International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
INTRODUCTION
TGF-β SIGNALING IN...
BMP SIGNALING IN OSTEOBLAST...
NEGATIVE REGULATION OF...
TGF-β/BMP SIGNALING IN...
SUMMARY AND PERSPECTIVE
ABBREVIATIONS
Acknowledgements
References
INTRODUCTION
TGF-β SIGNALING IN...
BMP SIGNALING IN OSTEOBLAST...
NEGATIVE REGULATION OF...
TGF-β/BMP SIGNALING IN...
SUMMARY AND PERSPECTIVE
ABBREVIATIONS
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(2):272-288. doi:10.7150/ijbs.2929 This issue Cite
Review
TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation
Guiqian Chen1,3, Chuxia Deng2 ![]() , Yi-Ping Li1,3
, Yi-Ping Li1,3 ![]()
1. Institute of Genetics, Life Science College, Zhejiang University, 388 Yuhang Road, Hangzhou 310058, China
2. Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA.
3. Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL, 35294-2182, USA.
Received 2011-4-25; Accepted 2011-12-29; Published 2012-1-21
Citation:
Chen G, Deng C, Li YP. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int J Biol Sci 2012; 8(2):272-288. doi:10.7150/ijbs.2929. https://www.ijbs.com/v08p0272.htm
Other stylesAbstract
Transforming growth factor-beta (TGF-β)/bone morphogenic protein (BMP) signaling is involved in a vast majority of cellular processes and is fundamentally important throughout life. TGF-β/BMPs have widely recognized roles in bone formation during mammalian development and exhibit versatile regulatory functions in the body. Signaling transduction by TGF-β/BMPs is specifically through both canonical Smad-dependent pathways (TGF-β/BMP ligands, receptors and Smads) and non-canonical Smad-independent signaling pathway (e.g. p38 mitogen-activated protein kinase pathway, MAPK). Following TGF-β/BMP induction, both the Smad and p38 MAPK pathways converge at the Runx2 gene to control mesenchymal precursor cell differentiation. The coordinated activity of Runx2 and TGF-β/BMP-activated Smads is critical for formation of the skeleton. Recent advances in molecular and genetic studies using gene targeting in mice enable a better understanding of TGF-β/BMP signaling in bone and in the signaling networks underlying osteoblast differentiation and bone formation. This review summarizes the recent advances in our understanding of TGF-β/BMP signaling in bone from studies of genetic mouse models and human diseases caused by the disruption of TGF-β/BMP signaling. This review also highlights the different modes of cross-talk between TGF-β/BMP signaling and the signaling pathways of MAPK, Wnt, Hedgehog, Notch, and FGF in osteoblast differentiation and bone formation.
Keywords: Osteoblasts, Bone, TGF signaling, BMP signaling, Smad, Runx2
INTRODUCTION
Bone is formed through two distinct phases: endochondral ossification, in which a cartilage model is replaced by bone, and intramembranous ossification, in which bones are shaped directly from condensations of mesenchymal cells without a cartilage intermediate (1). Bone is continuously remodeled throughout life and an imbalance in this process can result in bone disease. The integrity and function of bone are maintained by an exquisite balance between osteoblasts and osteoclasts, the two major bone cells involved in the bone remodeling process. Osteoblasts are responsible for bone formation, while osteoclasts are the bone-resorbing cells (1-2).
The transforming growth factor-beta (TGF-β) superfamily is comprised of over forty members, such as TGF-βs, Nodal, Activin, and bone morphogenetic proteins (BMPs) (3). TGF-β signaling first transmits signals across the plasma membrane through the formation of heteromeric complexes of specific type I and type II serine/threonine kinase receptors. The type I receptor is phosphorylated following the activation of specific type II receptor (4). Activated type I receptors initiate intracellular signaling through phosphorylation of specific Smad proteins, R-Smads. Activated R-Smads form a complex with co-Smad and Smad4 and then translocate into the nucleus to direct transcriptional response (5) (Figure 1, 2).
TGF-β/BMPs have widely recognized roles in bone formation during mammalian development and exhibit versatile functions in the body (6-7). For instance, a BMP morphogen gradient is established in a multicellular embryo, and BMP drives the differentiation of ectodermal cells and mediates dorsal patterning to establish dorsal-ventral axis (8). Disruptions of TGF-β/BMP signaling have been implicated in multiple bone diseases including tumor metastasis, brachydactyly type A2, and osteoarthritis (9-11). In this review, we will summarize novel discoveries from recent decades regarding the regulations of TGF-β/BMP signaling in bone as well as the different modes of cross-talk between TGF-β/BMP signaling and the signaling pathways of MAPK, Wnt, Hedgehog, Notch, and FGF. These new insights into TGF-β/BMP signaling on bone will open new prospects for generating novel therapies against clinical disorders.
Figure 1
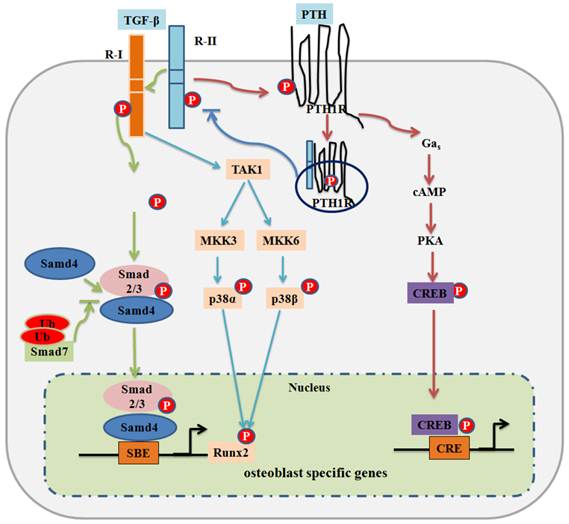
TGF-β signaling and negative regulation in bone formation. Canonical Smad-dependent TGF-β signaling first binds to receptor type II (R-II) and receptor type I (R-I), and then signaling transduces to their Smads. Activated Smads form a complex with Smad4 and then translocate into the nucleus where they interact with other transcription factors to trigger target gene expression. Smad7 disrupts the activated Smad2/3 to form a complex with Smad4. The non-Smad-dependent TAK1 signaling pathway also regulates bone formation. PTH binding activates PTH1R to stimulate several downstream effectors. PTH binding also drives internalization of PTH1R-TGFβRII complex, which attenuates both TGF-β and PTH signaling on bone development. Transcriptional factor cAMP response element binding protein (CREB) mediates PTH signaling in osteoblasts. P: phosphorylation; Ub: ubiquitination.
Figure 2
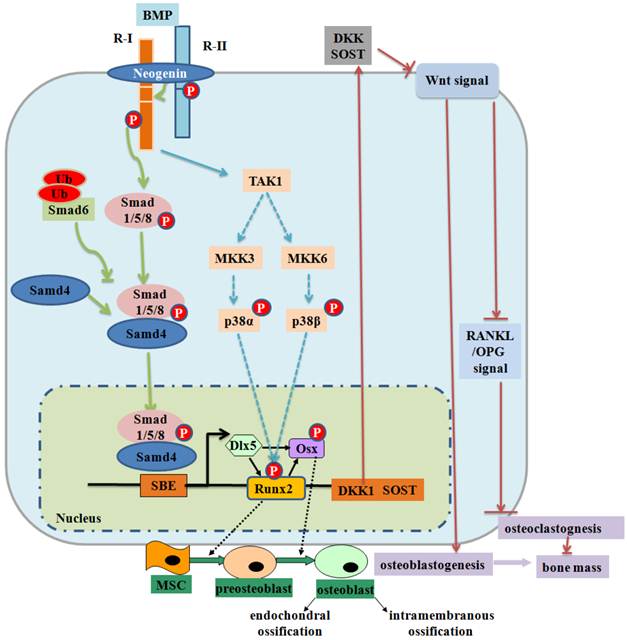
BMP signaling and negative regulation in bone formation. Smad-dependent-BMP signaling binds to receptor type II (R-II) and receptor type I (R-I) and then the signaling transduces to their Smads. Activated Smads form a complex with Smad4 and then translocate into the nucleus where they interact with other transcription factors to trigger target gene expression. Neogenin regulates BMP receptor association and Smad1/5/8 signaling. Activated Smads regulate expression of transcriptional factors and transcriptional coactivators important in osteoblasts (Dlx5, Runx2 and Osx). Smad6 binds type I BMP receptor and prevents Smad1/5/8 to be activated. Non-Smad-dependent TAK1 signaling pathway also regulates bone formation. The interplay between BMPs and Wnt signaling affects bone formation (99). BMPRIA signaling upregulates Sost expression primarily through Smad-dependent signaling, while it upregulates DKK1 through Smad-dependent and non-Smad-dependent signaling. Both Sost and DKK1 inhibit canonical Wnt signaling, leading to a decrease in bone mass. P: phosphorylation; Ub: ubiquitination.
TGF-β SIGNALING IN OSTEOBLAST DIFFERENTIATION AND BONE FORMATION
Autocrine and paracrine stimulation by TGF-β is important in the maintenance and expansion of the mesenchymal stem/progenitor cells, the progenitors of osteoblasts (12). Bone and cartilage contain large amounts of TGF-β and target cells for TGF-β activity. At earlier developmental stages, osteoblast-enriched populations from fetal bone are more sensitive to the mitogenic effect of TGF-β than similar populations from newborns (13). Furthermore, TGF-β signaling also promotes osteoprogenitor proliferation, early differentiation, and commitment to the osteoblastic lineage through the selective MAPKs and Smad2/3 pathways, and the cooperation between TGF-β and PTH, Wnt, BMP, as well as FGF signaling.
