Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structural and Functional...
Biological Functions of SRC-1
The Role of SRC-1 in Breast...
SRC-1 in Prostate Cancer
SRC-1 Expression in Other Cancers
SRC Proteins as Potential...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):470-485. doi:10.7150/ijbs.4125 This issue Cite
Review
The Function of Steroid Receptor Coactivator-1 in Normal Tissues and Cancer
Claire A. Walsh1, Li Qin2, Jean Ching-Yi Tien2, Leonie S. Young1, Jianming Xu2, 3, ![]()
1. Endocrine Oncology Research Group, Royal College of Surgeons in Ireland, Dublin, Ireland;
2. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas 77030, USA;
3. Luzhou Medical College, Luzhou, Sichuan 646000, China.
Received 2012-1-19; Accepted 2012-2-20; Published 2012-3-7
Abstract
In 1995, the steroid receptor coactivator-1 (SRC-1) was identified as the first authentic steroid receptor coactivator. Since then, the SRC proteins have remained at the epicenter of coregulator biology, molecular endocrinology and endocrine-related cancer. Cumulative works on SRC-1 have shown that it is primarily a nuclear receptor coregulator and functions to construct highly specific enzymatic protein complexes which can execute efficient and successful transcriptional activation of designated target genes. The versatile nature of SRC-1 enables it to respond to steroid dependent and steroid independent stimulation, allowing it to bind across many families of transcription factors to orchestrate and regulate complex physiological reactions. This review highlights the multiple functions of SRC-1 in the development and maintenance of normal tissue functions as well as its major role in mediating hormone receptor responsiveness. Insights from genetically manipulated mouse models and clinical data suggest SRC-1 is significantly overexpressed in many cancers, in particular, cancers of the reproductive tissues. SRC-1 has been associated with cellular proliferation and tumor growth but its major tumorigenic contributions are promotion and execution of breast cancer metastasis and mediation of resistance to endocrine therapies. The ability of SRC-1 to coordinate multiple signaling pathways makes it an important player in tumor cells' escape of targeted therapy.
Keywords: nuclear receptor, coactivator, SRC-1, transcription, development, cancer.
Introduction
Coregulator proteins were initially thought to be mere “acceptor proteins” that facilitated the interaction of specific ligand-bound nuclear receptors with general transcriptional machinery [1]. Over the last seventeen years, however, complex and essential roles for a cohort of over 350 coactivator proteins have steadily emerged [2]. Coregulator proteins have the uncanny ability to bind across unrelated families of transcription factors and coordinately facilitate the efficient and successful transcriptional activity of multiple target genes.
The nuclear receptors (NRs) comprise a large superfamily of proteins that bind as homo- or heterodimers to specific DNA elements in order to elicit transcriptional activation of target genes. Specifically, the steroid nuclear receptors or class I NRs are recruited to gene promoters upon the binding of a high-affinity ligand, which induce conformational changes in the NR essential for its activity. The significance of NR coregulator proteins came to light when in vitro experiments using purified NRs and basal transcription factors proved relatively incapable of inducing transcriptional activation on their own [3, 4]. Furthermore, NRs were also shown to compete with each other for these essential coregulators as overexpression of one NR appeared to inhibit the transactivation function of another [5].
The steroid receptor coactivator 1 (SRC-1, also known as NCOA1) was first discovered in 1995 in a yeast two-hybrid screen based on its interaction with the ligand binding domain (LBD) of progesterone receptor (PR) [6]. This work represented the first cloning of an authentic NR coactivator. SRC-1 had the ability to interact with and coactivate NRs in the presence of hormones. These SRC-1 coregulated NRs include PR, glucocorticoid receptor (GR), estrogen receptor alpha (ERα), thyroid receptor (TR), retinoid X receptor (RXR), hepatocyte nuclear factor 4 (HNF4α) and peroxisome proliferator-activated receptor γ (PPARγ) [6-8]. The binding affinity of SRC-1 for these NRs has been shown to vary depending on where it specifically binds the NR. SRC-1 can bind NRs via its central region or less commonly via its C-terminal domain. The central domain of SRC-1 has been shown to be unable to bind to AR and only exhibits a poor binding affinity for GR. In contrast, the C-terminus of SRC-1 exhibits a poor binding affinity for ER, VDR, RAR and TR, relative to its central domain [9]. Furthermore, fluorescence resonance energy transfer (FRET) experiments have shown that the complex formed between ERα and SRC-1 exhibited a particularly high affinity binding, compared to other SRC-1/NR complexes [10]. Importantly, SRC-1 coactivator activity is not limited to the transcriptional co-activation of NRs, SRC-1 is also capable of coactivating other non steroidal transcription factors such as AP-1, serum response factor, NFκβ, Ets2, PEA3 and HOXC11 [11-17].
SRC-1 is the founding member of the p160 SRC family which also includes SRC-2 (NCOA2, TIF2 or GRIP1) and SRC-3 (AIB1, p/CIP, ACTR, RAC3 or NCOA3) [18, 19]. Each member is approximately 160 kDa in size and their sequences are largely conserved across family members and also across species. The p160 SRC family members also have overlapping coactivator functions and transfection assays have shown that all three can coactivate GR, PR and ERα [6]. The potential for functional redundancy among the three members may serve to ensure a safety mechanism in the regulation of numerous important biological processes that are associated with NR signaling.
Structural and Functional Domains of SRC-1
NR coactivators are unable to bind directly to the DNA. Instead they form multiple contacts with the NR and with each other in multi-protein cooperative coactivator complexes. Initial investigations into coactivator complexes reported that steady-state SRC complexes consist of six to ten stably associated proteins and many more loosely-bound proteins [20]. The versatile structural domains of SRC-1 and the other SRC family members grant them a central position in such complexes, from which they regulate multiple biochemical processes critical for the successful execution of transcription.
1. The N-terminal domain
The SRC-1 protein structure is composed of several distinct functional domains. The N-terminus contains a basic helix-loop-helix-Per/Ah receptor nuclear translocation/Sim (bHLH/PAS) motif and is the most conserved region among the SRC family members with 75% similarity [4]. The bHLH/PAS domain is important for the protein-protein interactions that recruit secondary coactivators or co-coactivators to maximize the transcriptional activity of NRs (Figure 1). The domain is also important for the dimerization of SRC proteins and for the differential regulation of target genes [21, 22]. Nonetheless, cell-free chromatin transcription assays have shown that, although the bHLH/PAS domain can maximize the transcriptional potential of a complex, transcription can still occur in its absence. For instance, an N-terminal-deleted form of SRC-1 exhibits coactivation of PR-dependent transcription comparable to that of wild-type SRC-1 [23]. More recent studies have shown that the bHLH/PAS domain also has a bipartite nuclear localization signal (NLS) essential for the import and export of the SRC proteins between the nucleus and the cytoplasm. Mutation of two key residues Lys 17 (K17) and Arg 18 (R18) within this region of the bHLH/PAS domain prevented nuclear localization of all three p160 family members [24].
SRC-1-mediated coactivation of NRs. To exert its coactivation function in transcription, SRC-1 interacts with hormone (H)-bound nuclear receptors (NRs) to recruit other components of a large coactivator complex to the hormone response elements of a target gene. Specifically, SRC-1 binds NRs through one of its three LXXLL motifs (L1, L2 and L3) in the NR interaction domain (NRID) and interacts with CBP and p300 through its activation domain 1 (AD1), with CARM1 and PRMT1 through its AD2 and with SWI/SNF through its AD3. p/CAF is a p300/CBP-associated factor. CBP, p300 and p/CAF are histone acetyltransferases. CARM1 and PRMT1 are histone methyltransferases. RHA is a RNA helicase. SWI/SNF is an ATP-dependent chromatin remodeling complex. The formation of such a coactivator complex results in chromatin remodeling and bridges the hormone-activated NRs with the general transcription machinery for transcriptional activation of their specific target genes. bHLH/PAS: basic helix-loop-helix-Per/Ah receptor nuclear translocation/Sim motif; Ac: acetylation; Me: methylation; TBP, TATA-binding protein.
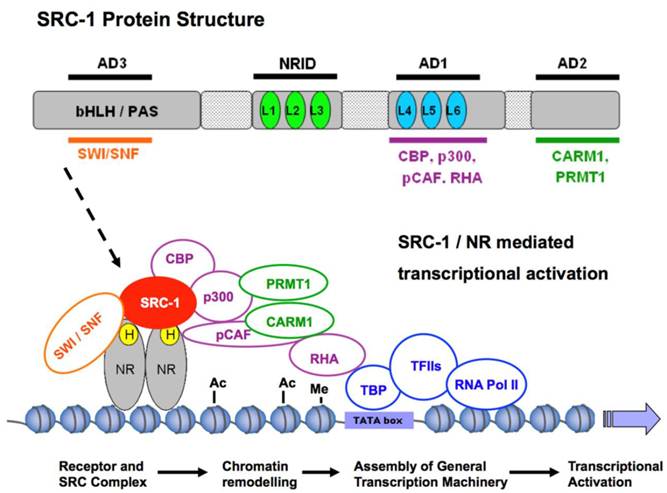
2. The nuclear receptor interaction domain (NRID)
The central region of the SRC-1 protein contains the nuclear receptor interaction domain (NRID). This domain contains three α-helical LXXLL (L, leucine; X, any amino acid) motifs which are essential for interaction of SRC-1 with its specific NRs (Figure 1) [25, 26]. These LXXLL motifs are the most common feature among the highly diverse group of coregulators [27]. Distinct LXXLL motifs along with their specific flanking sequences exhibit different binding affinities for different NRs inferring that NRs may prefer one LXXLL motif over another in the same coactivator or even prefer one coactivator over another [27, 28].
