathology scores and the number of MPO+ cells are summarized in the right panel of (B). ** P < 0.01, *** P < 0.001. Data are representative of three different experiments, 3~5 mice in each group.
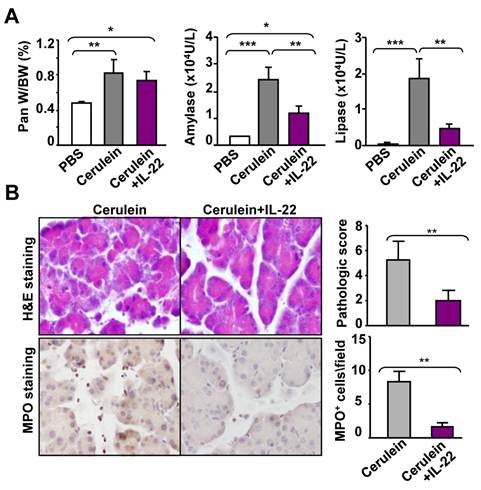
Treatment with recombinant mouse IL-22 (rmIL-22) or adeno-IL-22 protects mice from cerulein-induced acute and chronic pancreatitis
To evaluate the therapeutic potential of IL-22 in acute pancreatitis, we injected mice with rmIL-22 protein two hours before the first cerulein injection. As illustrated in Fig. 3A, pretreatment with rmIL-22 did not affect cerulein-mediated increase in the pancreas/body weight ratio, but markedly diminished the elevation of serum amylase and lipase levels. Consistently, cerulein-induced tissue damage and inflammatory cell infiltration were also greatly ameliorated by rmIL-22 treatment, as indicated by H&E staining and MPO staining (Fig. 3B).
Treatment with recombinant murine IL-22 (rmIL-22) protein ameliorates cerulein-induced acute pancreatitis in mice. Recombinant IL-22 protein was administrated to C57BL/6 mice two hours before the first of seven hourly cerulein. Mice were sacrificed one hour after the last cerulein injection. The ratio of pancreas weight to body weight and serum levels of amylase and lipase were measured (A). Representative examples of H&E and MPO staining are shown in the left panel of (B), and the pathology score and the number of MPO+ cells are summarized in the right panel of (B). **P<0.05, **P < 0.01, and ***P<0.001. Data are representative of three different experiments, 5~7 mice in each group.
The therapeutic efficacy of IL-22 was also tested in a cerulein-induced chronic pancreatitis model. Mice were repeatedly injected seven times with cerulein once a week for four weeks and received adenovirus-IL-22 (Ad-IL-22) or adenovirus-vector (Ad-vector) during the third week. Serum IL-22 levels were undetectable in the Ad-vector-treated mice but had risen to high levels (10 ng/ml) after Ad-IL-22 administration (Fig. 4A). Chronic cerulein treatment elevated serum amylase and lipase levels, which were markedly reduced by Ad-IL-22 treatment (Fig. 4A). H&E and MPO staining indicated that Ad-IL-22 treatment also markedly ameliorated chronic cerulein-induced pancreatic damage and MPO+ neutrophil infiltration compared to the Ad-vector-treated group (Fig. 4B).
In summary, the above data clearly indicate that IL-22 treatment ameliorates cerulein-induced acute and chronic pancreatitis.
The protective role of IL-22 in pancreatitis is mediated by the induction of Bcl-2 and Bcl-XL, which bind to beclin-1 and subsequently inhibit autophagy
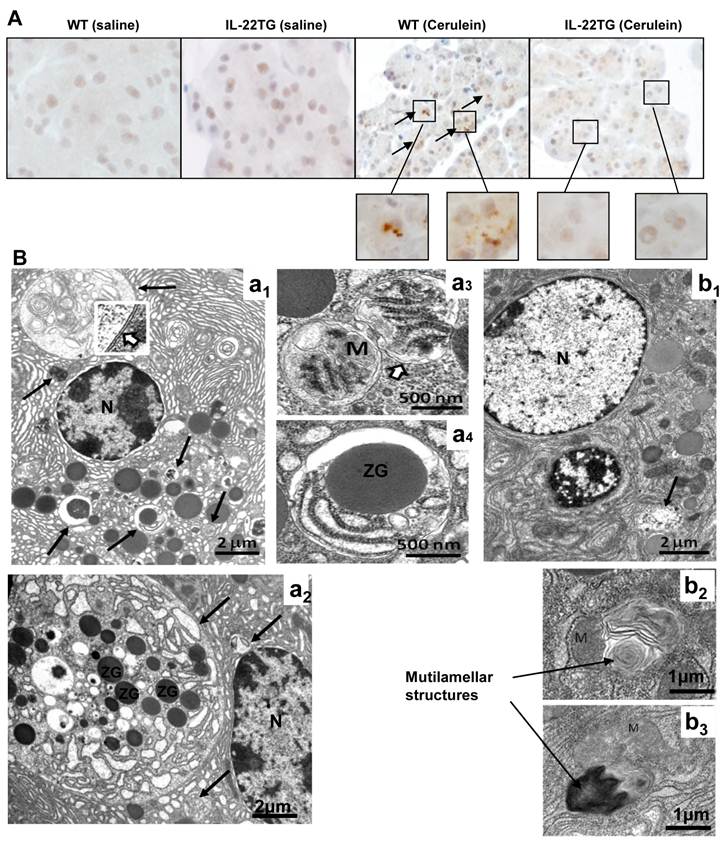
Although the mechanisms underlying pancreatitis are not fully understood, accumulating evidence has suggested that autophagosome formation and impaired autophagic flux due to deficient lysosomal degradation are important mechanisms contributing to the pathogenesis of pancreatitis [19, 21-23]. The accumulation of large vacuoles in acinar cells is a prominent observation in human pancreatitis samples and in various rodent models of pancreatitis [19]. The deletion of ATG5, an important component for autophagosome formation, prevents cerulein-induced acute pancreatitis, suggesting that excessive autophagy leads to intra-acinar