ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2012; 8(4):561-579. doi:10.7150/ijbs.3540 This issue Cite
Research Paper
Characterization of the Complete Mitochondrial Genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae)
Huan-Na Chai 1, Yu-Zhou Du 1 ![]() , Bao-Ping Zhai 2
, Bao-Ping Zhai 2
1. Institute of Applied Entomology, Yangzhou University, Yangzhou 225009, China;
2. Department of Entomology, Nanjing Agricultural University, Nanjing 210095, China.
Abstract
The complete mitochondrial genomes (mitogenomes) of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae) were determined and analyzed. The circular genomes were 15,388 bp long for C. medinalis and 15,395 bp long for C. suppressalis. Both mitogenomes contained 37 genes, with gene order similar to that of other lepidopterans. Notably, 12 protein-coding genes (PCGs) utilized the standard ATN, but the cox1 gene used CGA as the initiation codon; the cox1, cox2, and nad4 genes in the two mitogenomes had the truncated termination codons T, T, and TA, respectively, but the nad5 gene was found to use T as the termination codon only in the C. medinalis mitogenome. Additionally, the codon distribution and Relative Synonymous Codon Usage of the 13 PCGs in the C. medinalis mitogenome were very different from those in other pyralid moth mitogenomes. Most of the tRNA genes had typical cloverleaf secondary structures. However, the dihydrouridine (DHU) arm of the trnS1(AGN) gene did not form a stable stem-loop structure. Forty-nine helices in six domains, and 33 helices in three domains were present in the secondary structures of the rrnL and rrnS genes of the two mitogenomes, respectively. There were four major intergenic spacers, except for the A+T-rich region, spanning at least 12 bp in the two mitogenomes. The A+T-rich region contained an 'ATAGT(A)'-like motif followed by a poly-T stretch in the two mitogenomes. In addition, there were a potential stem-loop structure, a duplicated 25-bp repeat element, and a microsatellite '(TA)13' observed in the A+T-rich region of the C. medinalis mitogenome. A poly-T motif, a duplicated 31-bp repeat element, and a 19-bp triplication were found in the C. suppressalis mitogenome. However, there are many differences in the A+T-rich regions between the C. suppressalis mitogenome sequence in the present study and previous reports. Finally, the phylogenetic relationships of these insects were reconstructed based on amino acid sequences of mitochondrial 13 PCGs using Bayesian inference and maximum likelihood methods. These molecular-based phylogenies support the traditional morphologically based view of relationships within the Pyralidae.
Keywords: Mitochondrial genome, Cnaphalocrocis medinalis, Chilo suppressalis, Lepidoptera, Pyralidae, phylogenetic relationship.
Introduction
The mitochondrial genome (mitogenome), which is responsible for the oxidative reactions of the tricarboxylic acid cycle, as well as electron transfer and energy metabolism in cells, forms a unit of genetic information, evolving independently from the nuclear genome [1]. The mitogenome is characterized by its small size, maternal inheritance, stable and relatively short circular structure, lack of recombination, and frequent polymorphisms in most cells [2, 3]. In addition, it can provide sets of genome-level characters, such as the relative position of different genes, RNA secondary structures and modes of control of replication and transcription [4]. Thus, the mitogenome has been widely used as an informative molecular marker for diverse evolutionary studies of animals in the past several decades, including phylogenetics and population genetics [5, 6].
The insect mitogenome is a closed-circular duplex molecule, ranging from 13 to 20 kb in length, containing 13 protein-coding genes [PCGs: two subunits of the ATPase (atp6 and atp8), three cytochrome coxidase subunits (cox1, cox2, and cox3), one cytochrome B (cob), seven NADH dehydrogenase subunits (nad1, nad2, nad3, nad4, nad5, nad6, and nad4L), two ribosomal RNA genes (rRNAs: rrnL and rrnS), and 22 transfer RNA genes (tRNAs)]. Additionally, it has at least one sequence known in insect mitogenomes as the A+T-rich region, which includes some initiation sites for transcription and replication of the genome [7, 8]. The length of this region is highly variable in insects, due to the indels and the presence of variable copy numbers of tandem repeat elements [9].
To date, complete or near-complete mitogenomes have been sequenced from more than 241 species of insects. However, only 36 complete or near-complete mitogenomes are available for Lepidoptera, a large group of some 200,000 species. Sequences are only available from species in the Bombycoidea, Geometroidea, Papilionoidea, Noctuoidea, Tortricoidea, and Pyraloidea. Among the Pyraloidea, sequenced mitogenomes are available for six species: Cnaphalocrocis medinalis, Chilo suppressalis, Diatraea saccharalis, Maruca vitrata, Ostrinia furnacalis, and Ostrinia nubilalis [10-13]. However, data for three of these pyraloids, M. vitrata, O. furnacalis and O. nubilalis, lack some sequence information. Additional sequenced mitogenomes from the Lepidoptera can provide more detail about molecular phylogenetics of this important group.
The Asiatic leaf roller, C. medinalis, and the rice stem borer, C. suppressalis, are both well-known rice pests widely distributed in the main rice-growing regions of China, Japan, Korea, India, Australia, and other countries. In China, these rice pests are found from Heilongjiang to Taiwan and Hainan, and have caused serious yield losses in recent decades [14, 15]. In this study, we report the complete mitogenomes of C. medinalis and C. suppressalis and give a thorough description of their genome features, including gene order, nucleotide composition of protein-coding genes, secondary structures of tRNA and rRNA genes, and the A+T-rich region. In addition, we compare the C. suppressalis mitogenome sequence from this study with one from a previous study. Detailed genetic information on these two important rice pests may help in the development of methods for their control or prevention. Furthermore, information on C. medinalis, a migratory species, may provide insights into the energy supply mechanism used in its migration.
Materials and Methods
DNA sample extraction
Adults of C. medinalis and C. suppressalis were collected from rice paddies in Yangzhou (32o40.025N, 119o44.017E), Jiangsu Province, China, in July-October 2010. Samples were preserved in 100% ethanol and stored at -70oC until DNA extraction was performed. Whole genomic DNA was extracted from a single sample using the protocol of DNAVzol (Bioteke, Beijing, China) and then used for the PCR amplification.
PCR amplification, cloning and sequencing
The DNA samples from C. medinalis and C. suppressalis were amplified based on the primers from D. saccharalis, M. vitrata, O. furnacalis, O. nubilalis, and other lepidopteran sequences available in GenBank [11-13]. All these primer pairs were designed using Primer Premier 5.0 software. Conditions for PCR amplification were as follows: an initial denaturation for 5 min at 95 oC, followed by 35 cycles of denaturation for 1 min at 94 oC, annealing for 1 min at 45-50 oC, elongation for 1-3 min (depending on putative length of the fragments) at 68 oC, and a final extension step of 72 oC for 10 min. LA Taq polymerase (TaKaRa, Dalian, China) was used in PCR amplification, except for fragments less than 1.3 kb, which were amplified with Taq polymerase (TaKaRa) instead. All PCR reactions were performed in an ABI thermal cycler (PE Applied Biosystems, San Francisco, CA, USA).