The LXXLL core motifs in SRC-1 have shown distinct preferences for steroid and retinoid receptors. In particular, a hydrophobic residue at position -1 relative to the first conserved leucine and a nonhydrophobic residue at position +2 have a strong impact on the affinity and selectivity of the coactivator motifs to bind to certain NR LBDs [29].
Secondary structural analysis of the central region shows that LXXLL motifs form an amphipathic α-helix which binds to a hydrophobic cleft that forms in the ligand-binding domains of NRs once ligands have bound [30]. Interactions between the leucine residues of the LXXLL motif and the hydrophobic cleft stabilize the SRC-1/NR complex [31]. Mutation of these leucine residues have been shown to inhibit the binding of SRC-1 to the ERα LBD in vitro and SRC-1-mediated activation of ERα in vivo [26].
3. The C-terminal domain
The C terminal domain of SRC-1 contains two intrinsic transactivation domains: AD1 and AD2. The AD1 domain is required for the recruitment of secondary coactivators, such as cAMP response element binding protein (CREB)-binding protein (CBP) and the histone acetyltransferase p300 (Figure 1). Once SRC-1 has complexed with its ligand-bound receptor at the DNA, it must bring in additional proteins such as CBP and p300 to acetylate histone residues within the enhancer and promoter regions of the target gene so that transcription can successfully occur. In vitro transcription assays from chromatin-assembled templates have demonstrated that the interaction of p300 with the AD1 domain of SRC-1 is essential for SRC-1-mediated coactivation of ERα [32]. Furthermore, observations from the p300 protein structure have confirmed that it is the SRC-1 interacting domain and not the ERα-interacting domain, that is essential for p300-mediated coactivation of ERα [32]. Additional experiments have demonstrated that the C terminus of SRC-1 and also SRC-3 have intrinsic histone acetyltransferase activity (HAT); however, it remains unclear if such HAT activity is significant for target gene activation as the SRC-1 intrinsic HAT activity is much weaker than that in the CBP/p300 proteins [33, 34]. SRC-1 also uses its AD1 domain to interact directly or indirectly via CBP/p300 with another HAT protein known as p/CAF (Figure 1). p/CAF primarily acetylates histone H3 and H4 to further facilitate chromatin remodeling at the site of NR target genes [35].
The AD1 domain is also responsible for interactions with the general transcription machinery [36, 37]. The AD1 has three LXXLL-like motifs and if any of these motifs are mutated there is a consequential disruption of SRC-1 interaction with the general transcription machinery [38, 39]. Additionally, RNA helicase A (RHA) may be recruited to the SRC-1-CBP/p300 complex at the AD1 domain and interacts with RNA polymerase II, bringing NRs into touch with the general transcription machinery (Figure 1) [40].
The AD2 domain of SRC-1 recruits histone methyl transferases (HMTs) such as coactivator-associated arginine methyltransferase 1 (CARM1) and protein arginine N-methyltransferase 1 (PRMT1) (Figure 1). CARM1 is a histone H3 specific arginine methyltransferase, which specifically methylates histone H3 at arginines 2, 17, and 26. CARM1 can only enhance NR transcription in the presence of a SRC protein and mutation of its binding domain will reduce its HMT activity and ability to act as a co-coactivator [41]. There appears to be a cooperative effort at play between the recruited co-coactivators as CBP-mediated acetylation of H3K18 has been shown to promote CARM1-mediated methylation of H3K17 [42]. CARM1 and PRMT1 can also act in synergy to enhance transcription [41]. PRMT1 acts on a different substrate to CARM1 and specifically methylates H4 at arginine 3 [43]. Again cooperative crosstalk is observed between PRMT1 and p300 as PRMT1-mediated H4 methylation at arginine 3 facilitates the subsequent acetylation of histone H4 tails by p300 [44].
Recently, a third activation domain (AD3) has been identified in the bHLH/PAS domain. This region also binds co-coactivators for NR mediated transcription. Many of these AD3 co-coactivators function synergistically with the other co-coactivators that bind to the AD1 and AD2 domains of SRCs [45]. The co-coactivator BAF57, a subunit of the SWI/SNF ATP-dependent chromatin remodeling complex, binds to SRC-1 within its AD3 domain and potentiates SRC-mediated coactivation [46].
4. Post translational modification of SRC-1
SRC proteins are regulated by numerous post translational modifications (PTMs), which are crucial for determining protein stability, transcription factor interaction specificity and transcriptional activation [47]. In addition to ER, SRC-1 is regulated via alternative cellular signaling pathways which phosphorylate it at seven distinct sites in vivo. All of these phosphorylation sites contain a consensus sequence for proline-directed protein kinases. Phosphorylation of SRC-1 can also occur via Src kinase activity [48] and the mitogen-activated protein kinases (MAPK), ERK1 and ERK2 [49]{Rowan, 2000 #174;Shah, 2005 #56}. Growth factors such as EGF, interleukin-6 and cAMP can all induce phosphorylation of SRC-1 at specific consensus sites to increase its coactivator activity for ligand-bound and unliganded steroid receptor proteins. It has been shown that cAMP-induced phosphorylation of SRC-1 at Thr1179 and Ser1185 contribute to the steroid independent activation of both ERα and PR [50], whilst EGF-induced phosphorylation of SRC-1 can increase PR-dependent transcription [49].
SRC-3 has been previously associated with a specific PTM known as the glycogen synthase 3 (GSK3β) phospho-ubiquitin clock [51]. Initially, this PTM was believed to be specific to SRC-3 however a recent study has shown that SRC-1 is subject to similar regulation [52]. The phospho-ubiquitin clock refers to the phosphorylation of the SRC protein by GSK3β which increases the rate of turnover and the interaction of the SRC protein with a specific NR and then induces the ubiquitylation of the protein to ensure that the transcriptional event terminates at an appropriate time [53]. Current evidence suggests that the phospho-ubiquitin clock is conserved across all three p160 family members [52], this is an important mechanism through which the SRC proteins can maintain tight regulatory control over their many biological processes.
Biological Functions of SRC-1
1. The role of SRC-1 in reproduction and the uterus
Numerous studies over the years have accumulated evidence and shown that the SRC-1 protein is widely expressed in different tissues (Figure 2). SRC-1 knockout mice were generated to investigate the basic biological functions of SRC-1 in these tissues [54]. The targeted deletion of the SRC-1 gene in mice disrupted its nuclear receptor binding and transcriptional activation functions and presumably, its recruited HAT activity from the secondary coactivators such as CBP, p300 and p/CAF [54]. Despite these disruptions, SRC-1 null mice exhibited no obvious phenotype. Both male and female homozygotes were fertile and displayed a similar growth rate compared to the wild type mice [54]. However, further investigations revealed that steroid activity was partially impaired in SRC-1 null mice. Disruption of SRC-1 reduced estrogen-induced uterine growth; hence SRC-1 null mice achieved less than 60% uterine growth than that of the wild type mice when treated with estrogen. Furthermore, it was observed that SRC-1 is required for maximal uterine response to steroid hormones in vivo [54]. SRC-1 is required for estrogen and progesterone-induced activities of ER and PR in the uterus but not in the breast [55]. Furthermore, mitogen-inducible gene 6 (Mig6) has been identified as a downstream target of PR and SRC-1 in the uterus. Mig6 is required to maintain endometrial homeostasis in response to estrogen and has also been proposed as an important protein in the suppression of endometrial cancer [56].
The human tissues with SRC-1 protein expression.
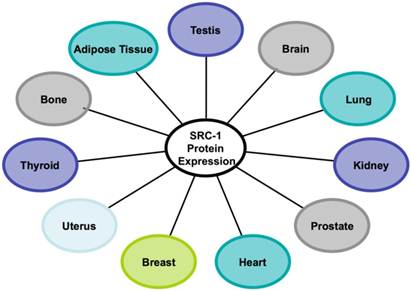
SRC-1 is also expressed in the glandular and stromal cells of the normal endometrium. Immunoprecipitation assays revealed that estrogen-induced SRC-1 interaction with endometrial ERα only occurred during the proliferative phase of the menstrual cycle, indicating that SRC-1 has a regulatory role specific to this cycle phase [57, 58].
2. The role of SRC-1 in the prostate and testis
SRC-1 is also a known coactivator of the androgen receptor (AR) and it is recruited alongside SWI3-related gene product (SRG3) for AR-mediated cell proliferation during prostate development and regeneration [59]. SRC-1 null male mice exhibit reduced responsiveness to androgen stimulation versus controls. SRC-1 knockout mice also show a reduction in prostate and urethra weight compared with wild type animals. Histological examination of testes in SRC-1 null mice showed smaller glands without any apparent structural abnormalities [54].