Canonical TGF-β signaling functions in bone (TGF-βs, receptors, Smads)
TGF-β isoforms and their receptors, type I receptor (TGFβRI or ALK5) and type II receptor (TGFβRII or Tgfbr2), play important roles in endochondral and intramembranous ossification (Table 1). TGF-β1 deficient mice display reduced bone growth and mineralization (14). TGF-β2 and TGF-β3 double knockout mice display a lack of distal parts of the rib (15). Mice carrying tissue-specific removal of TGFβRI using Dermo1-Cre have short and wide long bones, reduced bone collars, and reduced trabecular bone (16). Tgfbr2 exhibits functions in the maintenance of boundaries in the sclerotome and developing axial skeleton (17). Conditional transgenic mice with a dominant negative form of Tgfbr2 (dnTgfbr2) developed hypoplastic cartilage (18). Deletion of Tgfbr2 via Col2a1-Cre in mice causes multiple defects in the base of the skull and in the vertebrae (19). Removal of Tgfbr2 driven by Prx-Cre results in defects in the long bones, joints (20), and skull vault (21), indicating that Tgfbr2 has a critical role in both intramembranous bone formation and endochondral bone formation. Further evidence from Wnt1-Cre mediated tissue specific disruption, shows that the loss of Tgfbr2 signaling leads to defects in cranial neural crest cells (CNCC)-derived osteoprogenitor cells during intramembranous bone formation (22) (Table 1).
Table 1
Conditional knockout models on TGF-β/BMP signaling on bone formation
| Gene | Cre | Defects | Refer |
|---|---|---|---|
| Tgfbr2 | Col2a1-Cre | obvious defects in long bone formation | (18) |
| Wnt1-Cre | osteogenic cell proliferation and differentiation | (22) | |
| Prx1-Cre | short limbs and fusion of the joints in the phalanges | (20) | |
| Wnt1-Cre | severe defects in mandibular development | (170) | |
| Col2a-Cre | defects in the base of the skull and in the vertebrae | (19) | |
| ALK5 | Dermo1-Cre | the short bones and ectopic cartilaginous protrusions | (16) |
| TAK1 | Prx1-Cre | novel embryonic developmental cartilage defects | (171) |
| Osx-cre | clavicular hypoplasia and delayed fontanelle fusion | (82) | |
| Smad7 | Prx1-Cre | poor cartilage formation | (172) |
| BMP-7 | Prx1-Cre | normal postnatal limb growth and maintenance of bone mass | (49) |
| Bmp2/Bmp4 | Col2a1-Cre | a severe chondrodysplasia phenotype | (53) |
| Bmp2 | Prx1-Cre | spontaneous fractures | (52) |
| Bmp2/Bmp4 | Prx1-Cre | a severe impairment of osteogenesis | (50) |
| Bmp4 | Prx1-Cre | limb skeletogenesis occurs normally in the absence of BMP-4 | (51) |
| BMPR-II | Prx1-Cre | normal skeleton | (60) |
| BMPR-IA | Col1a1-Cre | increased bone volume | (61) |
| Col1-CreERTM | increased bone mass | (99) | |
| Prx1-Cre | shortened limbs and almost complete agenesis of the autopod | (62) | |
| Smad1 | Col1a1-Cre | osteopenic phenotype | (68) |
| Col2a1-Cre | calvarial bone development delay | (68) | |
| Smad1/5 | Col2a1-Cre | chondrodysplasia | (69) |
| Smad4 | TTR-Cre | died at E7.5-E9.5 without head-fold and anterior embryonic structures | (72) |
| Mu-Cre | misalignment of the cardiac outflow tract | (74) | |
| OC-Cre | lower bone mineral density, decreased bone volume, decreased bone formation rate | (26) | |
| Col2a1-Cre | dwarfism | (41) |
R-Smads that respond to TGF-β receptors are Smad2 and 3. Small C-terminal domain phosphatases (SCP) were reported to be good regulators of Smad2/3 activation. SCP1/2 knockdown inhibits TGF-β transcriptional responses by dephosphorylating Smad2/3 at the linker (inhibitory) but not the C-terminal (activating) site (23). Activation of Smad7 is another way to control the function of R-Smads, which targets the TGF-β receptor for degradation (24). Smad7 mutants that have impaired the ability for the recruitment of Smurf2 to the receptors are compromised in their inhibitory activity (25). Smad7 also competes with Smad2/3 to form complexes with Smad4, a common mediator for TGF-β signaling, which plays an essential role in coupling bone formation and bone resorption and maintaining normal postnatal bone homeostasis (26).
Recently, Smad2/3 were reported to directly associate with the TRAF6-TAB1-TAK1 molecular complex (27). Once TGF-β signaling is blocked, the TRAF6-TAB1-TAK1 molecular complex is not observed, indicating that TGF-β1 is indispensable in RANKL-induced osteoclastogenesis (28). TGF-β1 promotes matrix production and osteoblast differentiation while it reduces the ability of osteoblasts to secrete osteoclast differentiation factor RANKL, thereby TGF-β1 indirectly limits further osteoclast formation and may affect bone mass.
Non-canonical TGF-β signaling functions in bone
The non-Smad-dependent signaling pathway also contributes greatly to osteoblast differentiation and bone formation. The study of tamoxifen-inducible Cre-ER-mediated ALK5-deficient primary calvarial cell culture revealed that TGF-β signaling promotes osteoprogenitor proliferation, early differentiation, and commitment to the osteoblastic lineage through the selective MAPKs and Smad2/3 pathways (16). TGF-β activation kinase1 (TAK1) and TAK1 binding protein 1 (TAB1) play a pivotal role as upstream signal transducers by activating the MKK3-p38 MAPK signaling cascade that leads to the induction of type I collagen expression by TGF-β1. TAK1 has a novel function in the regulation of the steady-state protein levels of MKK3 and p38 MAPK (29) (Figure 1). Recent results demonstrate that following TGF-β induction, both the Smad and p38 MAPK pathways converge at the Runx2 gene to control mesenchymal precursor cell differentiation (30). In addition, ERK and p38 also differentially mediate TGF-β and BMP-2 function in osteoblasts (31). TGF-β2-induced activation of ERK-MAPK is an important signaling component that stimulates cell proliferation to enrich osteoprogenitor cells, thereby promoting their differentiation into osteoblasts to achieve a rapid calvarial bone expansion (32).
Interplay between TGF-β signaling and PTH, Wnt, FGF, as well as BMP signaling in bone
TGF-β and PTH signaling in bone
Mice with Tgfbr2 deleted in osteoblasts have increased bone mass due to the hyperactivition of PTH type I receptor (PTH1R) (33-34). Disruption of PTH signaling by injection of PTH (7-34) or ablation of PTH1R rescues the bone phenotype of these mutant mice (33). Molecular study shows that Tgfbr2 directly phosphorylates the PTH1R cytoplasmic domain. PTH couples the processes of bone resorption and formation by enforcing simultaneous internalization of Tgfbr2 and PTH1R (34) (Figure 1), which attenuates both TGF and PTH signaling in vivo. It is recognized that transcriptional factor cAMP response element binding protein (CREB) mediates PTH signaling in osteoblasts, and PTH-CREB signaling pathway acts as an effective activator of BMP-2 expression (35).
TGF-β and FGF signaling in bone
TGF-βs and FGF-2, -4, and -6 have been proven to be inducers of osteoblast proliferation (a higher extent for TGF-β and FGF-2) and inhibitors of alkaline phosphatase (ALP) activity and osteoblast mineralization (36), indicating potential application for in vitro bone growth induction in bone tissue engineering. In addition, FGF acts downstream of TGF-β signaling in regulating CNC cell proliferation and exogenous FGF-2 rescues the cell proliferation defect in the frontal primordium of Tgfbr2 mutants, demonstrating the biological significance of the TGF-β-mediated FGF signaling cascade in regulating frontal bone development (37). Reports also show that FGF/FGFR3 signals mediate some of the effects of TGF-β on embryonic bone formation (38) (Table 3).
TGF and Wnt signaling in osteoblast
TGF-β cooperates with Wnt signaling, and promotes osteoblast differentiation of human mesenchymal stem cells (hMSCs). Knockdown of β-catenin with siRNA stimulated ALP activity and antagonized the inhibitory effects of TGF-β1 on bone sialoprotein expression. TGF-β1 activates β-catenin signaling via ALK5, Smad3, PKA, and PI3K pathways, and modulates osteoblastogenesis via ALK5, PKA, and JNK pathways in hMSCs (39). Recent studies in differentiating osteoblasts indicate that Wnt pathway induction stabilizes β-catenin and increases TCF/LEF-dependent gene expression in parallel with β-catenin-independent complex formation between TCF-4 and Runx2. Activation of either Runx2 or TCF-4 coenhances TCF and Runx2 activity and increases TGFβRI expression. Overall, Wnt pathway induction has complex stimulatory and inhibitory effects on TGF-β activity (40) (Table 3).