PCR products were resolved by electrophoresis in a 1.0% agarose gel and extracted using a DNA Gel Extraction Kit (Bioteke, Beijing, China). Purified PCR products were ligated into T-vector (TaKaRa) before transformation into DH5α competent Escherichia coli cells. The positive recombinant clone was sequenced using upstream and downstream primers from both directions on ABI 3730XL Genetic Analyzer (PE Applied Biosystems) at least three times. The number of clones sequenced provided six-fold coverage of the mitogenome.
Genome annotation and secondary structures prediction
Protein-coding genes and rRNA genes were identified by comparison with other lepidopteran species previously sequenced [11-13]. Protein-coding genes were aligned using Clustal X version 2.0 [16]. Composition skew analysis was carried out with formulas AT skew = [A-T]/[A+T] and GC skew = [G-C]/[G+C], respectively [17]. The PCG nucleotide sequences without start and termination codons were translated on the basis of the Invertebrate Mitochondrial Genetic Code. The A+T content and codon usage were calculated using MEGA version 4.0 [18]. Transfer RNA genes were identified using tRNAscan-SE software available online at http://lowelab.ucsc.edu/tRNAscan-SE/ [19], and XRNA 1.2.0b was used to draw the secondary structure of tRNAs. The secondary structures of rrnL and rrnS genes were inferred based on models developed for other insect species [20-22]. To infer the secondary structures of tRNA and rRNA genes, we used a commonly accepted comparative approach to correct for unusual pairings with RNA-editing mechanisms that are well known in arthropod mitogenomes [20, 23]. A compensatory change was first defined as two substitutions occurring sequentially that maintained base pairing in a given position of a helix, then two or more Watson-Crick (or G-U) interactions at the same location in a putative helix indicated that the Watson-Crick process allowed to preserve correct base pairing in such a way that the structure conformed to the helical model. [24]. The entire A+T-rich region was subjected to a search for tandem repeats using the Tandem Repeats Finder program [25].
Phylogenetic analysis
The 34 complete or near-complete lepidopteran mitogenomes were downloaded from Genbank as references to determine phylogenetic relationships within this set of pyralids, using the mitogenome of Drosophila melanogaster as the outgroup [33] (Table 1). The amino acid sequences of each of the 13 PCGs were aligned with Clustal X version 2.0 using default settings and concatenation [16]. The concatenated set of amino acid sequences from all 13 PCGs was then used for phylogenetic analyses. The best-fitting model, by Modeltest [48] using likelihood ratio tests, was then used to perform Bayesian inferences (BI) and maximum likelihood (ML) analysis using the program MrBayes 3.1.2 (http://morphbank.ebc.uu.SE/mrbayes/) [49] and a PHYML online web server [50]. The BI analyses were conducted under the following conditions: 1,000,000 generations, four chains (one cold chain and three hot chains) and a burn-in step for the first 10,000 generations. The confidence values of the BI tree were expressed as the Bayesian posterior probabilities in percentages. The ML analysis was conducted using the proportion of invariable sites as “estimated,” the number of substitution rate categories as four, the gamma distribution parameter as “estimated,” and the starting tree as a BIONJ distance-based tree. The confidence values of the ML tree were evaluated via a bootstrap test with 100 iterations.
Source and information for the polygenomic analysis.
| Surperfamily | Family | Species | Length /bp | Accession Number | References |
|---|---|---|---|---|---|
| Bombycoidea | Saturniidae | Antheraea pernyi | 15575 | AY242996 | [26] |
| Saturniidae | Antheraea yamamai | 15338 | EU726630 | [27] | |
| Bombycidae | Bombyx mandarina | 15717 | FJ384796 | [28] | |
| Bombycidae | Bombyx mori | 15664 | AY048187 | [29] | |
| Saturniidae | Eriogyna pyretorum | 15327 | FJ685653 | [30] | |
| Sphingidae | Manduca sexta | 15516 | EU286785 | [31] | |
| Saturniidae | Saturnia boisduvalii | 15360 | EF622227 | [32] | |
| Drosophiloidea | Drosophilidae | Drosophila melanogaster | 19517 | U37541 | [33] |
| Geometroidea | Geometridae | Phthonandria atrilineata | 15499 | EU569764 | [34] |
| Noctuoidea | Noctuidae | Helicoverpa armigera | 15347 | GU188273 | [35] |
| Hypercompe | Hyphantria cunea | 15481 | GU592049 | [36] | |
| Lymantridae; | Lymantria dispar | 15569 | FJ617240 | [37] | |
| Notodontidae | Ochrogaster lunifer | 15593 | AM946601 | [38] | |
| Noctuidae | Sesamia inferens | 15413 | JN039362 | Unpublished | |
| Papilionoidea | Acraeidae | Acraea issoria | 15245 | GQ376195 | [39] |
| Pieridae | Artogeia melete | 15140 | EU597124 | [40] | |
| Nymphalidae | Apatura metis | 15236 | JF801742 | Unpublished | |
| Nymphalidae | Calinaga davidis | 15267 | HQ658143 | [41] | |
| Lycaenidae | Coreana raphaelis | 15314 | DQ102703 | [42] | |
| Nymphalidae | Hipparchia autonoe | 15489 | GQ868707 | [43] | |
| Papilionodae | Luehdorfia chinensis | 13860 | EU622524 | Unpublished | |
| Papilionodae | Papilio maraho | 16094 | FJ810212 | Unpublished | |
| Parnassiidae | Parnassius bremeri | 15389 | FJ871125 | [44] | |
| Papilionodae | Papilio xuthus | 13964 | EF621724 | [45] | |
| Nymphalidae | Sasakia charonda | 15244 | AP011824 | Unpublished | |
| Nymphalidae | Sasakia charonda uriyamaensis | 15222 | AP011825 | Unpublished | |
| Papilionodae | Teinopalpus aureus | 15242 | HM563681 | Unpublished | |
| Papilionodae | Troides aeacus | 15263 | EU625344 | Unpublished | |
| Pyraloidea | Pyralidae | Chilo suppressalis | 15395 | JF339041 | Unpublished |
| Pyralidae | Cnaphalocrocis medinalis | 15388 | JN246082 | Unpublished | |
| Pyralidae | Diatraea saccharalis | 15490 | FJ240227 | [11] | |
| Pyralidae | Maruca vitrata | 14054 | HM751150 | [12] | |
| Pyralidae | Ostrinia furnacalis | 14536 | AF467260 | [13] | |
| Pyralidae | Ostrinia nubilalis | 14535 | AF442957 | [13] | |
| Tortricoidea | Tortricidae | Adoxophyes honmai | 15680 | DQ073916 | [46] |
| Olethreutidae | Grapholita molesta | 15717 | HQ392511 | [24] | |
| Olethreutidae | Spilonota lechriaspis | 15368 | HM204705 | [47] |
Results
Genome structure and base composition
The complete mitogenomes of C. medinalis and C. suppressalis were found to be circular molecules 15,388 bp and 15,395 bp in size, respectively (Fig. 1). The sequences were submitted to GenBank under the accession numbers JN246082 (C. medinalis) and JF339041 (C. suppressalis). Both mitogenomes contained 37 typical mitochondrial genes (13 PCGs, 22 tRNA genes, and two rRNA genes), of which 23 were transcribed on the majority-coding strand (H-strand), and the remaining were transcribed on the minority-coding strand (L-strand) (Fig. 1 and Table 2).