3. The role of SRC-1 in the mammary gland
Mammary gland development is a tightly regulated, steroid dependent process. In wild type female mice, mammary gland ducts grow extensively and fill the fat pad by 8 weeks of age [60]. However, in the absence of SRC-1, ductal density is reduced and the number and extent of branching occupies much less of the fat pad area in mice of a similar age. SRC-1 is required for normal mammary duct elongation during puberty and alveolar development during pregnancy. Alveoli number and size were reduced in SRC-1 null mice compared with wild type mice of the same pregnant stage, although significantly the null mice did retain the capacity to produce milk [54].
4. The role of SRC-1 in other organs
SRC-1 is critical for appropriate brain development and function. In Purkinje cells (PCs) of the cerebellum, SRC-1 is more highly expressed than other SRC members. SRC-1 deficiency retards PC development during embryogenesis and neonatal stages. Adult SRC-1 null mice exhibit moderate motor learning deficiencies [61]. The delay of PC development at the embryonic and neonatal stages is mainly attributed to both late birth and slow migration of SRC-1 knockout PC precursors [61].
SRC-1 is a functional coactivator of the thyroid receptor (TR), and loss of SRC-1 activity causes partial resistance to thyroid hormone signaling [37, 62]. Simultaneous knockout of SRC-1 and thyroid hormone receptor proteins (SRC-1/TRα and SRC-1/TRβ knockout mice) caused more significant growth retardation than single knockout of either TRβ or TRα, suggesting SRC-1 is important for TRβ or TRα-mediated body growth [63].
In addition, SRC-1 deletion led to skeletal resistance to estrogen predominantly in the cancellous bones [64, 65]. Under both physiological and pathological conditions, SRC-1 was shown to play an important role in the maintenance of bone mineral density by sex hormones and is necessary for protection against osteopenia and osteoporosis [66]. Interestingly, a genetic polymorphic variant of the SRC-1 protein (SNP-1272S) was found to be associated with a decrease in bone mineral density [52, 67].
Finally, SRC-1 is an important mediator of the protective effects of estrogen in the cardiovascular system. SRC-1 is expressed in the endothelial cells and the vascular smooth muscle cells (VSMCs) of the normal arterial wall. SRC-1 expression is also found in the cells of neointima, induced by vascular injury. SRC-1-/- mice exhibit reduced vasoprotective capabilities in the vascular wall, which manifests as enhanced neointima formation and as a marked increase in VSMC proliferation in response to vascular injury [68]. Furthermore, SRC-1 is coexpressed with SRC-3 in the blood vessel wall and the roles of these two family members demonstrate considerable functional redundancy with regards to the execution of estrogen induced vasoprotective mechanisms [69].
5. Metabolic functions of SRC-1
The NR, PPARγ, is critical for the development of both white and brown adipose tissue [70]. SRC-1 activity in the adipose tissue is dependent on PPARγ and its coactivator, PPARγ coactivator-1 (PGC-1). When PGC-1 binds to PPARγ, it undergoes a conformational change and can complex with SRC-1 and CBP/p300, to significantly enhance the transcriptional activity of PPARγ in brown adipose tissue [71]. Inactivation of SRC-1 impairs the thermogenic activity of PGC-1 in the brown adipose tissue, it decreases energy expenditure and increases susceptibility to obesity following a high fat diet. The ratio of SRC-1 expression relative to SRC-2 also has a significant impact on energy homeostasis as a loss of SRC-2 enables SRC-1 to bind PGC-1 with little or no competition. As a result, SRC-2-/- mice have increased thermogenic activity, they are also protected against obesity and demonstrate high insulin sensitivity [72]. SRC-1 also acts in conjunction with SRC-3 to selectively regulate the expression of genes involved in brown adipose tissue development. SRC-1-/-/SRC-3-/- knockout mice have underdeveloped adipose tissue compartments, which are incapable of lipid storage and consequentially, demonstrate defective thermogenic response mechanisms [73].
Finally, in the liver, SRC-1 controls hepatic glucose production via its coactivation of the non steroidal C/EBPα transcription factor. C/EBPα signals through a feed-forward loop to transactivate pyruvate carboxylase, which is a crucial gene for the initiation of gluconeogenesis. SRC-1 null mice are hypoglycemic in both fed and fasting states, it is thus suggested that C/EBPα is unable to function in the absence of SRC-1. Hence, SRC-1 has a key role in the maintenance of glucose homeostasis in the liver [74]. SRC-1 also functions in the liver to coactivate the LXRα/RXR heterodimer. LXRα is commonly involved in lipid homeostasis and fatty acid synthesis in the hepatocytes. In this context, SRC-1 signals in conjunction with the LXRα/RXR heterodimer to upregulate transcription of the ABC transporter protein, ABCA1, which accelerates fecal cholesterol disposal by decreasing the efficiency of cholesterol absorption. DAX-1 is a member of the nuclear receptor superfamily that can repress other NR-mediated transcription. In the liver, DAX-1 directly interacts and competes with SRC-1 to repress LXRα transcriptional activity in the liver and control lipid homeostasis [75]. The ability of coactivators to compete and synergistically regulate transcription in response to homeostatic signals is an important aspect of their basic biological functions.
6. Redundant functions with other SRC proteins
Given the structural homology of the p160 family members, it is expected that SRC-1 would have certain redundant functions with SRC-2 and SRC-3 [76]. Genetic studies in mice have demonstrated partial functional redundancy between SRC-1 and SRC-2 with regards to postnatal survival and growth [77]. Furthermore, analysis of SRC-2 ablation in the testes results in abnormal spermatogenesis, age-dependent degeneration of seminiferous epithelium, and disorder of cholesterol homeostasis. However, this phenotype is significantly worsened in SRC-2 null mice with heterozygous SRC-1 knockout [77].
Further compensatory action was observed in the uterus of PRCre/+;SRC-2flox/flox mouse, where the additional loss of SRC-1 resulted in the complete absence of a decidual response. This indicates that the co-expressed SRC-2 and SRC-1 in the uterus cooperate in progesterone-initiated transcriptional programs to coordinate decidualization [78].
It was discovered that SRC-1 and SRC-3 both function to support labyrinth morphogenesis of the placenta. The labyrinth is an important component of the placenta which connects the maternal blood sinuses and fetal vascular networks. A double knockout of SRC-1 and SRC-3 increased proliferation of labyrinth trophoblasts and progenitor gene expression whilst down-regulating the expression of differentiation-related genes. Deficiency of SRC-1 and SRC-3 also affected the expression of several genes relevant to placental morphogenesis and glucose transportation. It was also noted that labyrinths of SRC-1/SRC-3 knockout embryos were smaller than wild type and single knockout embryos [79].
SRC-1 can also cooperate with SRC-3 to enable embryo survival. The majority of SRC-1 and SRC-3 double-knockout embryos die by E13.5. In contrast, SRC-1 single knockout embryos develop normally and SRC-3 single knockout embryos only exhibit partial lethality [79].
The Role of SRC-1 in Breast Cancer
SRCs are expressed at low levels in multiple tissues fundamental to the proper growth and development of these organs and SRCs often contribute to cancer in the same tissues. The general consensus as reported from both molecular and clinical studies of breast cancer is that SRC-1 has specific roles in the mediation of metastasis, in the switch from steroid dependent to steroid independent regulation and in the prediction of disease recurrence.
1. SRC-1 expression in human breast tumors
SRC-1 protein expression is very low in human mammary gland ductal epithelial cells although it is steadily detectable in the nuclei of mouse mammary ductal epithelial cells. However, SRC-1 protein was detected in 19% to 34% of human breast tumors. SRC-1 expression is significantly associated with large, high grade tumors, HER2 positivity, disease recurrence and resistance to endocrine therapy. SRC-1 expression also serves as an independent predictor of disease-free survival (Table 1) [14, 80, 81].
IHC Analysis of SRC-1 Protein Expression in Human Breast Cancer.
| References | SRC-1 Expression by IHC, (n) | Molecular Association | Pathological Association | Additional Observations |
|---|---|---|---|---|
| Hudelist et al. 2003 | 28% (25) | ERβ+, SRC-2+ | No association between SRC-1 and normal or malignant cancer | - |
| Fleming et al. 2004a | 29% (52) | ERα+ | Poor outcome Lymph node positive patients | 92% of recurrences were SRC-1 positive SRC-1 associated with resistance to endocrine therapy |
| Fleming et al. 2004b | 26% (70) | HER2+ PEA3+ | Reduced disease free survival Recurrence | Significant predictor of time to disease recurrence |
| Myers et al. 2004 | 19% (150) | HER2+ ERβ- | Larger tumors (>35mm) Reduced disease free survival | 90% of recurrences were SRC-1 positive Significant predictor of disease free survival |
| Myers et al. 2005 | 24% (134) | HER2+ Ets-1+ Ets-2+ | High grade tumors Recurrence | Both Ets-1 and Ets-2 colocalize with SRC-1 in tumor cells SRC-1 associates with phospho-Raf |
| Redmond et al. 2009 | 34% (560) | HER2+ COX2+ | Distant metastasis Reduced disease free survival | Significant predictor of time to disease recurrence Increased SRC-1 colocalization with ERα in Tamoxifen-treated recurrences |
| McIlroy et al. 2010 | HOXC11+ S100β+ | SRC-1 and HOXC11 are strong predictors of reduced disease free survival in a Tamoxifen treated population | ||
| McCartan et al. 2011 | ADAM22+ | SRC-1 and ADAM22 are independent predictors of disease recurrence |
IHC immunohistochemistry; n, number of patient population; (+), positive association; (-), negative association; ER, estrogen receptor.