trypsinogen activation [24]. In agreement with previous findings, immunohistochemical and electronic microscopic analyses indicate that repeated cerulein injection results in acute pancreatitis with accumulation of LC-3 staining and large autophagosomes in WT mice (Figs. 5A-B). In contrast, the number and size of autophagosomes are markedly reduced in IL-22TG mice (Figs. 5A-B). Interestingly, acinar cells from cerulein-treated IL-22TG mice often exhibited numerous multilamellar structures closely associated with or opposed to mitochondria which have been described as an early event in formation of autopagosomes [25]. An abundance of such structures along with the scarcity of their later, more mature stages and the lack of large autopagosome suggests that IL-22 might affect membrane biogenesis in autophagy, thereby blocking cerulein-induced autophagosome formation (b2 and b3 in Fig. 5B).
Reduced autophagosome formation in the pancreases of cerulein-treated IL-22TG mice. WT and IL-22TG mice received seven cerulein or PBS injections and were sacrificed one hour after the last injection. Pancreases were collected for immunohistochemical or electron microscopy analyses. (A) Representative immunohistochemical staining with anti-LC3 antibody. Arrows indicate the LC3-postive vesicles. (B) Electron micrographs of pancreatic tissues from WT (a1, a2, a3, a4) and IL-22TG (b1, b2, b3) mice treated with cerulein. (a1 and a2) Acinar cells from cerulein-treated WT mice contain one large and many small autophagosomes at different stages of maturation (long arrows). (a3 and a4) higher magnification of autophagosomes containing mitochondria (M) and other intracellular organelles such as zymogen (ZG). Please note the double membrane (insert in a1) surrounding autophagosomes. (b1) In contrast to WT, acinar cells from cerlulein-treated IL-22TG mice display only a few small autophagosomes (long arrow). (b2 and b3) mutilammellar structure can only be found in acinar cells from cerulein-treated IL-22TG mice.
Little is known about how repeated cerulein injections induce autophagosome formation, although several factors, such as VMP1, USP9x, and Beclin-1, have been implicated [26-28]. Beclin-1 has been reported to either stimulate autophagy (autophagosome formation) by interacting with hVps34 or inhibit it by interacting with Bcl-2, Bcl-XL, and Mcl-1 [29-31]. To explore the underlying mechanisms by which cerulein-induced autophagosome formation was inhibited in IL-22TG mice, we analyzed these authophagy-related proteins by Western blot analyses. As shown in Figure 6A, the expression levels of the pSTAT3, STAT3, Bcl-2, and Bcl-XL proteins were much higher in the pancreases from IL-22TG mice compared to WT mice. In contrast, the expression levels of the Mcl-1, VMP1, and USP9x proteins in the pancreas were similar between these two groups.
Upregulation of Bcl-2 and Bcl-XL, which bind Beclin-1, was associated reduced autophagosome formation in the pancreases of cerulein-treated IL-22TG mice. (A) Western blot analyses of pancreatic protein extracts from WT and IL-22TG mice. (B) Immunoprecipitation analyses of the interaction of Beclin-1 with Bcl-2 family proteins. (C) Scheme of the mechanisms underlying IL-22 inhibition of autophagy and pancreatitis. Data are representative of three different experiments.