TGF-β and Ihh signaling in bone
Expression of Indian hedgehog/parathyroid hormone-related protein (Ihh/PTHrP) signaling in the growth plate is decreased in Smad4 mutant mice. The cultured mutant metatarsal bones failed to respond to TGF-β1. These findings indicate that Smad4-mediated TGF-β signals are required for maintaining the normal organization of chondrocytes in the growth plate (41), which is important for normal endochondral ossification.
Interplay between TGF-β and BMP signaling in osteoblast and bone
TGF-β1 strongly enhances ectopic bone formation induced by BMP-2, with the resulting bone volume being five-fold greater than that induced by BMP-2 alone (42). Evidence shows that increased BMPR-IB by TGF-β1, FGF-2, and PDGF-AB significantly enhances BMP-2-induced osteogenic functions in vitro (43). Results demonstrate that BMPRII and ActRII are the functional type II TGF-β receptors in BMP-9-induced osteogenic differentiation of C3H10T1/2 cells (44), indicating a strong connection between TGF-β1 and BMP signaling in osteoblast differentiation (Table 3).
BMP SIGNALING IN OSTEOBLAST DIFFERENTIATION AND BONE FORMATION
BMPs have widely recognized roles in bone formation during mammalian development and exhibit versatile functions in the body, indicating potential for therapeutic use. Studies have elicited BMPs in mice to promote the rehabilitation of critical-size bone defects, which renders them useful in the field of tissue engineering and regeneration. Recent discoveries will shed light on better understanding of intrinsic BMP signaling and the crosstalk with Wnt, Notch, FGF and Hh signaling on osteoblast differentiation and bone formation.
Canonical BMP signaling in bone (BMPs, receptors, Smads)
BMP-2, 4, 5, 6, and 7 all have strong osteogenic capacity. Addition of BMP-2 vastly increases osteocalcin (45) and a short-term expression of the BMP-2 is necessary and sufficient to irreversibly induce bone formation (46). BMP-7 induced the expression of osteoblastic differentiation markers such as ALP activity and accelerated calcium mineralization (47-48). In vivo genetic studies using a Prx1-cre model demonstrated that the absence of locally produced BMP-7 has no effect on postnatal limb growth and maintenance of bone mass, indicating other BMPs present in adult bone are sufficient to compensate for the absence of BMP-7 (49). Loss of both BMP-2 and BMP-4 resulted in severe impairment of osteogenesis (50). However, limb skeletogenesis occurred normally despite the absence of BMP-4, suggesting that BMP-4 is not required for bone formation and function in the limb (51). Mice lacking the ability to produce BMP-2 in their limb bones have spontaneous fractures that do not resolve with time, other osteogenic stimuli cannot compensate for the absence of BMP-2 (52). BMP-2, not BMP-4, plays a crucial role for chondrocyte proliferation and maturation during endochondral bone development (53) (Table 1).
Trabecular bone volume of BMP-3-deficient mice is two-fold greater than that of wild-type mice (54). BMP-3 limits skeletal progenitor cell differentiation to mature osteoblasts, herein regulating adult bone mass (55). Overexpression of BMP-3 in chick wing bud reduces BMP signaling results in expanded skeletal elements (56). BMP-3 transgenic mice are subject to spontaneous rib fractures and have altered signaling through activin receptor type IIB (ActRIIB) in chondrocytes and the periosteum (57).
BMP receptors, such as BMPR-II, differentially modulate the responsiveness of target genes to BMP-2 (58). BMPR-II and ActR-IIB are able to compensate each other functionally in mediating BMP-2 signaling and BMP-2-induced osteoblast differentiation in 2T3 cells (59). Normal skeletons develop in mice with BMPR-II deletion via Prx1-Cre, indicating that BMPR-II is not required for limb development or that the loss of BMPR-II is compensated by BMP utilization of other type II BMP receptors (60). Conversely, mice with deletion of BMPR-IA through Col1-Cre have increased bone volume (61), shortened limbs, almost complete agenesis of the autopod (62), small body size, irregular calcification and low bone mass (63) (Table 1). Decreased osteoclastogenesis through the RANKL-OPG pathway was reported to contribute to the increased bone mass in trabecular bone (64).
Neogenin, a transmembranous protein, was reported to regulate BMP receptor association with lipid raft, where BMP induces canonical Smad1/5/8 phosphorylation (65-66). Overexpressing BMP signaling through ALK2 will lead to ectopic phosphorylation of Smad1/5/8 (67). Osteoblast-specific Smad1 conditional knockout mice developed an osteopenic phenotype and partial inhibition of BMP signaling (68). Combined loss of Smads 1/5/8 results in severe chondrodysplasia (69) (Table 1). If Smad1 is modified, it can modulate BMP-mediated osteogenesis (70) and the intensity of BMP signals can be determined by BMP receptors via Smad1 C-terminal phosphorylation (71). These finding demonstrated that Smad1/5/8 are intracellular signaling proteins that transduce signals elicited by members of BMP signaling in osteoblasts.
Smad4 is the only common Smad for both TGF-β and BMP signaling. Targeted disruption of Smad4 in mice results in numerous developmental defects and cancer formation in various tissues. Smad4-deficient mice died at E7.5-E9.5 without head-fold and anterior embryonic structures, and with impaired responsiveness to TGF-β-induced gene expression (72). Smad4-mediated TGF-β signaling is also reported to be important in the inhibition of oncogenesis (73). Myocardial deletion of Smad4 in mice causes misalignment of the cardiac outflow tract (74), highlighting essential roles of this co-Smad in mediating signaling of the TGF-β superfamily. Conditional deletion of Smad4 in osteoblast leads to lower bone mineral density, decreased bone volume, decreased bone formation rate, and a reduced number of osteoblasts (26). Control of Smad4 is a good way to regulate bone formation. FAM and Ectodermin/Tif1gamma (Ecto) were reported to respectively regulate the de-ubiquitination and ubiquitination of Smad4 (75-76). On the other hand, Smad6 competes with R-Smad and forms a non-functional complex with Smad4, which will inhibit BMP signaling in bone formation. Smad6 is involved in a negative feedback loop regulating BMP signaling (77) and is required to limit BMP signaling during endochondral bone formation (78). Smad6/Smurf1 double transgenic mice show severely delayed endochondral ossification compared to the Smad6 knockout (79). Smurf1, with its WW domain, specifically binds to the PY motif of Smad6 and transports Smad6 into the cytoplasm. When Smad6 is in its rest state, it is mainly localized in the nucleus (80).
Non-canonical BMP signaling function in bone
The importance of canonical BMP signaling is well established, but the necessity for non-canonical (Smad-independent) signaling during these processes is also important. TAK1 is a factor that is involved in the fine-tuning of BMP effects during osteogenic development (81). Osteoblast-specific deletion of Tak1 resulted in clavicular hypoplasia and delayed fontanelle fusion, a phenotype similar to the cleidocranial dysplasia observed in human haploinsufficient for Runx2 (82) (Table 1). Mechanistic analysis revealed that the TAK1-MKK3/6-p38 MAPK axis phosphorylated Runx2, promoting its association with the coactivator CREB-binding protein (CBP), which is required to regulate osteoblast genetic programs (82). Surprisingly, deletion of TAK1 seems to affect not only activation of the p38 MAPK signaling cascade, but also activation of the BMP-responsive Smad1/5/8 (83). Smad and MAPK pathways act synergistically in the BMP pathway controlling limb development (84). The coordinated activity of Runx2 and BMP/TGF-β-activated Smads is critical for the formation of the skeleton. TGF-β/BMP2 signaling, MAPK dependent phosphorylation, and Runx2 subnuclear targeting converge to induce the osteogenic phenotype (85).
Crosstalk between BMP and Notch, Hh, FGF, as well as Wnt signaling in osteoblasts and bone
Runx2, Dlx5, and Osx in osteoblasts and bone
Runx2 is a master transcription factor, and both endochondral and intramembranous bone formation are totally absent in Runx2-null mice (86). Autocrine BMP production is necessary for the Runx2 transcription factor to be active. Additionally, BMPs and Runx2 cooperatively interact to stimulate osteoblast gene expression (87), and direct evidence shows that Runx2 is essential for the execution and completion of BMP2 signaling for osteoblast differentiation (88). BMP-activated Smads interact with Runx2 to induce osteoblast-specific gene expression in C2C12 cells (86). Smad1 interacts with Runx2 on the promoter of target genes and controls osteoblast gene expression and differentiation (89). Mutations in Runx2 may lead to an autosomal-dominant human bone disease called cleidocranial dysplasia, which is related to impaired Smad signaling and the target the of Runx2 activity during bone formation (90).
However, BMP-Smads can also function independently of Runx2. Osterix (Osx), which is indispensable for preosteoblasts to differentiate into mature osteoblasts and form bone (91), was previously reported to be a Runx2-targeted gene. Osx induction by BMP-2 is completely abrogated by the antisense blocking of Dlx5, indicating BMP-2-induced Osx expression is mainly mediated by Dlx5, but not by Runx2, suggesting that Dlx5 may directly interact with Osx (92). On the other hand, Dlx5 can drive Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme (93), indicating that Dlx5 can work as an upstream gene of Runx2. Recently, Akt, a member of the serine/threonine-specific protein kinase, was found to phosphorylate Osx and Dlx5 (94). Akt activation increases protein stability, osteogenic activity, and transcriptional activity of Osx and Dlx5 (95), revealing that Osx regulates bone formation through different signaling pathways, including a novel mechanism involving Akt during osteoblast differentiation.