2. The SRC-1-/-;MMTV-PyMT model
The role of SRC-1 in mammary tumor initiation, progression and metastasis has been investigated by knocking out SRC-1 in the MMTV-PyMT (mouse mammary tumor virus-polyoma middle T) transgenic mice [82]. Analysis of mammary tumorigenesis in SRC-1-/-;MMTV-PyMT and MMTV-PyMT mice revealed that SRC-1 is not required for mammary tumor initiation and growth. However, SRC-1 does impact on the differentiation and polarized structuring of the mammary epithelial tumor cells in the three-dimensional culture [82]. Furthermore, SRC-1 activity has been strongly associated with mammary tumor cell intravasation and metastasis to the lungs. Blood samples taken from MMTV-PyMT mice showed a significantly higher number of mammary tumor cells in the circulation than that from the SRC-1-/-;MMTV-PyMT mice, indicating that SRC-1 promotes migration of breast cancer cells and invasion of these tumor cells into the vascular system.
SRC-1 expressed in the mammary tumor cells also upregulates the expression and secretion of colony stimulating factor-1 (CSF-1) to facilitate the recruitment of macrophages to the mammary tumor sites [82]. Macrophage recruitment has been associated with invasiveness and metastasis of breast tumor cells to local and distant sites [83]. At the tumor site, macrophages secrete factors such as EGF and TGF-β1 that stimulate proliferation and invasiveness of tumor cells [84]. Macrophages also induce angiogenesis via secretion of proangiogenic cytokines such as TNF-α and IL-8. In turn, these stimulate c-Jun-NH2-kinase and NFκβ signaling pathway to induce local release of the proinvasive angiogenesis-inducing MMP enzymes.
Importantly, the metastatic nature of SRC-1 is intrinsic to the tumor cells as demonstrated by reciprocal transplantation of MMTV-PyMT and SRC-1-/-;MMTV-PyMT tumors into SRC-1-/- and wild type recipient mice [82]. In the MMTV-PyMT model, loss of SRC-1 also correlated closely with reduced expression of the HER2 oncogene as well as with reduced levels of the steroid independent transcription factor Ets2, thus further confirming the associations between SRC-1 and growth factor signaling effectors in breast cancer [82].
3. The SRC-1;MMTV-neu model
The SRC-1-/-;MMTV-neu mouse breast cancer model was generated by crossing MMTV-neu transgenic mice with SRC-1 knockout mice. In this model, SRC-1 deficiency increased tumor latency and reduced tumor cell proliferation index and lung metastasis. As observed in the SRC-1-/-;MMTV-PyMT model, SRC-1-/-;MMTV-neu tumors were also more differentiated than the MMTV-neu tumors. Molecular analysis demonstrated an increase in the expression of several cell cycle inhibitors [85].
The findings from both SRC-1-/-;MMTV-PyMT and SRC-1-/-;MMTV-neu breast cancer models indicate that SRC-1 plays a crucial role in promotion of breast cancer metastasis, while in the MMTV-neu model SRC-1 also enhances tumor initiation and growth, suggesting that the role of SRC-1 on tumor growth may be specific to individual oncogene-induced tumorigenic pathways. In addition, since the tumors from both models are ER negative, SRC-1 likely promotes tumorigenesis and metastasis in these models through estrogen-independent pathways.
4. Growth factor signaling and SRC-1
SRC-1 is highly expressed in HER2 positive breast cancers [80, 81, 86] and has been shown to coactivate Ets2, a transcription factor activated by the HER2-MAPK signaling pathway. Under the influence of HER2, Ets2 and SRC-1 are recruited to the promoter of the c-myc oncogene. In turn, c-myc expression promotes breast tumor cell survival, metastasis and resistance to both Tamoxifen and aromatase inhibitors [87, 88].
Simultaneous overexpression of SRC-1 and HER2 frequently occurs and has important implications in tumor cell proliferation. SRC-1 mediates overexpression of stromal cell-derived factor alpha (SDF-1α) while HER2 signaling mechanisms mediate the stabilization of SDF-1α's specific receptor CXCR4 [89]. It has been reported that SDF-1a:CXCR4 signaling complex is required for the proliferation of cancer cells at sites of metastasis [90]. The formation of the SDF-1a:CXCR4 complex is one example of the ability of SRC-1 signaling to crosstalk with that of other pathways and therein promote disease progression.
5. Mechanisms of SRC-1 action in breast cancer metastasis
A role for SRC-1 in breast cancer metastasis has been established and to date, two distinct mechanisms have been elucidated. In the first instance, SRC-1 impacts on the epithelial to mesenchymal transition (EMT) and epithelial depolarization via regulation of the EMT transcription factor Twist. In the second, SRC-1 upregulates the expression of integrin α5 to promote cell migration and invasion.
Twist: EMT is characterized by the loss of epithelial differentiation and the gain of mesenchymal properties within the cellular population. This transition enables tumor cells to invade and survive in the stromal tissues. In essence, EMT may be an early step towards metastasis and is characterized by certain gene expression changes [17, 91]. In vitro experiments from the wild type and SRC-1 knockout cell lines show that SRC-1 inversely correlates with the major EMT hallmark protein, E-cadherin. Furthermore, SRC-1 regulates the EMT transcription factor, Twist, a known suppressor of E-cadherin. It serves as a master molecular switch of the EMT program [92].
Observations from HER2-positive cancer cells suggest that the MAPK pathway controls the direct phosphorylation, stabilization and accumulation of Twist protein expression. This was further confirmed in tumor samples where HER2 positively correlated with Twist phosphorylation, total Twist protein expression and activity of the c-Jun N-terminal kinase (JNK). JNK is a member of the MAPK family. Moreover, Twist is a transcriptional target for SRC-1 and the polyoma enhancer activator 3 (PEA3), a downstream MAPK effector [17]. Incidentally, these observations in HER2 positive tumors also correlate with negative PR status and are independent of ERα status. PR negative cancers are more aggressive and have associations with resistance to therapy and bone metastasis hence substantiating the tumorigenic potential role of Twist in breast cancer [93].
Integrin α5: SRC-1 can also mediate metastatic action in conjunction with the non-steroidal transcription factor AP-1 and the tumor microenvironment. Resident fibroblasts produce abundant extracellular matrix (ECM) proteins to provide anchorage for tumor cell adhesion and migration [94]. Heterodimeric integrin transmembrane receptors bind to ECM proteins to transport signals bidirectionally across the cell membrane, allowing cells to respond to environmental changes [94]. In breast cancer, these integrin receptors have been associated with tumor cell survival, growth and metastasis in an anchorage-independent manner. In particular, the heterodimeric receptor, α5β1, is increased in malignant breast cancer and is associated with poor prognosis [95]. SRC-1 serves as a coactivator for the transcription factor AP-1, the two complexing together at the promoter of the integrin α5 gene, enhances its transcription. In the absence of SRC-1 cell adhesion and migration via the ECM-integrin-focal adhesion kinase (FAK) pathway are reduced. The downstream signaling components of this pathway are also significantly reduced in the absence of SRC-1 and the integrin α5 [16], suggesting that integrin α5 is a key target gene of SRC-1 in its efforts to orchestrate the local invasion and metastatic survival of breast cancer tumor cells.
6. SRC-1 and resistance to Tamoxifen therapy
SRC-1 has been increasingly cited with regards to the development of resistance to endocrine therapy in breast cancer. Resistance to endocrine therapy represents a serious clinical dilemma and is an increasingly prevalent occurrence among breast cancer patients. Tamoxifen is a selective estrogen receptor modulator (SERM) and is the most common anti-estrogen therapy prescribed for pre-menopausal ERα-positive breast cancers. Studies carried out in HeLa cells where Tamoxifen is an agonist, have shown that overexpression of SRC-1 further increases the activity of Tamoxifen [96]. Similarly, in the Ishikawa endometrial cancer cell line where Tamoxifen is also agonistic, high levels of SRC-1 expression were reported compared to the MCF-7 breast cancer cell line [97]. Interestingly, src kinase activity has been shown to specifically enhance SRC-1 and CBP activity in these Ishikawa cells [48], thus associating high levels of SRC-1 protein and growth factor signaling pathways with tissues that are receptive to the agonistic properties of Tamoxifen [97]. In addition, a non-synonomous small nucleotide polymorphism (SNP-P1272S) was identified in the DNA sequence of SRC-1, which decreased SRC-1 activity. Specifically, this study also demonstrated that the SNP-P1272S had a negative impact on the agonistic effects of Tamoxifen on bone mineral density in post menopausal women [52].