The nucleotide compositions of the whole mitogenome of C. medinalis and C. suppressalis were as follows: (A) 40.3%, 40.6%; (T) 41.6%, 40.0%; (G) 7.5%, 7.5%; and (C) 10.6%, 11.9%, respectively. The whole mitogenome of C. medinalis was biased towards AT nucleotides (81.9%), similar to that of C. suppressalis (80.6%) (Table 3). Furthermore, GC% content as well as AT- and GC-skews were calculated for the mitogenomes of C. medinalis and C. suppressalis (Table 3). The results showed that the AT-skew of the C. medinalis mitogenome was -0.016 and was biased to use T rather than A, whereas the C. suppressalis mitogenome exhibited an AT-skew of 0.007. Meanwhile, GC-skews were similar in both the C. medinalis (-0.171) and C. suppressalis (-0.227) mitogenomes (Table 3).
Map of mitogenomes of Cnaphalocrocis medinalis and Chilo suppressalis. Transfer RNA genes are designated by single-letter amino acid codes. CR indicates A+T-rich region. Gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left.
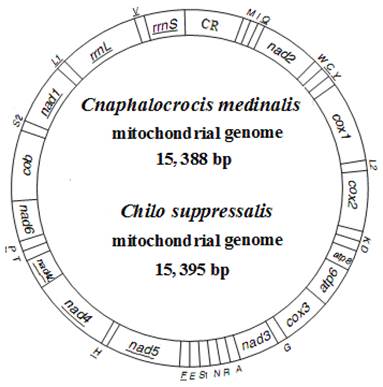
Annotion of the mitogenomes of Cnaphalocrocis medinalis (Cm) and Chilo suppressalis (Cs).
| Gene | Direction | Region | Size (bp) | IGN | Start codon | Stop codon | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | ||
| trnM | F | 1..68 | 1..68 | 68 | 68 | 0 | -1 | - | - | - | - |
| trnI | F | 69..133 | 68..133 | 65 | 66 | -3 | 0 | - | - | - | - |
| trnQ | R | 131..199 | 134..202 | 69 | 69 | 74 | 52 | - | - | - | - |
| nad2 | F | 274..1290 | 255..1268 | 1017 | 1014 | 5 | 7 | ATT | ATT | TAA | TAA |
| trnW | F | 1296..1363 | 1276..1343 | 68 | 68 | -8 | -8 | - | - | - | - |
| trnC | R | 1356..1423 | 1336..1402 | 68 | 67 | 5 | 30 | - | - | - | - |
| trnY | R | 1429..1495 | 1433..1500 | 67 | 68 | 41 | 6 | - | - | - | - |
| cox1 | F | 1537..3067 | 1507..3037 | 1531 | 1531 | 3 | 0 | CGA | CGA | T- | T- |
| trnL2(UUR) | F | 3068..3134 | 3038..3104 | 67 | 67 | 0 | 0 | - | - | - | - |
| cox2 | F | 3135..3816 | 3105..3786 | 682 | 682 | 0 | 0 | ATG | ATA | T- | T- |
| trnK | F | 3817..3887 | 3787..3857 | 71 | 71 | 11 | 0 | - | - | - | - |
| trnD | F | 3899..3964 | 3858..3925 | 66 | 68 | 0 | 0 | - | - | - | - |
| atp8 | F | 3965..4132 | 3926..4090 | 168 | 165 | -7 | -7 | ATT | ATC | TAA | TAA |
| atp6 | F | 4126..4800 | 4084..4764 | 675 | 681 | -1 | -1 | ATG | ATG | TAA | TAA |
| cox3 | F | 4800..5588 | 4764..5552 | 789 | 789 | 2 | 2 | ATG | ATG | TAA | TAA |
| trnG | F | 5591..5655 | 5555..5621 | 65 | 67 | 0 | 0 | - | - | - | - |
| nad3 | F | 5656..6009 | 5622..5975 | 354 | 354 | 46 | 101 | ATT | ATT | TAA | TAA |
| trnA | F | 6056..6121 | 6077..6144 | 66 | 68 | 1 | -1 | - | - | - | - |
| trnR | F | 6123..6189 | 6144..6212 | 67 | 69 | 0 | -1 | - | - | - | - |
| trnN | F | 6190..6256 | 6212..6278 | 67 | 67 | 2 | 1 | - | - | - | - |
| trnS1(AGN) | F | 6259..6324 | 6280..6345 | 66 | 66 | 1 | 0 | - | - | - | - |
| trnE | F | 6326..6393 | 6346..6414 | 68 | 69 | -2 | 0 | - | - | - | - |
| trnF | R | 6392..6460 | 6415.. 6481 | 69 | 67 | 0 | -17 | - | - | - | - |
| nad5 | R | 6461..8195 | 6465.. 8216 | 1735 | 1752 | 0 | 0 | ATT | ATT | T- | TAA |
| trnH | R | 8196..8261 | 8217.. 8281 | 66 | 65 | 0 | 0 | - | - | - | - |
| nad4 | R | 8262..9601 | 8282.. 9621 | 1340 | 1340 | -1 | 7 | ATG | ATG | TA- | TA- |
| nad4L | R | 9601..9894 | 9629.. 9922 | 294 | 294 | 2 | 2 | ATG | ATG | TAA | TAA |
| trnT | F | 9897..9961 | 9925.. 9989 | 65 | 65 | 0 | 0 | - | - | - | - |
| trnP | R | 9962..10027 | 9990..10055 | 66 | 66 | 2 | 2 | - | - | - | - |
| nad6 | F | 10030..10566 | 10058..10597 | 537 | 540 | 6 | -1 | ATT | ATT | TAA | TAA |
| cob | F | 10573..11721 | 10597..11742 | 1149 | 1146 | 1 | 3 | ATG | ATG | TAA | TAA |
| trnS2(UCN) | F | 11723..11790 | 11746..11811 | 68 | 66 | 16 | 12 | - | - | - | - |
| nad1 | R | 11807..12745 | 11824..12765 | 939 | 942 | 1 | 1 | ATG | ATG | TAA | TAA |
| trnL1(CUN) | R | 12747..12813 | 12767..12833 | 67 | 67 | -1 | -25 | - | - | - | - |
| rrnL | R | 12814..14202 | 12809..14191 | 1384 | 1383 | 0 | 0 | - | - | - | - |
| trnV | R | 14203..14268 | 14192..14259 | 66 | 68 | 0 | 0 | - | - | - | - |
| rrnS | R | 14269..15049 | 14260..15047 | 781 | 788 | 0 | 0 | - | - | - | - |
| A+T-rich region | 15050..15388 | 15048..15395 | 339 | 348 | 0 | 0 | - | - | - | - | |
IGN indicates intergenic nucleotide.