BMP and Wnt signaling in bone
Wnt3A and BMP-9 enhanced each other's ability to induce ALP in MSCs (96). In vitro deletion of the β-catenin gene inhibits osteoblast proliferation, alters osteoblast differentiation, and reduces the responsiveness of osteoblasts to BMP-2 treatment (97). Interactions between β-catenin and Runx2 play an important role in BMP-9-induced osteogenic differentiation of MSCs (96). On the other hand, BMPR1A specifically deleted in osteoblasts leads to a significant increase in bone mass, which is partially due to hyperactivated Wnt signaling (98) and increased expression of sclerostin (Wnt receptor antagonist) and Dkk1 activity. It has been shown that both sclerostin and DKK1 act physiologically as downstream molecules of BMP signaling to inhibit canonical Wnt signaling and therefore negatively regulate bone mass (99) (Figure 2). Loss-of-function of either DKK1 or SOST, which are downstream targets of BMPs, causes a high bone mass phenotype in humans and mice, suggesting an important role of DKK1 and SOST for bone mass regulation (100) (Table 3).
BMP and FGF signaling in bone
FGF signaling was reported to interplay with BMP signaling on bone formation. FGF-2 and FGF-9 increased expression of other osteogenic factors BMP-2 and TGF-β1, and endogenous FGF/FGFR signaling is a positive upstream regulator of the BMP-2 gene in calvarial osteoblasts (101). FGF-2 and BMP-2 have a synergistic effect on fracture healing: FGF-2 has a critical function at early stage while BMP-2 promotes mineralization at later stage (102). FGF-2 null mice have impaired nuclear accumulation of Runx2 and hindered BMP-2 induced bone formation and ALP activity (103). Runx2 is an important mediator of the expression of BMP-2 in response to FGF stimulation in cranial bone development (104). FGF and BMP synergy on osteogenesis may be modulated through Rnnx2 activation (Table 3).
BMP and Notch signaling in osteoblasts
Similar synergy is found in Notch and BMP crosstalk: activating Notch signaling enhanced BMP-induced ALP activity and formation of calcified nodules in vitro (105). Notch inhibition results in decreased ALP activity and decreased promoter activity of BMP target genes (106). Notch signal transduction pathway genes, Lfng, Hey1, and Hes1, are differentially regulated by BMP-2 and TGF-β. These genes might function as the focal point for interaction of Smad and Notch signaling during osteoblast differentiation (107). Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation (108), which may be beneficial to osteoblast differentiation.
BMP, Hh, and FGF signaling in osteoblasts and bone
Sonic hedgehog (Shh) is involved in osteoblast differentiation by cooperating with BMP-2 (109). Shh stimulates BMP-2 promoter activity and osteoblast differentiation. The effects of Shh are mediated by Gli2, a powerful activator of BMP-2 gene expression, which is required in turn for normal osteoblast differentiation (110). In the developing axial skeleton, sequential Shh and BMP signals are required for specification of a chondrogenic fate in presomitic tissue (111). On the other hand, BMP activity negatively regulates Shh transcription and a BMP-Shh negative-feedback loop serves to confine Shh expression during limb development (112) (Table 3).
Ihh is found to be required for BMP-induced osteogenesis of a limb-bud cell line in culture. Ihh signaling is directly required for the osteoblast lineage in developing long bones. Ihh functions in conjunction with other factors such as BMPs to induce osteoblast differentiation. In vivo, Ihh acts on potential progenitor cells to promote osteoblast differentiation and prevent chondrocyte differentiation (113). Ihh and BMP synergistically induce ALP activity. Evidence indicates a stimulatory role for osteoblast-expressed Ihh in bone formation in a positive feedback loop (114). Conditional knockout Smad1/5 mice developed chondrodysplasia and exhibited abnormal growth at the end of bones. The molecular mechanism underlying the defects in conditional knockout Smad1/5 mice appears to be an imbalance in the cross-talk between the BMP, FGF, and Ihh/PTHrP pathways (69), indicating that BMP, FGF, and Ihh/PTHrP is important for normal long bone development (Table 3).
NEGATIVE REGULATION OF TGF-β/BMP SIGNALING IN OSTEOBLAST DIFFERENTIATION AND BONE FORMATION
TGF-β and BMP signaling is under elaborate regulation to maintain correct patterning and morphogenesis of various tissues and organs. TGF/BMP signaling in bone is negatively regulated through a number of mechanisms, including extramembranous regulation (ligand antagonist), intracellular regulation (Smurf and inhibitory Smads), and transcriptional regulation (transcriptional repressors and epigenetic control).
Ligand antagonists
Antagonists targeting TGF-βs, such as Chordin (115) and Follistatin (116), and BMPs, such as Noggin (117), have been reported. Noggin, a BMP antagonist, negatively regulates BMP activities during vertebrate dorsal-ventral patterning, skeletogenesis, and joint formation (117). Noggin binds the domain that is required for BMP-7 to interact with BMP type I and type II receptors (118). Transgenic mice with overexpressed Noggin in the bone microenvironment are subject to osteopenia and fractures and exhbiti decreased trabecular bone volume and impaired osteoblastic function (119-120). Noggin suppression may be a novel strategy for the treatment of osteolytic bone metastases (121).
Ubiquitin ligase mediated degradation
Smurf1 and Smurf2 are E3 ubiquitin ligases known to suppress TGF-β signaling through degradation of Smads and receptors for TGF-β and BMPs (122). Smurf1 interacts with BMP-activated Smad1 and 5 and to mediate degradation of these Smad proteins as well as Runx2 (123). Smurf1-deficient mice exhibit an age-dependent increase of bone mass (124). Smurf2 has been reported to form a complex with Smad2 and then target SnoN for degradation (125). Smurf2-transgenic mice exhibited decreased articular cartilage area and subchondral sclerosis (126). In addition, some proteins (e.g. CHIP, carboxyl terminus of Hsc70-interacting protein) inhibit the signaling activities of Smad1/5 by recruiting Smad1/5 from the functional R-/Co-Smad complex and further promoting the ubiquitination and degradation of Smad1/5 in a chaperone-independent manner (127). Other proteins, such as the serine/threonine kinase Fused (Fu), can function in concert with the E3 ligase Smurf to regulate ubiquitination and proteolysis of the BMP receptor (128).
Transcriptional repressors
In the nucleus, transcriptional repressing factors Ski and SnoN play a negative role together with I-Smads (129) to disrupt the formation of the TGF-β-induced functional Smad-DNA complex and thereby inhibit target gene expression (130). Since the Ski-binding surface on Smad4 significantly overlaps with the surface required for binding R-Smad phosphorylated tails, binding to Ski or SnoN interferes with the interaction between Smad4 and phosphorylated R-Smads and disrupts the active heteromeric Smad4/R-Smad complex (131). Ski and SnoN also prevent Smads from binding to the transcriptional coactivator p300/CBP (131) which is important in skeletogenesis.
Epigenetic regulation
Epigenetics is an essential mechanism to control gene expression and fundamental cellular processes. DNA methylation is important in the regulation of ALPL expression through the osteoblast-osteocyte transition (132). Smad2-mediated transcription requires the histone acetyltransferase p300, which is required for the ability of Smad2 to mediate activin and TGF-β signaling (133). Mutation of lysine at Lys-19, Lys-20, and Lys-39 abolished Smad2 acetylation in vivo and prevented nuclear accumulation of Smad2 and subsequent TGF-β and activin responses (134). Smad7 interacts with specific histone deacetylases (HDACs), which are able to deacetylate Smad7 (135). This is significant since acetylation of Smad7 protects it against ubiquitination and degradation mediated by the ubiquitin ligase Smurf1 (136).
Runx2 is a global regulator of osteogenesis and is crucial for regulating the expression of bone-specific genes. Runx2 is controlled by a dynamic equilibrium of acetylation, deacetylation, and ubiquitination. BMP-2 signaling stimulates p300-mediated Runx2 acetylation, which increases transactivation activity and inhibits Smurf1-mediated degradation of Runx2. HDAC4 and HDAC5 deacetylate Runx2 and lead to a Smurf-mediated degradation (137). BMP-induced non-Smad ERK signaling pathway cooperatively regulates osteoblast differentiation, in part, through increasing the stability and transcriptional activity of Runx2 or increasing Runx2 acetylation by p300 (138). On the other hand, studies suggest that NFATc1 acts as a transcriptional co-repressor of osteocalcin promoter possibly in an HDAC-dependent manner (139). These findings demonstrate the importance of epigenetic regulation on bone formation and its important medical implications since TGF-β/BMPs and Runx2 are of great interest with regard to the development of therapeutic agents against bone diseases.