With regards to the development of resistance, it was reported that SRC-1 overexpression could convert Tamoxifen from a transcriptional repressor to a transcriptional activator in breast cancer [98]. It has been shown that SRC-3 could compete with the Tamoxifen-recruited transcriptional repressor PAX and result in upregulation of HER2 expression [99]. The stoichiometry of PAX and SRC-3 was thus shown to dictate the efficacy of Tamoxifen treatment in breast cancer as HER2 and SRC-3 upregulation have been positively associated with the development of Tamoxifen resistance in breast cancer. Similar effects have also been seen with NRs other than ER, as overexpression of SRC-1 is also capable of inducing PR activity in the presence of the PR antagonist RU486 [100].
As therapeutic resistance becomes more prevalent, the ability to predict positive patient response to Tamoxifen is becoming increasingly important. Treatment with Tamoxifen itself has been shown to increase the expression levels of both SRC-1 and SRC-3 in multiple studies [101, 102]. It was observed that the expression of SRC-1, SRC-3 and multiple cell cycle and cell adhesion genes was increased, while the expression of apoptotic genes and PR was concomitantly downregulated during the development of a Tamoxifen-resistant cell line. Loss of PR is indicative of an alteration in tumor steroid receptor profile, which is a common feature in the development of Tamoxifen resistance. Interestingly, the SRC-1 protein level was decreased in the resistant cells once Tamoxifen treatment was stopped for a 12 months period [103]. It was also reported that the co-association of SRC-1 and SRC-3 with ERα was increased in LY2 endocrine-resistant breast cancer cells following Tamoxifen treatment in comparison with endocrine-sensitive MCF-7 cells, and that the colocalization of SRC-1 and SRC-3 with ERα was significantly enhanced in patients who have relapsed on endocrine treatment in comparison with those patients who did not undergo recurrence. Overall, analysis of the clinical data suggest that SRC-1 is a strong independent predictor of reduced disease free survival and that the interactions of the p160 SRC proteins with ERα can predict the response of patients to endocrine therapy [104].
An aberrant up-regulation of growth factor pathways which target the activity of SRC proteins is a common feature in the development of an endocrine resistant phenotype. The HER2 pathway induces protein kinase A (PKA) to phosphorylate ERα at serine 305 in the presence of Tamoxifen. This phosphorylation alters the orientation of ERα and SRC-1 and switches Tamoxifen to an ERα agonist, resulting in RNA Polymerase II recruitment and transcriptional initiation [105]. These findings were supported by a separate study in which Tamoxifen resistance was also associated with ERα phosphorylation at serine 305 [106].
Additional clinical data showed an increased co-association of SRC-1 with MAPK-activated transcription factors PEA3 and Ets2 in a Tamoxifen treated patient population [14, 86]. As expected, both of these co-associations correlated positively with HER2 positivity in the same population. In agreement with the upregulation of c-myc by Ets2 and SRC-1 in cultured cells, this upregulation was also observed in the Tamoxifen-treated patient population.
More recent data has identified two more transcription factors that work with SRC-1. Firstly, HOXC11, a member of the homeobox family of developmental proteins was identified as a functional binding partner for SRC-1 in Tamoxifen-resistant LY2 cells. Hox family proteins have been implicated in EGF-mediated migration and adhesion as well with the agonistic properties of Tamoxifen. Immunohistochemical analysis of a Tamoxifen-treated patient population showed increased localization and expression of HOXC11 in the nuclei of tumor cells. Strong associations were also observed between HOXC11 and SRC-1 in the resistant LY2 cells compared to the Tamoxifen-sensitive MCF-7 cells. This observation was repeated in the patient population. Moreover, coexpression of HOXC11 with SRC-1 was noted as a superior predictor of poor clinical prognosis than any of the other classic parameters [15]. This study also recognized S100β as the transcriptional target for the HOXC11 and SRC-1 complex. In breast cancer patients, the elevated levels of S100β significantly predicted poor disease free survival. Since S100β is detectable in human serum, it can be potentially used as a biomarker to predict and monitor disease progression.
Secondly, SRC-1 interacts with the transcription factor MYB to directly regulate ADAM22, a non-protease member of the ADAM family of disintegrins [107]. Molecular analysis carried out in Tamoxifen-resistant cell lines discovered a role for ADAM22 in cellular migration and differentiation. In addition, expression of ADAM22 mRNA was increased in the Tamoxifen-resistant tumors in xenograft mouse models. Furthermore, ADAM22 expression was identified by immunohistochemical analysis in a clinical patient population as an independent predictor of poor disease free survival [107]. ADAM22 has also been proposed as a potential therapeutic target as its migratory action can be inhibited following treatment with its ligand LGI1 [107].
7. SRC-1 and resistance to aromatase inhibitor therapy
SRC-1 activity has been found to be associated with resistance to aromatase inhibitors (AIs). Like Tamoxifen resistance, AI resistance is also marked by a shift in cancer cell signaling from steroid dependency towards dependency on peptide growth factors. In AI resistant cell lines, loss of SRC-1 improved cells' ability to differentiate while reducing their migratory capacity. Interestingly, loss of ERα in these cells failed to impact on migration. This indicates that SRC-1 may also act in a steroid independent manner to promote AI resistance. In AI-resistant cells, SRC-1 also interacts with MAPK-activated Ets2 transcription factor. Specifically, SRC-1 and Ets2 are recruited to the c-myc and MMP9 promoters upon AI (Letrozole) treatment. Importantly, in an AI-treated patient population with two thirds of SRC-1-positive and one third of SRC-1-negative primary tumors, all AI-treated patients with disease relapse from original SRC-1-negative tumors presented AI-resistant SRC-1-positive tumors. This finding has important and exciting implications for the specific ability of SRC-1 to advance metastasis in AI-treated breast cancer [88].
SRC-1 in Prostate Cancer
SRC-1 has been linked to prostate cancer and extensively evaluated as an AR coactivator in both androgen-dependent and independent disease. In localized, androgen-dependent tumors, increased SRC-1 expression correlates with lymph node metastasis [108]. Ablation of SRC-1 in androgen-dependent LNCaP prostate cancer cells represses activation of AR target genes and reduces AR-dependent cellular proliferation [108]. Furthermore, in CV1 cells, SRC-1 overexpression increases the androstenedione-induced AR activity [109].
SRC-1 also has a role in androgen-independent prostate cancer. Although one clinical study found SRC-1 mRNA expression was lower in therapy-resistant prostate cancer compared with untreated patients [110], others evaluating tumors from resistant patients found SRC-1 expression significantly increased in hormone-refractory tumors compared to androgen-dependent tumors or benign prostatic hyperplasia [109, 111]. Re-activation of AR-mediated transcriptional programs in the absence of androgen is critical for the growth and survival of the majority of castration-resistant tumors. The elevated expression of SRC-1 in these prostate tumors raises a possibility that SRC-1 may support androgen-independent and AR-mediated transcription and promote hormone-refractory tumor growth. In support of this idea, CWR22 human prostate cancer cells xenografted into mice have decreased SRC-1 expression immediately following castration, but have significantly increased SRC-1 expression at the time of tumor relapse [109]. In androgen-independent C4-2 prostate cancer cells that depend on AR for proliferation, knockdown of SRC-1 significantly reduced growth and AR target gene expression. In contrast, knockdown of SRC-1 had no effect on the growth of the AR-negative PC-3 and DU145 prostate cancer cell lines [108]. Finally, SRC-1 is involved in ligand-independent activation of AR by IL-6, a cytokine that regulates proliferation, apoptosis and angiogenesis in prostate cancer [112].
The direct in vivo contribution of SRC-1 to prostate cancer was investigated through the use of genetically engineered mouse models. The SRC-1 gene was deleted in TRAMP mice, which harbor the oncogenic SV40 T/t antigen transgene driven by the prostate epithelium-specific probasin promoter [113]. Tumor mass, histology and biomarkers in cancers from these mice were assessed at various ages; however no significant difference in tumor initiation or progression between SRC1-/- and wild type TRAMP mice was found. Interestingly, expression of SRC-3, was significantly elevated thus indicating that, in TRAMP mice, SRC-1 and SRC-3 may have analogous functions and SRC-3 over-expression may be able to compensate for SRC-1 deficiency [113]. Although its contribution to tumourigenesis is non-essential in TRAMP mice, the significant findings in human samples and cell lines suggest that SRC-1 may still be a critical prostate cancer mediator.
SRC-1 Expression in Other Cancers
1. Endometrial cancer
SRC-1 dysregulation has been reported in endometrial cancer. SRC-1 mRNA levels are significantly increased in malignant endometrial cancer and the high expression of SRC-1 positively correlates with ERα and PR expression. However, no significant correlation was found between SRC-1 mRNA expression and grade, stage, or depth of myometrial invasion [114]. Conversely, SRC-1 expression was reported as reduced in the less aggressive endometrial hyperplasia and in endometrial carcinoma compared with normal glandular cells in the proliferative phase [115].