Skewed nucleotide composition in regions of Cnaphalocrocis medinalis (Cm) and Chilo suppressalis (Cs) mitogenomes.
| Region | A/% | G/% | T/% | C/% | A+T/% | G+C/% | AT skew/% | GC skew/% | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | Cm | Cs | |
| Whole mitogenome | 40.3 | 40.6 | 7.5 | 7.5 | 41.6 | 40.0 | 10.6 | 11.9 | 81.9 | 80.6 | 18.1 | 19.4 | -0.016 | 0.007 | -0.171 | -0.227 |
| PCGs | 34.7 | 33.9 | 10.2 | 10.8 | 45.8 | 45.0 | 9.3 | 10.3 | 80.5 | 78.9 | 19.5 | 21.2 | -0.138 | -0.140 | 0.046 | 0.024 |
| 1st codon position | 37.1 | 22.2 | 15.9 | 13.1 | 37.5 | 48.4 | 9.5 | 16.3 | 74.6 | 70.6 | 25.4 | 29.4 | -0.005 | -0.371 | 0.252 | -0.109 |
| 2nd codon position | 22.3 | 42.4 | 13.0 | 3.2 | 48.7 | 50.0 | 16.0 | 4.4 | 71.0 | 92.4 | 29.0 | 7.6 | -0.372 | -0.082 | -0.103 | -0.158 |
| 3rd codon position | 44.8 | 37.0 | 1.7 | 16.1 | 51.2 | 36.6 | 2.3 | 10.3 | 96.0 | 73.6 | 4.0 | 26.4 | -0.067 | 0.005 | -0.150 | 0.220 |
| tRNAs | 41.4 | 41.4 | 10.2 | 10.3 | 41.0 | 40.6 | 7.4 | 7.7 | 82.4 | 82.0 | 17.6 | 18.0 | 0.005 | 0.021 | 0.159 | 0.144 |
| rRNAs | 43.9 | 43.7 | 9.6 | 10.1 | 41.5 | 41.3 | 5.0 | 5.0 | 85.4 | 85.0 | 14.6 | 15.1 | 0.028 | 0.028 | 0.315 | 0.338 |
| A+T-rich region | 42.5 | 42.2 | 0.88 | 0.3 | 53.4 | 53.0 | 3.2 | 4.8 | 95.9 | 95.2 | 4.1 | 5.1 | -0.114 | -0.113 | -0.566 | -0.882 |
Protein-coding genes
The protein-coding gene (PCG) regions of the C. medinalis and C. suppressalis mitogenomes were consistent with those of other pyralid moth mitogenomes, both containing 13 PCGs (Table 2). All the genes were not coded by the same strand, but rather nine PCGs (nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, and cob) were coded by the H-strand, while the remaining four PCGs (nad1, nad4, nad4L, nad5) were coded by the L-strand in both the C. medinalis and C. suppressalis mitogenomes. The start and stop codons of the 13 PCGs are shown in Table 2, with 12 PCGs utilizing the standard ATN (ATA, ATC, ATG, and ATT) as observed in invertebrate mitogenomes [51]. However, the cox1 gene used CGA as the initiation codon in C. medinalis and C. suppressalis mitogenomes. Notably, the cox2 and atp8 genes started with ATG and ATT, respectively, in the C. medinalis mitogenome, whereas ATA were found to initiate the cox2 gene, and the atp8 gene started with ATC in the C. suppressalis mitogenome.
In the C. medinalis mitogenome, nine PCGs were terminated by the standard stop codon TAA, whereas the cox1, cox2 and nad5 genes used T, and the nad4 gene utilized TA as a truncated stop codon. However, in the C. suppressalis mitogenome, there were 10 PCGs with the standard stop codon TAA, while the cox1 and cox2 genes used T and the nad4 gene used TA as truncated termination codons.
The average A+T contents of PCGs (without start and stop codons) in the C. medinalis and C. suppressalis mitogenomes were 80.5 and 78.9%, respectively. The AT- and GC-skew values were calculated to analyze the AT and GC bias of the PCGs. The results demonstrated that AT-skews were negative (-0.138; -0.140), but the GC-skews were slightly positive (0.046; 0.024). The A+T content of the third codon position (96.0%) in the C. medinalis mitogenome was higher than that of the first (74.6%) and second (71.0%) positions. However, the A+T content of the second codon position (92.4%) in C. suppressalis mitogenome was much higher than that of the first (70.6%) and third (73.6%) positions.
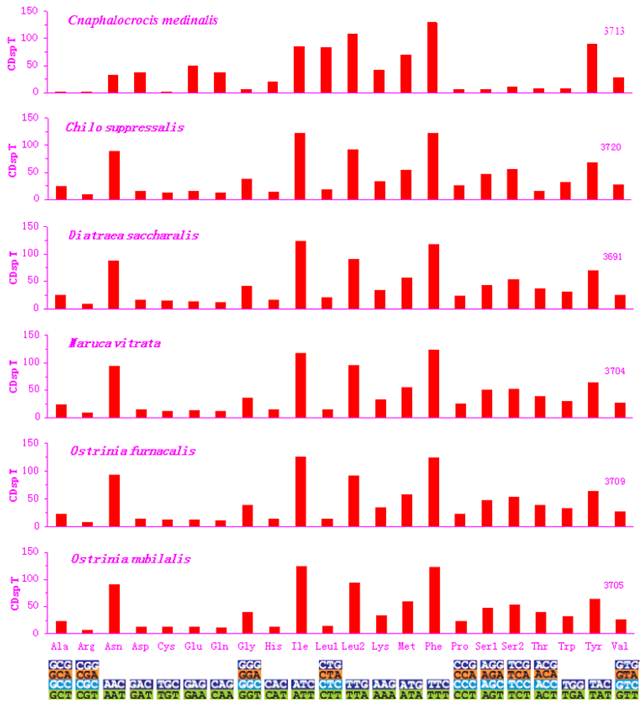
Excluding the start and termination codons, the 13 PCGs in the C. medinalis mitogenome consisted of 3,713 codons, similar to C. suppressalis (3,720). The behavior of codon families in the PCGs was determined (Figs. 2 and 3), with the start and stop codons excluded from the analysis to avoid biases due to unusual putative start and stop codons. The codon families were very different between the C. medinalis and C. suppressalis mitogenomes (Figs. 2 and 3).
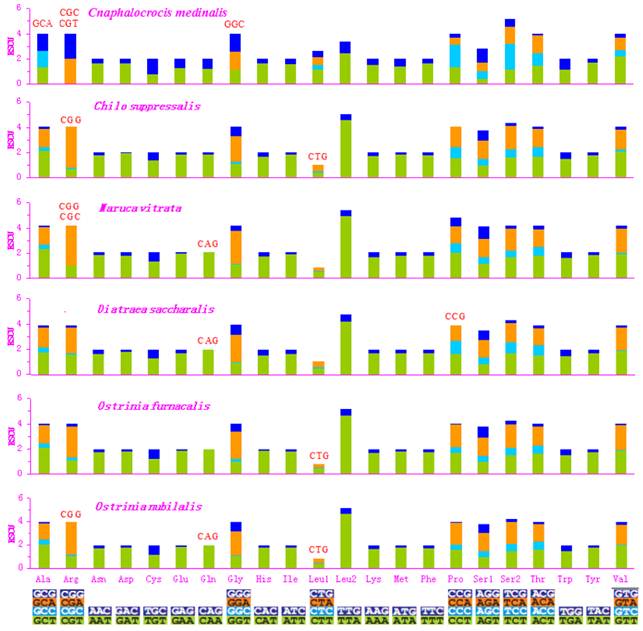
Asn, Ile, Leu, Phe, and Ser were the most abundant amino acids in D. saccharalis, M. vitrata, O. furnacalis and O. nubilalis analyzed [11-13]. However, the five most common codon families (Ile, Leu2, Met, Phe, and Tyr), each with at least 50 codons (CDs) per thousand CDs, were two-fold degenerate in codon usage and were rich in A and T in the C. medinalis mitogenome. Only Leu and Phe units with at least 100 CDs were found in the C. medinalis mitogenome, whereas Ile and Leu were found in C. suppressalis and other pyralid moth mitogenomes [11-13]. Nine codon families encoding Ala, Arg, Cys, Gly, Pro, Ser1, Ser2, Thr, and Trp displayed no more than 12 CDs in C. medinalis, lower than in C. suppressalis and other pyralids [11-13] (Fig. 2). In addition, the AT-rich CDs were favored over synonymous CDs with a lower A+T content, which was consistent with the results of Relative Synonymous Codon Usage (RSCU), especially for the Leu2 family, where the TTA codon accounted for the large majority of CDs in the family. Two- and four-fold degenerate codon usages were biased to use more A and T than G and C in the third position. The lost codons usually belonged to GC-rich codon-families (Fig. 3).