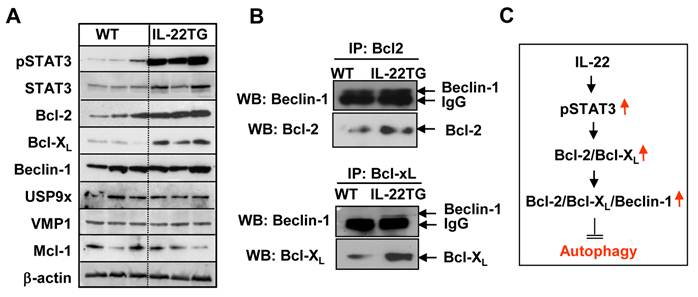
Moreover, immunoprecipitation assays showed that higher levels of Bcl-1 or Bcl-XL bind Beclin-1 in the pancreases from IL-22TG mice compared to WT mice (Fig. 6B), which is likely due to upregulated expression of Bcl-2 and Bcl-XL proteins in the IL-22TG mice. These findings suggest that IL-22 upregulates the expression of Bcl-2 and Bcl-XL and subsequently enhances the interaction of Bcl-2 or Bcl-XL with Beclin-1, thereby inhibiting autophagy (autophagosome formation) and pancreatitis.
Discussion
In this study, we showed that IL-22 effectively protected mice from cerulein induced acute or chronic pancreatitis. This is likely mediated via the induction of Bcl-2 and Bcl-XL gene expression in pancreas and subsequent reduction of autophagosome formation, a critical step in the initialization of pancreatitis [19, 21-23].
Accumulating evidence suggests that autophagy in acinar cells is a critical process in the pathogenesis of pancreatitis. Autophagy comprises two key sequential steps: first the formation of autophagosomes that sequester the organelles destined for degradation; and second the fusion of autophagosomes with lysosomes to form autolysosomes where materials are degraded by hydrolases including the cathepsin family of proteases. Recent studies have suggested that impaired autophagic influx due to deficient lysosomal degradation is an important mechanism that contributes to intra-acinar trypsin activation and pancreatitis [19, 21, 23]. In addition, mice deficient in the ability to form autophagosomes are resistant to acute pancreatitis [24], suggesting that formation of autophagosomes is also an important step to develop pancreatitis. In our study, we found that cerulein-induced autophagosime formation was markedly diminished in IL-22TG mice compared with WT mice (Figs. 5A-B), indicating that the protective effect of IL-22 on cerulein-induced pancreatitis is mediated by inhibiting autophagosome formation.
Next we asked how IL-22 inhibits autophagy in cerulein-induced pancreatitis. To date, three major pathways that regulate autophagy have been identified. These include the inhibitory mammalian target of rapamycin (mTOR) pathway, the stimulatory beclin-1 (also called ATG6) pathway, and the stimulatory LC3-II/Atg5-Atg12-Atg16 pathway [22]. Recent studies have suggested that Bcl-2 and Bcl-XL play an important role in inhibiting the stimulatory beclin-1 pathway by binding to beclin-1 and subsequently inhibiting autophagy [30].
Several previous studies have demonstrated that STAT3 activation induces the expression of Bcl-2 and Bcl-XL in a variety of cell types via transcriptional upregulation of these genes [32]. Thus, we speculated that upregulated Bcl-2 and Bcl-XL in the pancreases of IL-22TG mice are likely due to the enhanced IL-22 activation of STAT3. This hypothesis was supported by our experimental data that expression of pSTAT3, Bcl-2 and Bcl-XL levels was much higher in the pancreas of IL-22TG mice than that in WT mice (Fig. 6A). Moreover, immunoprecipitation assays in Fig. 6B showed that binding of Bcl-2 or Bcl-XL with Beclin 1 was enhanced in the pancreas from IL-22TG mice compared with WT mice. Collectively, our findings suggest that IL-22 activates STAT3 and then upregulates the STAT3 downstream genes such as Bcl-2 and Bcl-XL. Increased Bcl-2 and Bcl-XL subsequently binds to Beclin1 and inhibits autophagosome formation, thereby ameliorating acinar cell damage in cerulein-induced pancreatitis (Fig. 6C).
Chronic alcohol drinking is a leading cause for both acute and chronic pancreatitis [1-3]. Thus, further studies of the protective effects of IL-22 on alcoholic pancreatitis are urgently needed. Alcoholic pancreatitis is attributed to its non-oxidative metabolism by phospholipase D (PLD) to fatty acid ethyl esters (FAEEs) generated in the pancreatic acinar cells [33, 34]. It will be interesting to determine whether IL-22 affects the interaction between PLD activation and formation of FAEEs (or phosphatidylethanol), thereby ameliorating alcoholic pancreatitis. In addition, rats fed ethanol diet for 14 weeks, then received a single lipopolysaccharide injection showed depletion of several lysosomal proteins including lysosomal-associated membrane protein-2 (Lamp-2), an essential lysosomal membrane protein that is required for the proper fusion of autophagosomes with lysosomes [35]. Accordingly, Lamp-2 depletion is reported to inhibit the fusion of lysosomes and autophagosomes, thus causing the accumulation of autophagosomes [36, 37]. Importantly, patients with alcoholic pancreatitis also exhibit local Lamp-2 depletion, which highlights the important role of Lamp-2 in inhibiting autophagy and pancreatic acinar cell death [35]. It will be important to examine whether IL-22 regulates Lamp-2 gene expression and subsequently ameliorates alcoholic pancreatitis.