2. Thyroid cancer
In thyroid cancer, SRC-1 is expressed in epithelial tumor cells. In non-anaplastic tumors, SRC-1 expression is significantly associated with HER2 expression, and together their levels correlate with poor cellular differentiation, capsular invasion and disease progression. In anaplastic thyroid tumors, SRC-1 is expressed in 87% of samples and again positively correlates with reduced survival rate (p<0.001) [116] .
3. Cutaneous melanoma
Analogous to that observed in breast cancer, SRC-1 also serves as a coactivator for HOXC11 to upregulate S100β in cutaneous melanomas. Serum levels of S100β have been proposed as a marker of tumor burden, predicted poor response to treatment and poor prognosis in melanoma patients [117, 118]. Higher expression levels and co-association of SRC-1 and HOXC11 were reported in malignant melanoma compared to benign nevi. The same was also observed in a metastatic melanoma cell line. Interestingly, disruption of HOXC11 and SRC-1-regulated S100β in malignant cells using the dual Src/Abl inhibitor, Desatinib, can interfere with the phosphorylation of SRC-1 by Src kinase [119]. This may represent a new therapeutic approach for the treatment of advanced melanoma.
SRC Proteins as Potential Therapeutic Targets
Given the wide-reaching oncogenic effects of the SRC proteins, particularly SRC-1 and SRC-3, significant strides are being made to elucidate their potential as effective biological therapeutic targets. Previous reluctance to pursue SRCs as therapeutic targets arose from their large size, lack of structural conformity and the absence of any high affinity ligand-binding domains. However, new proof of principle experiments has challenged this view and shown that small molecule inhibitors (SMIs) such as gossypol, a natural polyphenol, are capable of reducing SRC-1 and SRC-3 proteins in MCF-7 and other cancer cell lines. Interestingly, SRC-2 was not susceptible to the inhibitory effects of gossypol treatment. Gossypol was shown to physically interfere with the binding between the SRC-3 RID and the ERα ligand binding domain. However, the resultant degradation of SRC-1 and SRC-3 occurred independently of a proteasomal mechanism. This ability of gossypol to reduce SRC-1 and SRC-3 proteins was also maintained independently of the various ER ligands such as estrogen, Tamoxifen and ICI 182,780. Moreover, the gossypol action is independent of ERα as it also degrades SRC-3 in ERα negative cancer cells. Treatment of cancer cells with gossypol also resensitized the cells to MEK, EGF and IGF pathway inhibitors. Of significant importance, this form of SMI treatment is preferentially toxic to cancerous cells and is relatively selective for SRC-1 and SRC-3 [120]. This work is the first to show that SRC proteins are accessible to SMI-based chemotherapy. Continued research into this area could have significant implications for treatment of cancers, especially those resistant to current therapies.
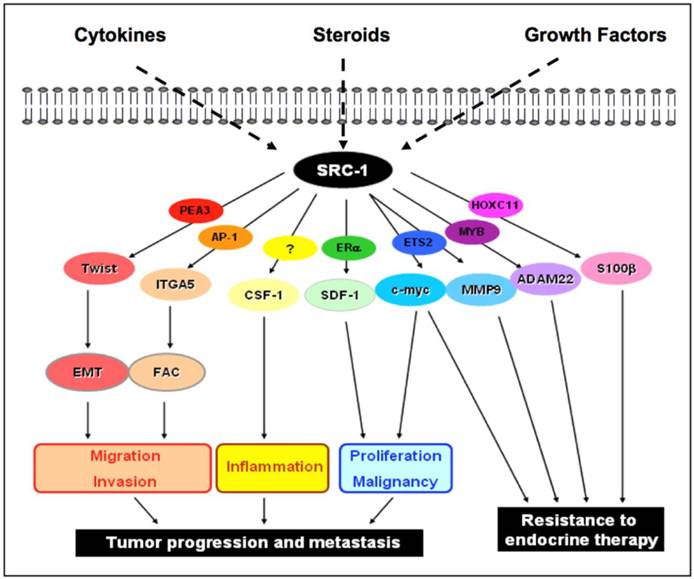
SRC-1 coregulator activity in breast cancer. SRC-1 is susceptible to activation by the cytokine, steroid or growth factor pathways. These pathways encode the SRC -1 protein to coactivate a specific transcription factor in order to mediate specific transcriptional events within the nucleus. The schematic diagram illustrates the potential transcriptional binding partners of SRC-1 and their respective downstream effector targets. Under the influence of either steroid dependent or steroid independent signaling networks, SRC-1 will interact with different transcription factors to form varying complexes for transcriptional regulation of tumor cell processes that will promote an increasingly aggressive and resistant tumor phenotype. EMT: epithelial mesenchymal transition; FAC: focal adhesion complex.
Conclusion
The discovery and characterization of the p160 SRC family have revolutionized the field of molecular endocrinology and endocrine-related oncology. SRCs have been described as “the powerhouses of transcription” and have been rightfully reclassified as “master genes” on the basis of their ability to bind across unrelated families of transcription factors and coordinate the regulation of multiple genes in multiple complex physiological states [121]. SRC-1 epitomizes all of these master qualities and the recent work outlined in this review highlights this transcriptional coactivator as a key player in normal development, multiple cellular signaling crosstalk, promoting resistance to endocrine treatments and coordination of tumor cell metastasis.
Acknowledgements
This work is partially supported by Cancer Prevention and Research Institute of Texas grant RP101251-P4 and National Institutes of Health grants R01 CA112403 and R01 DK058242 to J.X., Department of Defense Prostate Cancer Research Program Predoctoral Scholarship PC102004 to J.C.Y.T., and grants from Science Foundation Ireland (B1853) and Health Research Board, Ireland (PHD/2007/11) to L.S.Y. and C.A.W.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Spelsberg TC, Steggles AW, O'Malley BW. Progesterone-binding Components of Chick Oviduct. J Biol Chem. 1971;246:4188-97
2. York B, O'Malley BW. Steroid Receptor Coactivator (SRC) Family: Masters of Systems Biology. J Biol Chem. 2010;285:38743-50
3. Klein-Hitpass L, Tsai SY, Weigel NL. et al. The progesterone receptor stimulates cell-free transcription by enhancing the formation of a stable preinitiation complex. Cell. 1990;60:247-57
4. Kim J, Stallcup M. P160 Coactivators: Critical Mediators of Transcriptional Activation by Nuclear Receptors. Singapore: World Scientific Publishing Co Pte Ltd. 2008
5. Meyer M-E, Gronemeyer H, Turcotte B. et al. Steroid hormone receptors compete for factors that mediate their enhancer function. Cell. 1989;57:433-42
6. Onate S, Tsai S, Tsai MJ. et al. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;24:1354-7
7. Wang J-C, Stafford JM, Granner DK. SRC-1 and GRIP1 Coactivate Transcription with Hepatocyte Nuclear Factor 4. J Biol Chem. 1998;273:30847-50
8. Zhu Y, Qi C, Calandra C. et al. Cloning and identification of mouse steroid receptor coactivator-1 (mSRC-1), as a coactivator of peroxisome proliferator-activated receptor gamma. Gene Expr. 1996;6:185-95
9. Ding XF, Anderson CM, Ma H. et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol Endocrinol. 1998;12:302-13
10. Zhou G, Cummings R, Li Y. et al. Nuclear receptors have distinct affinities for coactivators: characterization by fluorescence resonance energy transfer. Mol Endocrinol. 1998;12:1594-604
11. Lee S-K, Kim H-J, Na S-Y. et al. Steroid Receptor Coactivator-1 Coactivates Activating Protein-1-mediated Transactivations through Interaction with the c-Jun and c-Fos Subunits. J Biol Chem. 1998;273:16651-4
12. Kim H-J, Kim JH, Lee JW. Steroid Receptor Coactivator-1 Interacts with Serum Response Factor and Coactivates Serum Response Element-mediated Transactivations. J Biol Chem. 1998;273:28564-7
13. Na S-Y, Lee S-K, Han S-J. et al. Steroid Receptor Coactivator-1 Interacts with the p50 Subunit and Coactivates Nuclear Factor κB-mediated Transactivations. J Biol Chem. 1998;273:10831-4
14. Myers E, Hill ADK, Kelly G. et al. Associations and Interactions between Ets-1 and Ets-2 and Coregulatory Proteins, SRC-1, AIB1, and NCoR in Breast Cancer. Clin Cancer Res. 2005;11:2111-22
15. McIlroy M, McCartan D, Early S. et al. Interaction of Developmental Transcription Factor HOXC11 with Steroid Receptor Coactivator SRC-1 Mediates Resistance to Endocrine Therapy in Breast Cancer. Cancer Res. 2010;70:1585-94
16. Qin L, Chen X, Wu Y. et al. Steroid Receptor Coactivator-1 Upregulates Integrin α5 Expression to Promote Breast Cancer Cell Adhesion and Migration. Cancer Res. 2011;71:1742-51
17. Qin L, Liu Z, Chen H. et al. The Steroid Receptor Coactivator-1 Regulates Twist Expression and Promotes Breast Cancer Metastasis. Cancer Res. 2009;69:3819-27
18. Hong H, Kohli K, Trivedi A. et al. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci U S A. 1996;93:4948-52
19. Torchia J, Rose DW, Inostroza J. et al. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677-84