Codon distribution in pyralid moth mitogenomes. Numbers to the left refer to the total number of codon. CDsp T, codons per thousand codons. Codon families are given on the x axis.
Relative Synonymous Codon Usage (RSCU) in pyralid moth mitogenomes. Codon families are given on the x axis. Codons that are not present in the genome are indicated in red.
Transfer RNA genes
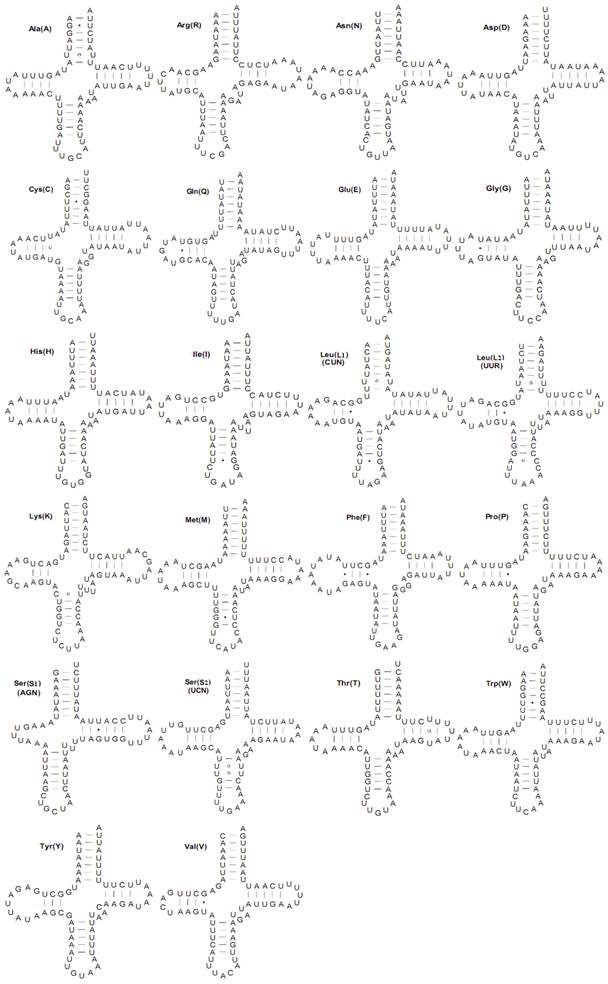
Both the C. medinalis and C. suppressalis mitogenomes contained the set of 22 tRNAs found in other pyralid moth mitogenomes that are typical of animal mitogenomes [1]. Twenty-two tRNA genes (totaling 1,474 bp and 1,482 bp and ranging in size from 65 to 71 bp) were interspersed with rRNAs or PCGs and had 82.4 and 82.0% A+T content in the C. medinalis and C. suppressalis mitogenomes, respectively. Fourteen tRNAs were codified by the H-strand and eight by the L-strand. All tRNA genes had the typical cloverleaf secondary structures with respective anticodons, except for the trnS1(AGN) gene, in which a simple loop was substituted for a dihydrouridine (DHU) arm in the C. medinalis and C. suppressalis mitogenomes (Fig. 4).
The tRNA genes usually contained a 7-bp amino acid acceptor (AA) stem, where most nucleotide substitutions were compensatory. The anticodon (AC) stem and the loop (7 bp) were both conserved in all tRNAs, where two U-U pairs were usually located at the second and third couplets in the stem. The length of DHU was 3-4 bp, except for trnS1(AGN). The TΨC arm was usually 3-6 bp in length. Compared to other lepidopteran species, there were 24 mismatched base pairs in 16 tRNAs secondary structures in the mitogenome of C. medinalis, including 15 G-U pairs, seven U-U pairs, one A-C pair, and one C-U pair. However, a total of 23 mismatched base pairs existed in the 16 C. suppressalis mitochondrial tRNA secondary structures, with 16 G-U pairs, four U-U pairs, one A-A pair, one A-C pair, and one C-C pair (Supplementary Material: Fig. S1).
Inferred secondary structures for 22 typical tRNAs of the Cnaphalocrocis medinalis mitogenome. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Base-pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and other non-canonical pairs by circles.
Ribosomal RNA genes
As in the mitogenomes of other insects, there were two ribosomal genes, a 1,384 bp/1,383 bp rrnL gene and a 781 bp/788 bp rrnS gene in the C. medinalis and C. suppressalis mitogenomes, respectively. The locations of rRNAs in the two species were the same as in other pyralid moth mitogenomes. The rrnL gene resided between trnL1(CUN) and trnV, and the rrnS gene between trnV and the A+T-rich region.