In summary, IL-22 treatment appears to have beneficial effects on acute and chronic pancreatitis with the added benefit of potentially few side effects due to the restricted expression of IL-22R [38]. Clinical trials examining IL-22 treatment for pancreatitis are therefore a reasonable next step.
Abbreviations
adeno-IL-22: IL-22 adenovirus; adeno-vector: control vector adenovirus; IL-22TG: IL-22 transgenic mice; LC3: Microtubule-associated protein 1A/1B-light chain 3; EM: Electron microscope; FAEEs: fatty acid ethyl esters; MPO: myeloperoxidase; STAT3: signal transducer and activator of transcription 3; PLD: phospholipase D
Acknowledgements
We thank the Electron Microscopy Facility at NINDS, NIH for technical support.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Harper SJ, Cheslyn-Curtis S. Acute pancreatitis. Ann Clin Biochem. 2011;48:23-37
2. Braganza JM, Lee SH, McCloy RF, McMahon MJ. Chronic pancreatitis. Lancet. 2011;377:1184-1197
3. Apte MV, Pirola RC, Wilson JS. Mechanisms of alcoholic pancreatitis. J Gastroenterol Hepatol. 2010;25:1816-1826
4. Lankisch PG, Breuer N, Bruns A, Weber-Dany B, Lowenfels AB, Maisonneuve P. Natural history of acute pancreatitis: a long-term population-based study. Am J Gastroenterol. 2009;104:2797-2805
5. Dervenis C, Johnson CD, Bassi C, Bradley E, Imrie CW, McMahon MJ. et al. Diagnosis, objective assessment of severity, and management of acute pancreatitis. Santorini consensus conference. Int J Pancreatol. 1999;25:195-210
6. Petrov MS, Shanbhag S, Chakraborty M, Phillips AR, Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology. 2010;139:813-820
7. Gaisano HY, Gorelick FS. New insights into the mechanisms of pancreatitis. Gastroenterology. 2009;136:2040-2044
8. Dumoutier L, Louahed J, Renauld JC. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol. 2000;164:1814-1819
9. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17-31
10. Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol. 2011;23:159-163
11. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454-467
12. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332-1342
13. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647-659
14. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S. et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. 2011;54:252-261
15. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947-957
16. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534-544
17. Aggarwal S, Xie MH, Maruoka M, Foster J, Gurney AL. Acinar cells of the pancreas are a target of interleukin-22. J Interferon Cytokine Res. 2001;21:1047-1053
18. Jiang R TZ, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900-909
19. Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P. et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340-3355
20. Wang H, Park O, Lafdil F, Shen K, Horiguchi N, Yin S. et al. Interplay of hepatic and myeloid signal transducer and activator of transcription 3 in facilitating liver regeneration via tempering innate immunity. Hepatology. 2010;51:1354-1362
21. Fortunato F, Kroemer G. Impaired autophagosome-lysosome fusion in the pathogenesis of pancreatitis. Autophagy. 2009;5:850-853
22. Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895-1908
23. Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy. 2010;6:428-429
24. Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S. et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181:1065-1072
25. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656-667
26. Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL. et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem. 2011;286:8308-8324
27. Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A. et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem. 2007;282:37124-37133
28. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672-676
29. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927-939
30. Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600-606
31. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P. et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527-2539
32. Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97-105
33. Werner J, Laposata M, Fernandez-del Castillo C, Saghir M, Iozzo RV, Lewandrowski KB. et al. Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology. 1997;113:286-294
34. Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y. et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706-716
35. Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G. et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350-360
36. Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. Embo J. 2007;26:313-324
37. Saftig P, Tanaka Y, Lullmann-Rauch R, von Figura K. Disease model: LAMP-2 enlightens Danon disease. Trends Mol Med. 2001;7:37-39
38. Wolk K, Sabat R. Interleukin-22: A novel T- and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. 2006;17:367-380
Author contact
![]() Corresponding author: Bin Gao, M.D., Ph.D., NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892. E-mail: bgaonih.gov. This work was supported by the intramural program of NIAAA, NIH.
Corresponding author: Bin Gao, M.D., Ph.D., NIAAA/NIH, 5625 Fishers Lane, Bethesda, MD 20892. E-mail: bgaonih.gov. This work was supported by the intramural program of NIAAA, NIH.