20. Lonard DM, O'Malley BW. Nuclear Receptor Coregulators: Judges, Juries, and Executioners of Cellular Regulation. Mol Cell. 2007;27:691-700
21. Zhang H, Yi X, Sun X. et al. Differential gene regulation by the SRC family of coactivators. Genes Dev. 2004;18:1753-65
22. Kim JH, Li H, Stallcup MR. CoCoA, a Nuclear Receptor Coactivator which Acts through an N-Terminal Activation Domain of p160 Coactivators. Mol cell. 2003;12:1537-49
23. Liu Z, Wong J, Tsai SY. et al. Sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc Natl Acad Sci U S A. 2001;98:12426-31
24. Li C, Wu R-C, Amazit L. et al. Specific Amino Acid Residues in the Basic Helix-Loop-Helix Domain of SRC-3 Are Essential for Its Nuclear Localization and Proteasome-Dependent Turnover. Mol Cell Biol. 2007;27:1296-308
25. Chang C-y, Norris JD, Grøn H. et al. Dissection of the LXXLL Nuclear Receptor-Coactivator Interaction Motif Using Combinatorial Peptide Libraries: Discovery of Peptide Antagonists of Estrogen Receptors α and β. Mol Cell Biol. 1999;19:8226-39
26. Heery DM, Kalkhoven E, Hoare S. et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733-6
27. Lanz RB, Lonard DM, O'Malley BW. Nuclear Receptor Coregulators in Human Diseases. Singapore: World Scientific Publishing Co Pte Ltd. 2008
28. Coulthard VH, Matsuda S, Heery DM. An Extended LXXLL Motif Sequence Determines the Nuclear Receptor Binding Specificity of TRAP220. J Biol Chem. 2003;278:10942-51
29. Heery DM, Hoare S, Hussain S. et al. Core LXXLL motif sequences in CREB-binding protein, SRC1, and RIP140 define affinity and selectivity for steroid and retinoid receptors. J Biol Chem. 2001;276:6695-702
30. Shiau AK, Barstad D, Loria PM. et al. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95:927-37
31. Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000;21:381-8
32. Kraus WL, Manning ET, Kadonaga JT. Biochemical Analysis of Distinct Activation Functions in p300 That Enhance Transcription Initiation with Chromatin Templates. Mol Cell Biol. 1999;19:8123-35
33. Chen H, Lin RJ, Schiltz RL. et al. Nuclear Receptor Coactivator ACTR Is a Novel Histone Acetyltransferase and Forms a Multimeric Activation Complex with P/CAF and CBP/p300. Cell. 1997;90:569-80
34. Spencer TE, Jenster G, Burcin MM. et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194-8
35. Yang X-J, Ogryzko VV, Nishikawa J-i. et al. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319-24
36. Voegel J, Heine M, Zechel C. et al. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667-75
37. Onate SA, Boonyaratanakornkit V, Spencer TE. et al. The Steroid Receptor Coactivator-1 Contains Multiple Receptor Interacting and Activation Domains That Cooperatively Enhance the Activation Function 1 (AF1) and AF2 Domains of Steroid Receptors. J Biol Chem. 1998;273:12101-8
38. Voegel JJ, Heine MJS, Tini M. et al. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J. 1998;17:507-19
39. Li J, O'Malley BW, Wong J. p300 Requires Its Histone Acetyltransferase Activity and SRC-1 Interaction Domain To Facilitate Thyroid Hormone Receptor Activation in Chromatin. Mol Cell Biol. 2000;20:2031-42
40. Nakajima T, Uchida C, Anderson SF. et al. RNA Helicase A Mediates Association of CBP with RNA Polymerase II. Cell. 1997;90:1107-12
41. Koh SS, Chen D, Lee Y-H. et al. Synergistic Enhancement of Nuclear Receptor Function by p160 Coactivators and Two Coactivators with Protein Methyltransferase Activities. J Biol Chem. 2001;276:1089-98
42. Daujat S, Bauer U-M, Shah V. et al. Crosstalk between CARM1 Methylation and CBP Acetylation on Histone H3. Curr Biol. 2002;12:2090-7
43. Lee DY, Teyssier C, Strahl BD. et al. Role of Protein Methylation in Regulation of Transcription. Endocr Rev. 2005;26:147-70
44. Wang H, Huang Z, Xia L. et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853-7
45. Johnson AB, O'Malley BW. Steroid receptor coactivators 1, 2, and 3: Critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol. 2012;348:430-9
46. Belandia B, Orford RL, Hurst HC. et al. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002;21:4094-103
47. Xu J, Wu R-C, O'Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615-30
48. Shah YM, Rowan BG. The Src Kinase Pathway Promotes Tamoxifen Agonist Action in Ishikawa Endometrial Cells through Phosphorylation-Dependent Stabilization of Estrogen Receptor α Promoter Interaction and Elevated Steroid Receptor Coactivator 1 Activity. Mol Endocrinol. 2005;19:732-48
49. Rowan BG, Weigel NL, O'Malley BW. Phosphorylation of Steroid Receptor Coactivator-1. J Biol Chem. 2000;275:4475-83
50. Coleman KM, Dutertre M, El-Gharbawy A. et al. Mechanistic Differences in the Activation of Estrogen Receptor-α (ERα)- and ERβ-dependent Gene Expression by cAMP Signaling Pathway(s). J Biol Chem. 2003;278:12834-45
51. Wu R-C, Feng Q, Lonard DM. et al. SRC-3 Coactivator Functional Lifetime Is Regulated by a Phospho-Dependent Ubiquitin Time Clock. Cell. 2007;129:1125-40
52. Hartmaier RJ, Richter AS, Gillihan RM. et al. A SNP in Steroid Receptor Coactivator-1 Disrupts a GSK3beta Phosphorylation Site and Is Associated with Altered Tamoxifen Response in Bone. Mol Endocrinol. 2012;26:220-7
53. O'Malley BW. The “Fourth Dimension” of Gene Transcription. Mol Endocrinol. 2009;23:587-9
54. Xu J, Qiu Y, DeMayo FJ. et al. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science. 1998;279:1922-5
55. Han SJ, DeMayo FJ, Xu J. et al. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol Endocrinol. 2006;20:45-55
56. Jeong JW, Lee HS, Lee KY. et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc Natl Acad Sci U S A. 2009;106:8677-82
57. Shiozawa T, Shih HC, Miyamoto T. et al. Cyclic changes in the expression of steroid receptor coactivators and corepressors in the normal human endometrium. J Clin Endocrinol Metab. 2003;88:871-8
58. Wieser F, Schneeberger C, Hudelist G. et al. Endometrial nuclear receptor co-factors SRC-1 and N-CoR are increased in human endometrium during menstruation. Mol Hum Reprod. 2002;8:644-50
59. Hong CY, Suh JH, Kim K. et al. Modulation of androgen receptor transactivation by the SWI3-related gene product (SRG3) in multiple ways. Mol Cell Biol. 2005;25:4841-52
60. Topper YJ, Freeman CS. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 1980;60:1049-106
61. Nishihara E, Yoshida-Komiya H, Chan CS. et al. SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J Neurosci. 2003;23:213-22
62. Weiss RE, Xu J, Ning G. et al. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J. 1999;18:1900-4
63. Sadow PM, Koo E, Chassande O. et al. Thyroid hormone receptor-specific interactions with steroid receptor coactivator-1 in the pituitary. Mol Endocrinol. 2003;17:882-94
64. Modder UI, Sanyal A, Kearns AE. et al. Effects of loss of steroid receptor coactivator-1 on the skeletal response to estrogen in mice. Endocrinology. 2004;145:913-21
65. Mödder UI, Sanyal A, Xu J. et al. The skeletal response to estrogen is impaired in female but not in male steroid receptor coactivator (SRC)-1 knock out mice. Bone. 2008;42:414-21
66. Yamada T, Kawano H, Sekine K. et al. SRC-1 is necessary for skeletal responses to sex hormones in both males and females. J Bone Miner Res. 2004;19:1452-61
67. Hartmaier R, Tchatchou S, Richter A. et al. Nuclear receptor coregulator SNP discovery and impact on breast cancer risk. BMC Cancer. 2009;9:438
68. Yuan Y, Xu J. Loss-of-Function Deletion of the Steroid Receptor Coactivator-1 Gene in Mice Reduces Estrogen Effect on the Vascular Injury Response. Arterioscler Thromb Vasc Biol. 2007;27:1521-7
69. Yuan Y, Liao L, Tulis DA. et al. Steroid Receptor Coactivator-3 Is Required for Inhibition of Neointima Formation by Estrogen. Circulation. 2002;105:2653-9
70. Barak Y, Nelson MC, Ong ES. et al. PPAR³ Is Required for Placental, Cardiac, and Adipose Tissue Development. Mol Cell. 1999;4:585-95
71. Puigserver P, Adelmant G, Wu Z. et al. Activation of the PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368-71
72. Picard F, Géhin M, Annicotte J-S. et al. SRC-1 and TIF2 Control Energy Balance between White and Brown Adipose Tissues. Cell. 2002;111:931-41