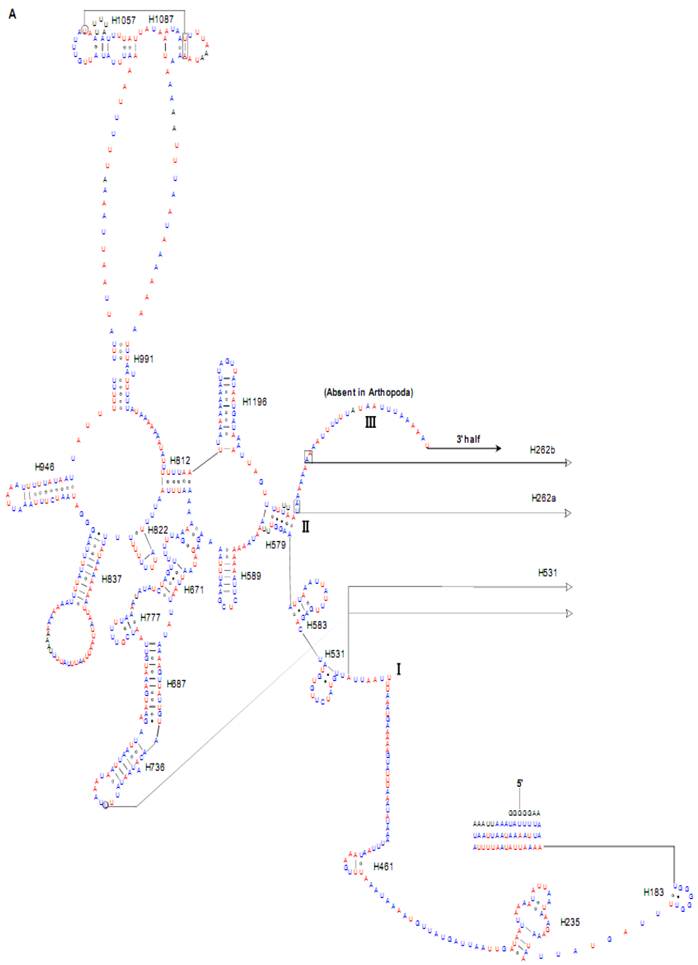
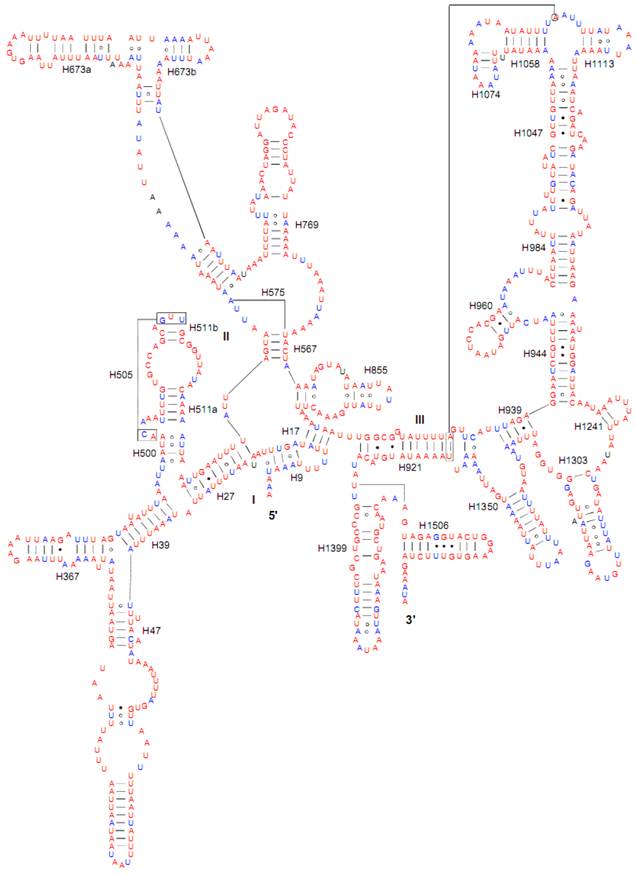
Both of the secondary structures of the rrnL and rrnS genes were inferred from models proposed for other insects [20-22, 31]. Six domains with 49 helices were present in the C. medinalis rrnL and C. suppressalis rrnL genes (Supplementary Material: Fig. S2) as in Apis mellifera [21] and Manduca sexta [31] (Fig. 5). Meanwhile, there were 33 helices in the C. medinalis rrnS and C. suppressalis rrnS genes (Supplementary Material: Fig. S3) in three domains (labeled I, II, III) (Fig. 6), which were largely in agreement with those proposed for A. mellifera, M. sexta, and other insect species [20-22, 31].
Non-coding and overlapping regions
The mitogenomes of C. medinalis and C. suppressalis contained 219 bp and 221 bp of intergenic spacer sequences, respectively (Table 2). Additionally, there were four major intergenic spacers (S1, S2, S3, and S4) with at least 12 bp in C. medinalis and C. suppressalis, all of which were rich in A and T. The features of the S1-S4 spacers are illustrated below (Fig. 7).
The S1 spacer (74 bp), located between trnQ and nad2, appeared to be the result of a duplicated 20-bp segment and a poly-T motif within the last 16 bp with minor changes in C. medinalis. In addition, there was a triplication and a similar poly-T motif within the last 19 bp in C. suppressalis. The S2 spacer (41 bp) located between the trnY and cox1 genes was derived from the microsatellite '(TA)17' in C. medinalis. However, the S2 spacer (30 bp) found between the trnC and trnY genes was derived from a duplicated segment with minor changes in C. suppressalis. Spacer S3 (46 bp) was present between nad3 and trnA and contained a microsatellite '(AT)18' in C. medinalis. Furthermore, the S3 spacer (101 bp) in C. suppressalis featured a microsatellite '(AT)19' and a duplicated segment. Along with the S4 spacer, there was a 7 bp motif 'ATACTAA' in C. medinalis and C. suppressalis between trnS2(UCN) and nad1.
In addition, there were overlapping nucleotides 23 bp in length scattered over seven locations in C. medinalis, and ones 62 bp in length scattered over nine locations in C. suppressalis (Table 2). The overlaps of the gene coding sequence between trnW and trnC (8 bp), atp8 and atp6 (7 bp), and atp6 and cox3 (1 bp) in the two mitogenomes indicate that the mature transcripts remain intact in order to preserve their respective coding sequences. There was a 7 bp motif 'AGCCTTA' between trnW and trnC in C. medinalis and C. suppressalis. An overlap of seven nucleotides 'ATGATAA' was observed between atp8 and atp6 in both C. medinalis and C. suppressalis, which is a common feature of many other insect mitogenomes. Furthermore, there was a 17 bp motif 'TTATAAGCTATTTAAAT' between trnF and nad5 in C. suppressalis.
The A+T-rich region
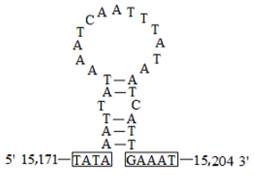
The A+T-rich region, flanked by the rrnS and trnM genes, was 339 bp in length with a 95.9% A+T content in C. medinalis and 348 bp in length with a 95.2% A+T content in C. suppressalis. There was a conserved structure that consisted of the motif 'ATAGT(A)' and a poly-T stretch in C. medinalis (Fig. 8A). A very similar pattern occurred in C. suppressalis, where the sequence 'ATAGA' was followed by a poly-T stretch (Fig. 8B). A duplicated 25-bp repeat element was found in C. medinalis, which was similar to that in C. suppressalis (a duplicated 31-bp repeat element). There was also a stem-and-loop structure in C. medinalis (Figs. 8A and 9), but not in C. suppressalis. Furthermore, a microsatellite '(TA)13' was observed in C. medinalis, while a poly-T motif and a triplicated 19-bp repeat element were detected in C. suppressalis (Fig. 8B).