TGF-β/BMP SIGNALING IN DISEASES AND CLINICAL APPLICATIONS
TGF-β/BMP signaling in human diseases
Basic research has contributed greatly to our understanding of human skeletal diseases caused by mutations related to TGF-β/BMP signaling (Table 2). Fibrodysplasia ossificans progressiva (FOP) is a rare disabling disease caused by mutations in ALK2 (140) and characterized by heterotopic ossification (141). Mutations in BMPR1B were recently demonstrated in two affected families with brachydactyly type A2 (BDA2) (142). BDA2 is characterized by hypoplasia/aplasia of the second middle phalanx of the index finger and sometimes the little finger. BDA2 was first described by Mohr and Wriedt in a large Danish/Norwegian kindred and GDF5 (growth and differentiation factor 5) was identified as a novel BDA2 causing gene (142-143). Furthermore, two mutations (N445K, T) of GDF5 in patients developed synostosis syndrome (144). An additional functional mechanism for the pathogenesis of BDA2 is duplication of a regulatory element that affects the expression of BMP2 in the developing limb (145). Mutations in BMP antagonist NOGGIN cause brachydactyly type B (BDB), which is characterized by terminal deficiency of fingers and toes (146). Mutations of Sost (antagonist of BMPs) are associated with sclerosteosis which is a disorder featuring increased bone density (147). Mutations of the TGF-β1 gene on chromosome 19q13.1-q13.3 was reported to be the cause for camurati-engelmann disease (CED) characterized by bone pain and osteosclerosis affecting the diaphysis of long bones (148). Recently, a new TGF-β1 mutation (E169K) in exon 2 was identified in a Chinese family (149) which developed CED (Table 2).
Except for the skeletal disorder, disruptions of TGF-β/BMP signaling also cause other human diseases. BMP-15 defects are involved in the pathogenesis of hypergonadotropic ovarian failure in humans, which leads to female infertility (150). Mutation of BMPRII is linked to the development of primary pulmonary hypertension (PPH) and features the widespread occlusion of small pulmonary arteries, which leads to sustained elevation of pulmonary arterial pressure (151) (Table 2). Germline mutations of the gene encoding BMPR-IA results in juvenile polyposis, an autosomal dominant gastrointestinal hamartomatous polyposis syndrome in which patients are at risk for developing gastrointestinal cancers (152). Moreover, BMPs have been implicated in periodontal disease (153), osteoarthritis (154), and the tumor metastasis. Indeed, BMP-2 may facilitate bone metastasis in gastric cancer (155). These findings indicate the importance of TGF-β/BMPs signaling in bone development and homeostasis.
It is hoped that basic research on disease and its underlying mechanism may open new avenues for the generation of antagonists, small inhibitory molecules, or novel delivery systems that target bone diseases. A small molecule inhibitor of BMP type I receptor activity has been demonstrated to be useful in treating FOP and heterotopic ossification syndromes (156). TGF-β type I receptor kinase inhibitor downregulates rheumatoid synoviocytes and prevents the arthritis (157). TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation, so modulation of TGF-β1 activity could be an effective treatment for bone remodeling diseases (158). The delivery of TGF-β3 with an injectable calcium-phosphate matrix at the supraspinatus tendon footprint has promise to improve healing after soft tissue repair (159).
Table 2
TGF-β/BMP mutations involved in human diseases
| Gene | Disease | Defects | Refer |
|---|---|---|---|
| ALK2 | fibrodysplasia ossification progressive (FOP) | ectopic bone formation | (173) (140) |
| NOGGIN(NOG) | Brachydactyly type B (BDB) | terminal deficiency of fingers and toes | (146) |
| SOST | sclerosteosis | increased bone density | (147) |
| BMP15 | hypergonadotropic ovarian failure | a common cause of female infertility | (150) |
| BMPR-II | primary pulmonary hypertension (PPH) | obstruction of pre-capillary pulmonary arteries | (151) |
| GDF5 | brachydactyly type A2 (BDA2) | hypoplasia/aplasia of the second middle phalanx of the index finger and sometimes the little finger. | (142) |
| BMPR1B | (142-143) | ||
| conserved regulatory element downstream of BMP2 | (145) | ||
| BMPRIA | juvenile polyposis | early developing gastrointestinal cancers | (152) |
| TGF-β 1 | camurati-engelmann disease (CED) | characterized by bone pain and osteosclerosis affecting the diaphysis of long bones | (148-149) |
Table 3
Crosstalk between TGF-β/BMP signaling and other signaling molecules in osteoblast and bone
| Gene | Crosstalk signaling | Results | Refer |
|---|---|---|---|
| Tgfbr2↓ | PTH type I receptor↑ | increased bone mass | (33) |
| PTH-CREB→ | BMP-2 expression | osteoblastogenesis | (35) |
| FGF2↑ | Tgfbr2 mutant→normal | regulates frontal bone | (37) |
| FGF-- FGFR3 | TGF-beta | mediates embryonic bone formation | (37) |
| TGF-β 1→ | beta-catenin↑ | osteoblastogenesis↑ | (39) |
| Wnt and TGF-β | TCF,Runx2↑ TβRI ↑ | Runx2↑ osteoblast maturation↑ | (40) |
| Smad4 mutant | Ihh/PTHrP↓ | ×→TGF-β 1 response | (41) |
| TGF-β1→ | ↑BMP-2 | → ectopic bone formation | (42) |
| Wnt3A→ | ↑ BMP-9 | ↑ ALP | (96) |
| β-catenin ↓ | ↓responsiveness to BMP-2 | alters osteoblast differentiation | (97) |
| β-catenin and Runx2→ | ↑ BMP-9-induced | osteogenic differentiation | (96) |
| BMPR1A↓ | ↑ Wnt (SOST, Dkk1) | bone mass↑ | (99) |
| FGF-2,-9 →FGF/FgfR→ | ↑ BMP-2 and TGFbeta-1 | ↑osteogenic expression | (101) |
| Notch | ↑BMP-induced ALP | →Smad and Notch | (105) |
| Shh (Gli2)→ | ↑ BMP-2 promoter activity | normal osteoblast differentiation | (110) |
| Ihh→ | ↑BMP-induced osteogenesis | bone formation | (113) |
| Ihh and BMP→ | ↑ALP, Ihh expression | long bone development | (114) |
Note: ↓decrease; ↑increase; → stimulate; × block
TGF-β /BMP signaling in clinical applications
Currently, two BMP products have been approved by the Food and Drug Administration (FDA) for clinical applications to treat fractures of long bones and improve intervertebral disk regeneration through a purified collagen matrix respectively infused with BMP-2 (Medtronic) or OP-1 BMP-7 (Stryker Biotech) and implanted at the site of the fracture. BMP treatment for acute open tibial fractures may be more favorable economically (160). Combination of BMP-7 with a type-one collagen carrier has been the subject of increasing interest. BMP-7 in combination with osteosynthesis revision and bone grafting, or with bone grafting alone, shows that there is no perioperative or postoperative complications in patients (161). The application of BMP-7 in a total of 19 joint fusions (ankle, subtalar, talonavicular, pubic and sacroiliac) resulted in healing rates of 90% and satisfactory subjective functional outcome in 70% of cases (162).
With the use of BMPs increasingly accepted in spinal fusion surgeries, other therapeutic approaches targeting BMP signaling are emerging beyond applications to skeletal disorders. BMP-7 is also regarded as a strong candidate for the clinical treatment of chronic kidney disease (CKD) (163). Administration of BMP-7 prevents the development of adynamic bone disease in a preclinical model of chronic kidney failure (164). TGF-β/BMPs have also been used as the prognostic biomarkers. For instance, GDF-15 has emerged as a prognostic biomarker in acute coronary syndrome trial populations (165) and in cardiac and vascular dysfunction and disease (166). GDF-11 may be a novel diagnostic and prognostic biomarker in patients with colorectal cancer (167). Additionally, GDF-5 has emerged as a therapeutic target for rheumatic diseases (168). Recent applications graft BMP peptides corresponding to residues 73-92, 89-117, and 68-87 of BMP-2, BMP-7, and BMP-9 as adhesion peptides (GRGDSPC) onto polyethylene terephthatalate (PET) surfaces to enhance osteogenic differentiation and mineralization of pre-osteoblastic cells (169). These engineered biomaterials for enhanced bone regneration are in the initial trial stage of development.
SUMMARY AND PERSPECTIVE
Our understanding of bone is rapidly advancing with the use of new technology, such as conditional knockout mice, high-throughput screening, and the discovery of newly recognized cross-talk between TGF-β/BMP signaling and many other major signaling pathways such as MAPK, Wnt, Hedgehog, Notch, and FGF. TGF-β/BMP signaling plays critical regulatory functions in osteoblast differentiation and bone formation. The signaling relays in each stage (ligands, receptors, Smads) are responsible for the final target gene expression. Perturbations of TGF-β/BMP signaling result in various clinical disorders including cancers, bone diseases, and vascular diseases. Thus, there is great potential for the clinical applications of TGF-β/BMP molecules for the treatment of bone-related diseases, such as FOP, chronic kidney disease, brachydactyly type A2, and osteoporosis. More importantly, with the aging population expected to double over the next decade, the number of people suffering from osteoporosis is likely to increase dramatically and so is the cost of Medicare. The current cost of medical care associated with osteoporosis (especially hip fractures) has been estimated at more than $17 billion a year. As such, there is increased pressure to elucidate the pathophysiology of bone diseases and the molecular mechanisms of skeletal remodeling in health and disease.