73. Wang Z, Qi C, Krones A. et al. Critical roles of the p160 transcriptional coactivators p/CIP and SRC-1 in energy balance. Cell Metab. 2006;3:111-22
74. Louet J-F, Chopra AR, Sagen JV. et al. The Coactivator SRC-1 Is an Essential Coordinator of Hepatic Glucose Production. Cell Metab. 2010;12:606-18
75. Nedumaran B, Kim GS, Hong S. et al. Orphan nuclear receptor DAX-1 acts as a novel corepressor of liver X receptor alpha and inhibits hepatic lipogenesis. J Biol Chem. 2010;285:9221-32
76. Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681-92
77. Mark M, Yoshida-Komiya H, Gehin M. et al. Partially redundant functions of SRC-1 and TIF2 in postnatal survival and male reproduction. Proc Natl Acad Sci U S A. 2004;101:4453-8
78. Mukherjee A, Soyal SM, Fernandez-Valdivia R. et al. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol Cell Biol. 2006;26:6571-83
79. Chen X, Liu Z, Xu J. The Cooperative Function of Nuclear Receptor Coactivator 1 (NCOA1) and NCOA3 in Placental Development and Embryo Survival. Mol Endocrinol. 2010;24:1917-34
80. Hudelist G, Czerwenka K, Kubista E. et al. Expression of Sex Steroid Receptors and their Co-Factors in Normal and Malignant Breast Tissue: AIB1 is a Carcinoma-Specific Co-Activator. Breast Cancer Res Treat. 2003;78:193-204
81. Fleming FJ, Hill ADK, McDermott EW. et al. Differential Recruitment of Coregulator Proteins Steroid Receptor Coactivator-1 and Silencing Mediator for Retinoid and Thyroid Receptors to the Estrogen Receptor-Estrogen Response Element by β-Estradiol and 4-Hydroxytamoxifen in Human Breast Cancer. J Clin Endocrinol Metab. 2004;89:375-83
82. Wang S, Yuan Y, Liao L. et al. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Natl Acad Sci U S A. 2009;106:151-6
83. Leek RD, Lewis CE, Whitehouse R. et al. Association of Macrophage Infiltration with Angiogenesis and Prognosis in Invasive Breast Carcinoma. Cancer Res. 1996;56:4625-9
84. Wyckoff J, Wang W, Lin EY. et al. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004;64:7022-9
85. Han J, Crowe D. Steroid receptor coactivator 1 deficiency increases MMTV-neu mediated tumor latency and differentiation specific gene expression, decreases metastasis, and inhibits response to PPAR ligands. BMC Cancer. 2010;10:629
86. Fleming FJ, Myers E, Kelly G. et al. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol. 2004;57:1069-74
87. Al-azawi D, McIlroy M, Kelly G. et al. Ets-2 and p160 proteins collaborate to regulate c-Myc in endocrine resistant breast cancer. Oncogene. 2008;27:3021-31
88. McBryan J, Theissen SM, Byrne C. et al. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 2012;72:548-59
89. Kishimoto H, Wang Z, Bhat-Nakshatri P. et al. The p160 family coactivators regulate breast cancer cell proliferation and invasion through autocrine/paracrine activity of SDF-1α/CXCL12. Carcinogenesis. 2005;26:1706-15
90. Li YM, Pan Y, Wei Y. et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer cell. 2004;6:459-69
91. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
92. Yang J, Mani SA, Donaher JL. et al. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell. 2004;117:927-39
93. Hong J, Zhou J, Fu J. et al. Phosphorylation of Serine 68 of Twist1 by MAPKs Stabilizes Twist1 Protein and Promotes Breast Cancer Cell Invasiveness. Cancer Res. 2011;71:3980-90
94. Luo B-H, Carman CV, Springer TA. Structural Basis of Integrin Regulation and Signaling. Annu Rev Immunol. 2007;25:619-47
95. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9-22
96. Smith CL, Nawaz Z, O'Malley BW. Coactivator and Corepressor Regulation of the Agonist/Antagonist Activity of the Mixed Antiestrogen, 4-Hydroxytamoxifen. Mol Endocrinol. 1997;11:657-66
97. Shang Y, Brown M. Molecular Determinants for the tissue specificty of SERMs. Science. 2002;295:2465-8
98. Romano A, Adriaens M, Kuenen S. et al. Identification of novel ER-α target genes in breast cancer cells: Gene- and cell-selective co-regulator recruitment at target promoters determines the response to 17β-estradiol and tamoxifen. Mol Cell Endocrinol. 2010;314:90-100
99. Hurtado A, Holmes KA, Geistlinger TR. et al. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663-6
100. Liu Z, Auboeuf D, Wong J. et al. Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc Natl Acad Sci U S A. 2002;99:7940-4
101. Lonard DM, Tsai SY, O'Malley BW. Selective Estrogen Receptor Modulators 4-Hydroxytamoxifen and Raloxifene Impact the Stability and Function of SRC-1 and SRC-3 Coactivator Proteins. Mol Cell Biol. 2004;24:14-24
102. Haugan Moi LL, Hauglid Flågeng M, Gandini S. et al. Effect of Low-Dose Tamoxifen on Steroid Receptor Coactivator 3/Amplified in Breast Cancer 1 in Normal and Malignant Human Breast Tissue. Clin Cancer Res. 2010;16:2176-86
103. Scott D, Parkes A, Ponchel F. et al. Changes in expression of steroid receptors, their downstream target genes, and their associated co-regulators during the sequential acquisition of tamoxifen resistance in vitro. Int J Oncol. 2007;31:557-65
104. Redmond AM, Bane FT, Stafford AT. et al. Coassociation of Estrogen Receptor and p160 Proteins Predicts Resistance to Endocrine Treatment; SRC-1 is an Independent Predictor of Breast Cancer Recurrence. Clin Cancer Res. 2009;15:2098-106
105. Zwart W, Griekspoor A, Berno V. et al. PKA-induced resistance to tamoxifen is associated with an altered orientation of ER[alpha] towards co-activator SRC-1. EMBO J. 2007;26:3534-44
106. Holm C, Rayala S, Jirström K. et al. Association Between Pak1 Expression and Subcellular Localization and Tamoxifen Resistance in Breast Cancer Patients. J Natl Cancer Inst. 2006;98:671-80
107. McCartan D, Bolger J, Fagan A. et al. Global characterization of the SRC-1 transcriptome identifies ADAM22 as an ER-independent mediator of endocrine resistant breast cancer. Cancer Res. 2012;72:220-9
108. Agoulnik IU, Vaid A, Bingman WE. et al. Role of SRC-1 in the Promotion of Prostate Cancer Cell Growth and Tumor Progression. Cancer Res. 2005;65:7959-67
109. Gregory CW, He B, Johnson RT. et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315-9
110. Linja MJ, Porkka KP, Kang Z. et al. Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res. 2004;10:1032-40
111. Maki HE, Waltering KK, Wallen MJ. et al. Screening of genetic and expression alterations of SRC1 gene in prostate cancer. Prostate. 2006;66:1391-8
112. Ueda T, Mawji NR, Bruchovsky N. et al. Ligand-independent Activation of the Androgen Receptor by Interleukin-6 and the Role of Steroid Receptor Coactivator-1 in Prostate Cancer Cells. J Biol Chem. 2002;277:38087-94
113. Tien JC, Zhou S, Xu J. The role of SRC-1 in murine prostate cancinogenesis is nonessential due to a possible compensation of SRC-3/AIB1 overexpression. Int J Biol Sci. 2009;5:256-64
114. Kershah SM, Desouki MM, Koterba KL. et al. Expression of estrogen receptor coregulators in normal and malignant human endometrium. Gynecol Oncol. 2004;92:304-13
115. Uchikawa J, Shiozawa T, Shih HC. et al. Expression of steroid receptor coactivators and corepressors in human endometrial hyperplasia and carcinoma with relevance to steroid receptors and Ki-67 expression. Cancer. 2003;98:2207-13
116. Kavanagh DO, McIlroy M, Myers E. et al. The role of oestrogen receptor α in human thyroid cancer: contributions from coregulatory proteins and the tyrosine kinase receptor HER2. Endocr Relat Cancer. 2010;17:255-64
117. Donato R, Sorci G, Riuzzi F. et al. S100B's double life: Intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793:1008-22
118. Mocellin S, Zavagno G, Nitti D. The prognostic value of serum S100B in patients with cutaneous melanoma: A meta-analysis. Int J Cancer. 2008;123:2370-6
119. deBlacam C, Byrne C, Hughes E. et al. HOXC11-SRC-1 regulation of S100beta in cutaneous melanoma: new targets for the kinase inhibitor dasatinib. Br J Cancer. 2011;105:118-23
120. Wang Y, Lonard DM, Yu Y. et al. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 2011;25:2041-53
121. O'Malley BW. Masters of the genome. Nat Rev Mol Cell Biol. 2010;11:311
Author contact
![]() Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine. E-mail: jxuedu.
Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine. E-mail: jxuedu.