Phylogenetic analysis
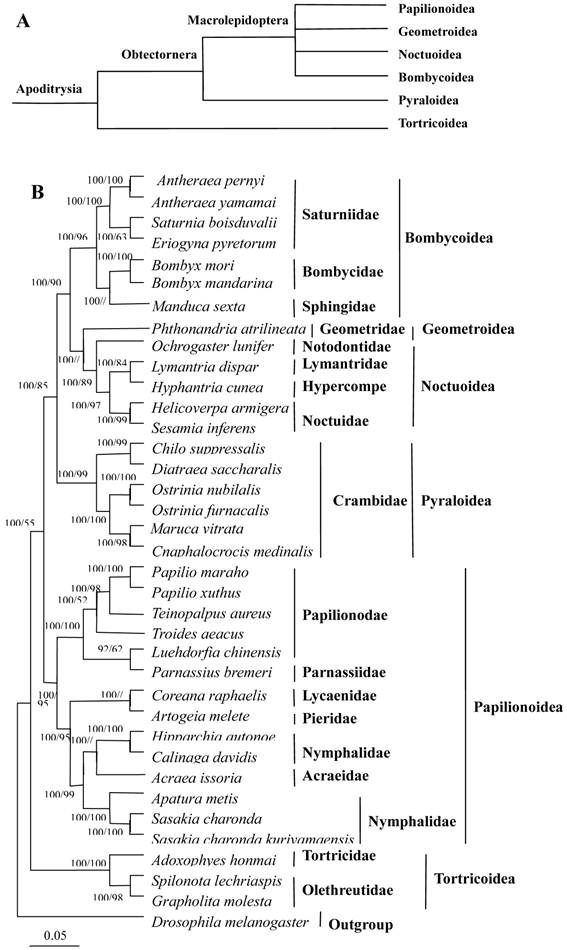
In this study, the amino acid sequences of the 13 PCGs were concatenated to reconstruct phylogenetic relationships. Based on morphological analysis and mitochondrial analysis of the species studied here, and other species from the literature, the relationships among six superfamilies in Lepidoptera were recontstructed, as shown in Fig. 10 [52].
The C. medinalis and C. suppressalis mitogenomes were grouped with those of D. saccharalis, M. vitrata, O. furnacalis, and O. nubilalis in the family Pyralidae (Pyraloidea) and clustered with other surperfamilies, including the Papillionoidea (14 taxa), Tortricoidea (three taxa), Bombycoidea (seven taxa), Geometroidea (one taxon), and Noctuoidea (five taxa). The phylogenetic analyses indicated a close relationship between C. medinalis, C. suppressalis, and other pyralid moths, with a 100% bootstrapping value, which was consistent with the morphological classification. Bombycidae (two taxa), Saturniidae (four taxa), and Sphingoidae (one taxon) were clustered in one branch in the phylogenetic tree. Geometroidea was the superfamily most closely related to the Noctuoidea, a finding consistent with morphological analysis. Tortricoidea was the sister superfamily to the remaining lepidopteran superfamilies, in agreement with the tree topology [53, 54].
Predicted secondary structure of the rrnL gene in the Cnaphalocrocis medinalis mitogenome. Tertiary interactions and base triples are shown connected by continuous lines. A represents the 5' half of rrnL, with the remaining 3' half in B. Base-pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and other non-canonical pairs by circles.
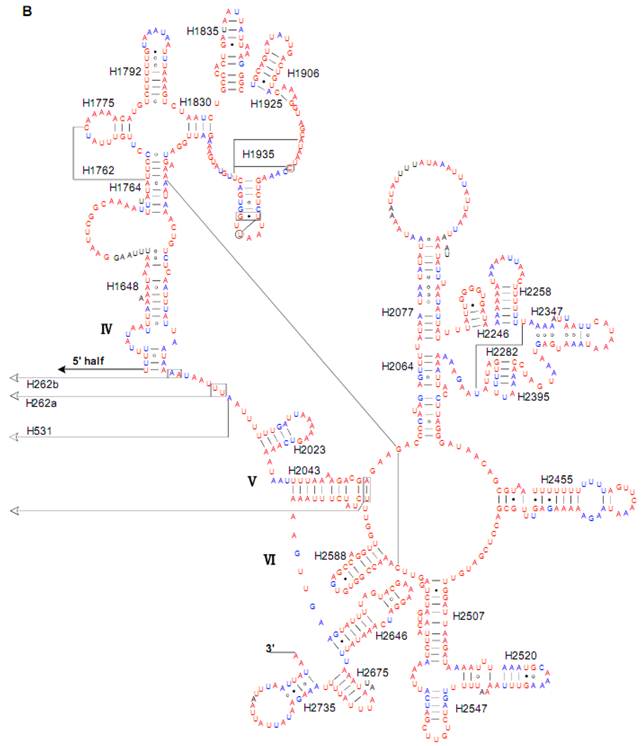
Predicted secondary structure of the rrnS gene in the Cnaphalocrocis medinalis mitogenome. Tertiary interactions and base triples are shown connected by continuous lines. Base-pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and other non-canonical pairs by circles.
Intergenic spacer sequences in the mitogenomes of Cnaphalocrocis medinalis (A) and Chilo suppressalis (B).

Structures of the A+T-rich regions of Cnaphalocrocis medinalis (A) and Chilo suppressalis (B) mitogenomes.

The potential stem-loop structure found in the Cnaphalocrocis medinalis A+T-rich region. Boxed sequences in the flanking region of the stem-loop structures are the motifs, possessing 3' flanking 'GAAAT' and the 5' flanking 'TATA' (nucleotides in box) as described in Zhang et al. [5].
Phylogeny of lepidopteran insects. (A) Current hypothesis of lepidopteran superfamily relationships after Kristensen and Skalski (1999) [54]. (B) Inferred phylogenetic relationships among Lepidoptera based on amino acid sequences of mitochondrial 13 PCGs using Bayesian inference (BI) and maximum likelihood (ML). Numbers at each node indicate bootstrap support; percentages of Bayesian posterior probabilities (first value) and ML bootstrap support values (second value), respectively. The dipteran D. melanogaster was used as an outgroup [33]. The scale bar indicates the number of substitutions per site.
Discussion
The mitogenome of C. medinalis was reported for the first time in this study. Although the mitogenome of C. suppressalis was reported earlier [10], we independently sequenced the complete mitogenome as part of this study. We not only described the mitogenome features of C. medinalis and C. suppressalis, but also compared the two. Because the evolutionary aspects of the C. suppressalis mitogenome were not discussed in detail in the previous report [10], we presented our mitogenome sequence for C. suppressalis in the context of an evolutionary analysis of the genus, to provide an insight into species diversity.
The sizes of the C. medinalis and C. suppressalis mitogenomes from our study were similar to those of other pyralid moths, with all of the 37 typical mitochondrial genes identified in both mitogenomes. The genome organization and order seen in C. medinalis and C. suppressalis were typical for insect mitogenomes (Table 1), and are the same as those of other pyralid moths [11-13].
Four mitochondrial DNA gene fragments (cox1, cox2, nad1, and rrnL) and four microsatellite DNA markers [(AC)n, (GT)n, and two (CA)n loci] were previously used to perform a phylogenetic analysis of C. suppressalis from 18 localities in China, and it was found that C. suppressalis was highly differentiated among the different geographical populations [55]. Although the mitogenome of C. suppressalis from our study had only a 94% sequence homology with the published C. suppressalis mitogenome [10] with a 15,465 bp length, its genomic organization and order were very similar. Also, the nucleotide composition and the A+T content of all components were similar in the two C. suppressalis mitogenomes, with a nucleotide composition of 80.6% and 79.7%, PCGs (78.9%, 77.7%), tRNAs (82.0%, 84.7%), rRNAs (85.0%, 81.2%), and A+T-rich region (95.2%, 94.2%), respectively. But for some components, the AT- and GC-skews were different between the two mitogenome sequences. It is possible that these selective nucleotide compositional biases can be attributed to natural selection and/or different mutational pressures [56].