So far, BMP-2-and BMP-7-containing osteogenic implants have been used in over one million patients worldwide for the treatment of long bone nonunions, spinal fusions, and acute fractures. Apart from their recognized role in bone regeneration, BMPs have been used systemically to improve skeletal volume, kidney regeneration, glucose, and iron metabolism. There are still much to discover, including small molecule inhibitors with special target sites and better effectiveness, as well as better delivery systems and reliable TGF-β/BMPs retention at target sites in vivo.
ABBREVIATIONS
PTH: parathyroid hormone; ActR-II: Activin type II receptor; ActR-IIB: Activin type II B receptor; TβR-II, Tgfbr2: TGF-β type II receptor; BMPR-II: BMP type II receptor; AMHR-II: AMH type II receptor; ALK: Activin receptor-like kinase; BMPR-IA: Type I receptor of ALK-3; BMPR-IB: Type I receptor of ALK-6; TAK1: TGF-β activation kinase1; TAB1: TAK1 binding protein 1.
Acknowledgements
We would like to thank Ms. Christie Paulson and Ms. Mengrui Wu for their excellent assistance with the manuscript. We apologize to the many researchers whose work could not be cited due to space limitations. Work in our laboratory is supported by grants by NIH grant AR44741 (Y.-P. Li) and AR055307 (Y.-P. Li.).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Soltanoff CS, Yang S, Chen W, Li YP. Signaling networks that control the lineage commitment and differentiation of bone cells. Crit Rev Eukaryot Gene Expr. 2009;19:1-46
2. Huang W, Yang S, Shao J, Li YP. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci. 2007;12:3068-92
3. Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71-88
4. Wagner DO, Sieber C, Bhushan R, Borgermann JH, Graf D, Knaus P. BMPs: from bone to body morphogenetic proteins. Sci Signal. 2010;3:mr1
5. Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. TGF-beta signaling specifies axons during brain development. Cell. 2010;142:144-57
6. Katagiri T, Takahashi N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis. 2002;8:147-59
7. Urist MR. Bone: formation by autoinduction. Science. 1965;150:893-9
8. Lapraz F, Besnardeau L, Lepage T. Patterning of the dorsal-ventral axis in echinoderms: insights into the evolution of the BMP-chordin signaling network. PLoS Biol. 2009;7:e1000248
9. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-21
10. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16:251-63
11. Papachroni KK, Karatzas DN, Papavassiliou KA, Basdra EK, Papavassiliou AG. Mechanotransduction in osteoblast regulation and bone disease. Trends Mol Med. 2009;15:208-16
12. Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9:1000-4
13. Centrella M, McCarthy TL, Canalis E. Skeletal tissue and transforming growth factor beta. FASEB J. 1988;2:3066-73
14. Geiser AG, Hummel CW, Draper MW, Henck JW, Cohen IR, Rudmann DG. et al. A new selective estrogen receptor modulator with potent uterine antagonist activity, agonist activity in bone, and minimal ovarian stimulation. Endocrinology. 2005;146:4524-35
15. Dunker N, Krieglstein K. Tgfbeta2 -/- Tgfbeta3 -/- double knockout mice display severe midline fusion defects and early embryonic lethality. Anat Embryol (Berl). 2002;206:73-83
16. Matsunobu T, Torigoe K, Ishikawa M, de Vega S, Kulkarni AB, Iwamoto Y. et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev Biol. 2009;332:325-38
17. Baffi MO, Moran MA, Serra R. Tgfbr2 regulates the maintenance of boundaries in the axial skeleton. Dev Biol. 2006;296:363-74
18. Hiramatsu K, Iwai T, Yoshikawa H, Tsumaki N. Expression of dominant negative TGF-beta receptors inhibits cartilage formation in conditional transgenic mice. J Bone Miner Metab. 2011;29:493-500
19. Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276:124-42
20. Seo HS, Serra R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev Biol. 2007;310:304-16
21. Seo HS, Serra R. Tgfbr2 is required for development of the skull vault. Dev Biol. 2009;334:481-90
22. Iwata J, Hosokawa R, Sanchez-Lara PA, Urata M, Slavkin H, Chai Y. Transforming growth factor-beta regulates basal transcriptional regulatory machinery to control cell proliferation and differentiation in cranial neural crest-derived osteoprogenitor cells. J Biol Chem. 2010;285:4975-82
23. Sapkota G, Knockaert M, Alarcon C, Montalvo E, Brivanlou AH, Massague J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281:40412-9
24. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
25. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
26. Tan X, Weng T, Zhang J, Wang J, Li W, Wan H. et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J Cell Sci. 2007;120:2162-70
27. Yasui T, Kadono Y, Nakamura M, Oshima Y, Matsumoto T, Masuda H. et al. Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and Traf6. J Bone Miner Res. 2011;26:1447-56
28. Yasui T, Kadono Y, Nakamura M, Oshima Y, Matsumoto T, Masuda H. et al. Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and TRAF6. J Bone Miner Res. 2011Jul;26(7):1447-56
29. Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292:F1471-8
30. Lee KS, Hong SH, Bae SC. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene. 2002;21:7156-63
31. Lai CF, Cheng SL. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J Biol Chem. 2002;277:15514-22
32. Lee SW, Choi KY, Cho JY, Jung SH, Song KB, Park EK. et al. TGF-beta2 stimulates cranial suture closure through activation of the Erk-MAPK pathway. J Cell Biochem. 2006;98:981-91
33. Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol. 2010;12:224-34
34. Atfi A, Baron R. PTH battles TGF-beta in bone. Nat Cell Biol. 2010;12:205-7
35. Zhang R, Edwards JR, Ko SY, Dong S, Liu H, Oyajobi BO. et al. Transcriptional regulation of BMP2 expression by the PTH-CREB signaling pathway in osteoblasts. PLoS One. 2011;6:e20780
36. Bosetti M, Boccafoschi F, Leigheb M, Cannas MF. Effect of different growth factors on human osteoblasts activities: a possible application in bone regeneration for tissue engineering. Biomol Eng. 2007;24:613-8
37. Sasaki T, Ito Y, Bringas P Jr, Chou S, Urata MM, Slavkin H. et al. TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development. 2006;133:371-81
38. Mukherjee A, Dong SS, Clemens T, Alvarez J, Serra R. Co-ordination of TGF-beta and FGF signaling pathways in bone organ cultures. Mech Dev. 2005;122:557-71
39. Zhou S. TGF-beta regulates beta-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J Cell Biochem. 2011;112:1651-60
40. McCarthy TL, Centrella M. Novel links among Wnt and TGF-beta signaling and Runx2. Mol Endocrinol. 2010;24:587-97
41. Zhang J, Tan X, Li W, Wang Y, Wang J, Cheng X. et al. Smad4 is required for the normal organization of the cartilage growth plate. Dev Biol. 2005;284:311-22
42. Tachi K, Takami M, Sato H, Mochizuki A, Zhao B, Miyamoto Y. et al. Enhancement of bone morphogenetic protein-2-induced ectopic bone formation by transforming growth factor-beta1. Tissue Eng Part A. 2011;17:597-606
43. Singhatanadgit W, Salih V, Olsen I. Up-regulation of bone morphogenetic protein receptor IB by growth factors enhances BMP-2-induced human bone cell functions. J Cell Physiol. 2006;209:912-22
44. Wu N, Zhao Y, Yin Y, Zhang Y, Luo J. Identification and analysis of type II TGF-beta receptors in BMP-9-induced osteogenic differentiation of C3H10T1/2 mesenchymal stem cells. Acta Biochim Biophys Sin (Shanghai). 2010;42:699-708
45. Huang Z, Ren PG, Ma T, Smith RL, Goodman SB. Modulating osteogenesis of mesenchymal stem cells by modifying growth factor availability. Cytokine. 2010;51:305-10
46. Noel D, Gazit D, Bouquet C, Apparailly F, Bony C, Plence P. et al. Short-term BMP-2 expression is sufficient for in vivo osteochondral differentiation of mesenchymal stem cells. Stem Cells. 2004;22:74-85
47. Gu K, Zhang L, Jin T, Rutherford RB. Identification of potential modifiers of Runx2/Cbfa1 activity in C2C12 cells in response to bone morphogenetic protein-7. Cells Tissues Organs. 2004;176:28-40
48. Shen B, Wei A, Whittaker S, Williams LA, Tao H, Ma DD. et al. The role of BMP-7 in chondrogenic and osteogenic differentiation of human bone marrow multipotent mesenchymal stromal cells in vitro. J Cell Biochem. 2010;109:406-16
49. Tsuji K, Cox K, Gamer L, Graf D, Economides A, Rosen V. Conditional deletion of BMP7 from the limb skeleton does not affect bone formation or fracture repair. J Orthop Res. 2010;28:384-9
50. Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216
51. Tsuji K, Cox K, Bandyopadhyay A, Harfe BD, Tabin CJ, Rosen V. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J Bone Joint Surg Am. 2008;90(Suppl 1):14-8
52. Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L. et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38:1424-9
53. Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W. et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124:3428-40
54. Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL. et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet. 2001;27:84-8
55. Kokabu S, Gamer L, Cox K, Lowery J, Tsuji K, Raz R. et al. BMP3 Suppresses Osteoblast Differentiation of Bone Marrow Stromal Cells via Interaction with Acvr2b. Mol Endocrinol. 2012Jan;26(1):87-94
56. Gamer LW, Ho V, Cox K, Rosen V. Expression and function of BMP3 during chick limb development. Dev Dyn. 2008;237:1691-8
57. Gamer LW, Cox K, Carlo JM, Rosen V. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev Dyn. 2009;238:2374-81
58. Kudo TA, Kanetaka H, Watanabe A, Okumoto A, Asano M, Zhang Y. et al. Investigating bone morphogenetic protein (BMP) signaling in a newly established human cell line expressing BMP receptor type II. Tohoku J Exp Med. 2010;222:121-9
59. Liu H, Zhang R, Chen D, Oyajobi BO, Zhao M. Functional redundancy of type II BMP receptor and type IIB activin receptor in BMP2-induced osteoblast differentiation. J Cell Physiol. 2011
60. Gamer LW, Tsuji K, Cox K, Capelo LP, Lowery J, Beppu H. et al. BMPR-II is dispensable for formation of the limb skeleton. Genesis. 2011;49:719-24
61. Okamoto M, Murai J, Imai Y, Ikegami D, Kamiya N, Kato S. et al. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J Bone Miner Res. 2011;26:2511-22
62. Ovchinnikov DA, Selever J, Wang Y, Chen YT, Mishina Y, Martin JF. et al. BMP receptor type IA in limb bud mesenchyme regulates distal outgrowth and patterning. Dev Biol. 2006;295:103-15
63. Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG. et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279:27560-6
64. Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M. et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007-17
65. Zhou Z, Xie J, Lee D, Liu Y, Jung J, Zhou L. et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev Cell. 2010;19:90-102
66. Podkowa M, Attisano L. A skeleton in the closet: neogenin guides bone development. Dev Cell. 2010;19:1-2
67. Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK. et al. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis. 2006;44:159-67
68. Wang M, Jin H, Tang D, Huang S, Zuscik MJ, Chen D. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage. 2011;19:751-62
69. Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093-104
70. Skarzynska J, Damulewicz M, Filipowska J, Madej W, Leboy PS, Osyczka AM. Modification of Smad1 Linker Modulates BMP-Mediated Osteogenesis of Adult Human MSC. Connect Tissue Res. 2011;52:408-14
71. Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM. et al. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell. 2007;131:980-93
72. Li C, Li YP, Fu XY, Deng CX. Anterior visceral endoderm SMAD4 signaling specifies anterior embryonic patterning and head induction in mice. Int J Biol Sci. 2010;6:569-83
73. Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6:1-8
74. Azhar M, Wang PY, Frugier T, Koishi K, Deng C, Noakes PG. et al. Myocardial deletion of Smad4 using a novel alpha skeletal muscle actin Cre recombinase transgenic mouse causes misalignment of the cardiac outflow tract. Int J Biol Sci. 2010;6:546-55
75. Wrana JL. The secret life of Smad4. Cell. 2009;136:13-4
76. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M. et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123-35
77. Kang M, Bok J, Deocaris CC, Park HW, Kim MH. Hoxc8 represses BMP-induced expression of Smad6. Mol Cells. 2010;29:29-33
78. Estrada KD, Retting KN, Chin AM, Lyons KM. Smad6 is essential to limit BMP signaling during cartilage development. J Bone Miner Res. 2011;26:2498-510
79. Horiki M, Imamura T, Okamoto M, Hayashi M, Murai J, Myoui A. et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J Cell Biol. 2004;165:433-45
80. Shen R, Chen M, Wang YJ, Kaneki H, Xing L, O'Keefe R J. et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem. 2006;281:3569-76
81. Hoffmann A, Preobrazhenska O, Wodarczyk C, Medler Y, Winkel A, Shahab S. et al. Transforming growth factor-beta-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J Biol Chem. 2005;280:27271-83
82. Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D. et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120:2457-73
83. Greenblatt MB, Shim JH, Glimcher LH. TAK1 mediates BMP signaling in cartilage. Ann N Y Acad Sci. 2010;1192:385-90
84. Zuzarte-Luis V, Montero JA, Rodriguez-Leon J, Merino R, Rodriguez-Rey JC, Hurle JM. A new role for BMP5 during limb development acting through the synergic activation of Smad and MAPK pathways. Dev Biol. 2004;272:39-52
85. Afzal F, Pratap J, Ito K, Ito Y, Stein JL, van Wijnen AJ. et al. Smad function and intranuclear targeting share a Runx2 motif required for osteogenic lineage induction and BMP2 responsive transcription. J Cell Physiol. 2005;204:63-72
86. Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T. et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol. 2000;20:8783-92
87. Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Miner Res. 2006;21:637-46
88. Javed A, Afzal F, Bae JS, Gutierrez S, Zaidi K, Pratap J. et al. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs. 2009;189:133-7
89. Jonason JH, Xiao G, Zhang M, Xing L, Chen D. Post-translational Regulation of Runx2 in Bone and Cartilage. J Dent Res. 2009;88:693-703
90. Zhang YW, Yasui N, Ito K, Huang G, Fujii M, Hanai J. et al. A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc Natl Acad Sci U S A. 2000;97:10549-54
91. Zhang C. Transcriptional regulation of bone formation by the osteoblast-specific transcription factor Osx. J Orthop Surg Res. 2010;5:37
92. Lee MH, Kwon TG, Park HS, Wozney JM, Ryoo HM. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem Biophys Res Commun. 2003;309:689-94
93. Holleville N, Mateos S, Bontoux M, Bollerot K, Monsoro-Burq AH. Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev Biol. 2007;304:860-74
94. Jeong HM, Jin YH, Kim YJ, Yum J, Choi YH, Yeo CY. et al. Akt phosphorylates and regulates the function of Dlx5. Biochem Biophys Res Commun. 2011;409:681-6
95. Choi YH, Jeong HM, Jin YH, Li H, Yeo CY, Lee KY. Akt phosphorylates and regulates the osteogenic activity of Osterix. Biochem Biophys Res Commun. 2011;411:637-41
96. Tang N, Song WX, Luo J, Luo X, Chen J, Sharff KA. et al. BMP-9-induced osteogenic differentiation of mesenchymal progenitors requires functional canonical Wnt/beta-catenin signalling. J Cell Mol Med. 2009;13:2448-64
97. Zhang M, Yan Y, Lim YB, Tang D, Xie R, Chen A. et al. BMP-2 modulates beta-catenin signaling through stimulation of Lrp5 expression and inhibition of beta-TrCP expression in osteoblasts. J Cell Biochem. 2009;108:896-905
98. Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM. et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development. 2008;135:3801-11
99. Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM. et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res. 2010;25:200-10
100. Kamiya N. The Role of BMPs in Bone Anabolism and their Potential Targets SOST and DKK1. Curr Mol Pharmacol. 2011
101. Fakhry A, Ratisoontorn C, Vedhachalam C, Salhab I, Koyama E, Leboy P. et al. Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone. 2005;36:254-66
102. Hughes-Fulford M, Li CF. The role of FGF-2 and BMP-2 in regulation of gene induction, cell proliferation and mineralization. J Orthop Surg Res. 2011;6:8
103. Naganawa T, Xiao L, Coffin JD, Doetschman T, Sabbieti MG, Agas D. et al. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J Cell Biochem. 2008;103:1975-88
104. Choi KY, Kim HJ, Lee MH, Kwon TG, Nah HD, Furuichi T. et al. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev Dyn. 2005;233:115-21
105. Tezuka K, Yasuda M, Watanabe N, Morimura N, Kuroda K, Miyatani S. et al. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res. 2002;17:231-9
106. Nobta M, Tsukazaki T, Shibata Y, Xin C, Moriishi T, Sakano S. et al. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J Biol Chem. 2005;280:15842-8
107. de Jong DS, Steegenga WT, Hendriks JM, van Zoelen EJ, Olijve W, Dechering KJ. Regulation of Notch signaling genes during BMP2-induced differentiation of osteoblast precursor cells. Biochem Biophys Res Commun. 2004;320:100-7
108. Dahlqvist C, Blokzijl A, Chapman G, Falk A, Dannaeus K, Ibanez CF. et al. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 2003;130:6089-99
109. Yuasa T, Kataoka H, Kinto N, Iwamoto M, Enomoto-Iwamoto M, Iemura S. et al. Sonic hedgehog is involved in osteoblast differentiation by cooperating with BMP-2. J Cell Physiol. 2002;193:225-32
110. Zhao M, Qiao M, Harris SE, Chen D, Oyajobi BO, Mundy GR. The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Mol Cell Biol. 2006;26:6197-208
111. Karamboulas K, Dranse HJ, Underhill TM. Regulation of BMP-dependent chondrogenesis in early limb mesenchyme by TGFbeta signals. J Cell Sci. 2010;123:2068-76
112. Bastida MF, Sheth R, Ros MA. A BMP-Shh negative-feedback loop restricts Shh expression during limb development. Development. 2009;136:3779-89
113. Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131:1309-18
114. van der Horst G, Farih-Sips H, Lowik CW, Karperien M. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone. 2003;33:899-910
115. Harland RM. A protein scaffold plays matchmaker for chordin. Cell. 2008;134:718-9
116. Gajos-Michniewicz A, Piastowska AW, Russell JA, Ochedalski T. Follistatin as a potent regulator of bone metabolism. Biomarkers. 2010Nov;15(7):563-74
117. Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713-26
118. Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W. et al. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636-42
119. Devlin RD, Du Z, Pereira RC, Kimble RB, Economides AN, Jorgetti V. et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology. 2003;144:1972-8
120. Wu XB, Li Y, Schneider A, Yu W, Rajendren G, Iqbal J. et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112:924-34
121. Secondini C, Wetterwald A, Schwaninger R, Thalmann GN, Cecchini MG. The role of the BMP signaling antagonist noggin in the development of prostate cancer osteolytic bone metastasis. PLoS One. 2011;6:e16078
122. Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19:176-84
123. Zhao M, Qiao M, Harris SE, Oyajobi BO, Mundy GR, Chen D. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J Biol Chem. 2004;279:12854-9
124. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX. et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101-13
125. Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K. et al. TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587-95
126. Wu Q, Kim KO, Sampson ER, Chen D, Awad H, O'Brien T. et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheum. 2008;58:3132-44
127. Wang L, Liu YT, Hao R, Chen L, Chang Z, Wang HR. et al. Molecular Mechanism of the Negative Regulation of Smad1/5 Protein by Carboxyl Terminus of Hsc70-interacting Protein (CHIP). J Biol Chem. 2011;286:15883-94
128. Xia L, Jia S, Huang S, Wang H, Zhu Y, Mu Y. et al. The Fused/Smurf complex controls the fate of Drosophila germline stem cells by generating a gradient BMP response. Cell. 2010;143:978-90
129. Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science. 1999;286:771-4
130. Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y. et al. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol. 2007;27:4488-99
131. Wu JW, Krawitz AR, Chai J, Li W, Zhang F, Luo K. et al. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling. Cell. 2002;111:357-67
132. Delgado-Calle J, Sanudo C, Sanchez-Verde L, Garcia-Renedo RJ, Arozamena J, Riancho JA. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone. 2011;49:830-8
133. Tu AW, Luo K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J Biol Chem. 2007;282:21187-96
134. Ross S, Cheung E, Petrakis TG, Howell M, Kraus WL, Hill CS. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006;25:4490-502
135. Simonsson M, Heldin CH, Ericsson J, Gronroos E. The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem. 2005;280:21797-803
136. Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483-93
137. Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH. et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. 2006;281:16502-11
138. Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS, Ryoo HM. et al. BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J Biol Chem. 2010;285:36410-9
139. Choo MK, Yeo H, Zayzafoon M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone. 2009;45:579-89
140. Kaplan FS, Pignolo RJ, Shore EM. The FOP metamorphogene encodes a novel type I receptor that dysregulates BMP signaling. Cytokine Growth Factor Rev. 2009;20:399-407
141. Katagiri T. [Genetic basis for skeletal disease. Establishment of novel treatments for fibrodysplasia ossificans progressiva (FOP)]. Clin Calcium. 2010;20:1204-11
142. Kjaer KW, Eiberg H, Hansen L, van der Hagen CB, Rosendahl K, Tommerup N. et al. A mutation in the receptor binding site of GDF5 causes Mohr-Wriedt brachydactyly type A2. J Med Genet. 2006;43:225-31
143. Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K. et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest. 2005;115:2373-81
144. Seemann P, Brehm A, Konig J, Reissner C, Stricker S, Kuss P. et al. Mutations in GDF5 reveal a key residue mediating BMP inhibition by NOGGIN. PLoS Genet. 2009;5:e1000747
145. Dathe K, Kjaer KW, Brehm A, Meinecke P, Nurnberg P, Neto JC. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet. 2009;84:483-92
146. Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW. et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007;81:388-96
147. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S. et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577-89
148. Campos-Xavier B, Saraiva JM, Savarirayan R, Verloes A, Feingold J, Faivre L. et al. Phenotypic variability at the TGF-beta1 locus in Camurati-Engelmann disease. Hum Genet. 2001;109:653-8
149. Wu S, Liang S, Yan Y, Wang Y, Li F, Deng Y. et al. A novel mutation of TGF beta1 in a Chinese family with Camurati-Engelmann disease. Bone. 2007;40:1630-4
150. Di Pasquale E, Beck-Peccoz P, Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am J Hum Genet. 2004;75:106-11
151. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE. et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81-4
152. Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM. et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184-7
153. Li Y, Messina C, Bendaoud M, Fine DH, Schreiner H, Tsiagbe VK. Adaptive immune response in osteoclastic bone resorption induced by orally administered Aggregatibacter actinomycetemcomitans in a rat model of periodontal disease. Mol Oral Microbiol. 2010;25:275-92
154. Valdes AM, Spector TD. The clinical relevance of genetic susceptibility to osteoarthritis. Best Pract Res Clin Rheumatol. 2010;24:3-14
155. Park Y, Kim JW, Kim DS, Kim EB, Park SJ, Park JY. et al. The Bone Morphogenesis Protein-2 (BMP-2) is associated with progression to metastatic disease in gastric cancer. Cancer Res Treat. 2008;40:127-32
156. Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML. et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008;14:1363-9
157. Sakuma M, Hatsushika K, Koyama K, Katoh R, Ando T, Watanabe Y. et al. TGF-beta type I receptor kinase inhibitor down-regulates rheumatoid synoviocytes and prevents the arthritis induced by type II collagen antibody. Int Immunol. 2007;19:117-26
158. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757-65
159. Kovacevic D, Fox AJ, Bedi A, Ying L, Deng XH, Warren RF. et al. Calcium-phosphate matrix with or without TGF-beta3 improves tendon-bone healing after rotator cuff repair. Am J Sports Med. 2011;39:811-9
160. Garrison KR, Shemilt I, Donell S, Ryder JJ, Mugford M, Harvey I. et al. Bone morphogenetic protein (BMP) for fracture healing in adults. Cochrane Database Syst Rev. 2010:CD006950
161. Moghaddam A, Elleser C, Biglari B, Wentzensen A, Zimmermann G. Clinical application of BMP 7 in long bone non-unions. Arch Orthop Trauma Surg. 2010;130:71-6
162. Kanakaris NK, Mallina R, Calori GM, Kontakis G, Giannoudis PV. Use of bone morphogenetic proteins in arthrodesis: clinical results. Injury. 2009;40(Suppl 3):S62-6
163. Mathew S, Davies M, Lund R, Saab G, Hruska KA. Function and effect of bone morphogenetic protein-7 in kidney bone and the bone-vascular links in chronic kidney disease. Eur J Clin Invest. 2006;36(Suppl 2):43-50
164. Zeisberg M, Kalluri R. Reversal of experimental renal fibrosis by BMP7 provides insights into novel therapeutic strategies for chronic kidney disease. Pediatr Nephrol. 2008;23:1395-8
165. Kempf T, Sinning JM, Quint A, Bickel C, Sinning C, Wild PS. et al. Growth-differentiation factor-15 for risk stratification in patients with stable and unstable coronary heart disease: results from the AtheroGene study. Circ Cardiovasc Genet. 2009;2:286-92
166. Kempf T, Wollert KC. Growth differentiation factor-15: a new biomarker in cardiovascular disease. Herz. 2009;34:594-9
167. Yokoe T, Ohmachi T, Inoue H, Mimori K, Tanaka F, Kusunoki M. et al. Clinical significance of growth differentiation factor 11 in colorectal cancer. Int J Oncol. 2007;31:1097-101
168. Lories RJ, Luyten FP. Bone morphogenetic protein signaling in joint homeostasis and disease. Cytokine Growth Factor Rev. 2005;16:287-98
169. Zouani OF, Chollet C, Guillotin B, Durrieu MC. Differentiation of pre-osteoblast cells on poly(ethylene terephthalate) grafted with RGD and/or BMPs mimetic peptides. Biomaterials. 2010Nov;31(32):8245-53
170. Oka K, Oka S, Sasaki T, Ito Y, Bringas P Jr, Nonaka K. et al. The role of TGF-beta signaling in regulating chondrogenesis and osteogenesis during mandibular development. Dev Biol. 2007;303:391-404
171. Gunnell LM, Jonason JH, Loiselle AE, Kohn A, Schwarz EM, Hilton MJ. et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res. 2010;25:1784-97
172. Iwai T, Murai J, Yoshikawa H, Tsumaki N. Smad7 Inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J Biol Chem. 2008;283:27154-64
173. Fiori JL, Billings PC, de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res. 2006;21:902-9
Author contact
![]() Corresponding author: Yi-Ping Li, Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA. Tel: 205-975-2606, Fax: 205-975-4919, E-mail: ypliedu. Chuxia Deng, Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA. Tel: 301-402-7225, Fax: 301-480-113, E-mail: Chuxiadniddk.nih.gov
Corresponding author: Yi-Ping Li, Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA. Tel: 205-975-2606, Fax: 205-975-4919, E-mail: ypliedu. Chuxia Deng, Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA. Tel: 301-402-7225, Fax: 301-480-113, E-mail: Chuxiadniddk.nih.gov
Citation styles
APA
Chen, G., Deng, C., Li, Y.P. (2012). TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. International Journal of Biological Sciences, 8(2), 272-288. https://doi.org/10.7150/ijbs.2929.
ACS
Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8 (2), 272-288. DOI: 10.7150/ijbs.2929.
NLM
Chen G, Deng C, Li YP. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int J Biol Sci 2012; 8(2):272-288. doi:10.7150/ijbs.2929. https://www.ijbs.com/v08p0272.htm
CSE
Chen G, Deng C, Li YP. 2012. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int J Biol Sci. 8(2):272-288.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.