In this study, the start codons of the 13 PCGs in the C. suppressalis mitogenome were identical to the previously reported sequence [10], expect for the nad6 gene. The cox1 gene usually has the start codon CGA in most lepidopterans [24]. The cox1 gene is considered to be one of more conserved mitochondrial genes, but its start codon is variable, which has been extensively discussed in relation to various insect and arthropod species [10, 33, 57]. Some PCGs, especially the cox1 and cox2 genes, have incomplete stop codons in lepidopteran species, as previously reported in other insects [58-60]. It is commonly believed that the TAA terminator results from post- transcriptional polyadenylation [61].
The codon families with high CDs have a prevalence of A and T in the third position, which might reflect selection for optimal tRNA usage, genome bias, and speed and efficacy of the genome/DNA repair mechanisms [62]. The sequences of the 13 PCGs have very high similarity between C. medinalis and C. suppressalis, but the codon distribution and RSCU of codon families are different in the two species. All codons were present in the C. medinalis PCGs, except for the GCA (Alanine), CGC (Arginine), CGT (Arginine), and GGC (Glycine) codons. Meanwhile, CGG (Arginine) and CTG (Leucine) were not present in the C. suppressalis PCGs, reflecting the influence of a strongly biased codon usage [63]. This codon bias may have resulted from genetic mutation, selection pressures, or genetic drift [56]. Another explanation may be the migratory habits of C. medinalis. The mitogenome is responsible for the oxidative reactions in cells, so it plays an important role during migration to provide adequate supplies of energy to meet migration demands. However, the PCGs within the C. suppressalis sequence in the present study had 3,721 codons, with a similar codon distribution and RSCU of codon families to those of the previously reported C. suppressalis mitogenome [10].
The size and AT-content of the 22 tRNA genes in C. suppressalis were also similar to those of the published mitogenome (1,478 bp long with 84.7% A+T content) [10]. There were a total of 26 mismatched base pairs in the previously published C. suppressalis mitogenome, including 14 G-U pairs, three U-U pairs, five A-C pairs, one A-A pair, one A-G pair, one C-C pair, and one C-U pair [10]. Such mismatches are common in arthropod mitogenomes, and are mostly located in the acceptor and anticodon stems [64].
The sizes of both the rrnL and rrnS genes in C. suppressalis from this study were shorter than the rRNAs (1,442 bp, 789 bp) reported previously in the literature [10]. We also used a commonly accepted comparative approach to construct the secondary structure of rRNAs [65]. The effect of base pairing is not perfect in the 5' half of the rrnL gene in C. medinalis (Fig. 5A). The mis-pairings were observed in the stem regions of H991 and other helices under the criteria of Watson-Crick pairs, but the secondary structure of rrnL proved that the effect of base pairing is profound in the C. suppressalis mitogenome (Supplementary Material: Fig. S2). The structures of the rRNAs we presented were based mainly on sequence comparison and mathematical methods, but the reason for high occurrence of mis-pairings in C. medinalis is not clear. In the secondary structure of the rrnS gene, the H47 portion has a small loop in C. medinalis and C. suppressalis compared to M. sexta, which seems to be variable among species and therefore useful to predict phylogenetic relationships with H39 and H367 [8, 31]. The helices of H673, H1047, H1068, and H1074 in C. medinalis and C. suppressalis are different in length and structure from those in Alloeorhynchus bakeri (Hemiptera) and M. sexta, but similar to Grapholita molesta, Liriomyza trifolii (Diptera) and Libelloides macaronius (Neuroptera) [22, 31, 66-68]. Nucleotide variability among domains and helices is unevenly distributed in the secondary structures of the rrnL and rrnS genes. It is very interesting that some helices in the secondary structures of rrnL and rrnS are different among lepidopteran species, but very similar to those of other orders of insects.
Although there is a slight difference in the sizes of three major intergenic spacers in C. suppressalis mitogenomes between the present study and the previously reported one [10], the spacers have similar genomic locations, with similar motifs. For example, the longest spacer is located between the trnC and trnY genes, which contains the microsatellite repeat '(AT)n' commonly observed in other insects [10, 11, 28, 29]. In spacer S4, there is a 7 bp motif 'ATACTAA' in both C. medinalis and C. suppressalis. Similarly, in this region there is a 5 bp motif 'TACTA' that is conserved in Coleoptera [69], and a 6 bp conserved motif 'THACWW' found in Hymenoptera [8]. Obviously, the similar motif found here is the most conserved one in Lepidoptera. The nucleotide overlap 'AGCCTTA' between the trnW and trnC genes was detected in both C. suppressalis mitogenome sequences and in other lepidopteran species [10, 13]. Furthermore, a similar nucleotide overlap 'TTATAAGCTATTTAAAT' in the C. suppressalis mitogenome between trnF and nad5 was observed in the O. furnacalis (with 'TTATAAGCTATTTA') and O. nubilalis mitogenomes (with 'TTATAAGCTATTTAAA') [13], all of which suggests that spacer sequences can potentially be utilized for studying higher level phylogeny.
In the mitogenome there is often a non-coding region rich in A and T (A+T-rich region), which varies considerably in length among insect species, or even within the same species [70]. This region includes various copy numbers of certain tandem-repeat elements scattered through the entire region, such as TA repeat, poly A, and poly T. The combined structural motif 'ATAGA(T)' and a poly-T stretch have been widely observed in other lepidopteran mitogenomes, which might be the origin of light-strand replication [10, 70]. The stem-and-loop structure, with the conserved 3' flanking 'G(A)nT' and the 5' flanking 'TATA' sequences, was observed in the C. medinalis mitogenome and is considered to be the site of the initiation of secondary strand synthesis [5, 70, 71] (Fig. 8A and 9). However, the stem-and loop structure often found in the A+T-rich region of insects was not present in C. suppressalis [10], but is found in several insect orders, including Orthoptera, Lepidoptera, Diptera, Plecoptera, and Hymenoptera [5, 40, 70, 72].
The A+T-rich regions had many differences between the two C. suppressalis mitogenomes, probably due to the high variability. However, both of the mitogenomes contained the motif 'ATAGA,' followed by a 19 bp poly-T stretch. In this study, the phylogenetic tree that was reconstructed was in disagreement with previous research on six lepidopteran superfamilies [12, 30, 36, 41]. The reason for this may be explained by our having access to more lepidopteran mitogenomes than did past studies. Such a larger set of entities might better explain the underlying phylogenetic relationships in the Lepidoptera than did previous studies. Our results provide some clarification about the placement of the Bombycoidea but were still broadly consistent with previous morphological classification of the Lepidoptera [53, 54]. However, the addition of the C. medinalis and C. suppressalis mitogenomes to the literature did not resolve uncertainties about the position of Pyralidae. It will likely require the availability of still more mitogenomes to fully resolve the relationships among these surperfamilies of Lepidoptera.
Supplementary Material
Fig. S1: Inferred secondary structures for 22 typical tRNAs of the Chilo suppressalis mitogenome. Fig. S2: Predicted secondary structure of the rrnL gene in the Chilo suppressalis mitogenome. Fig. S3: Predicted secondary structure of the rrnS gene in the Chilo suppressalis mitogenome.
Acknowledgements
This work was supported by the Special Fund for Agro-Scientific Research in the Public Interest of China (200903051) and the National Basic Research Program of China (973 Program) (2012CB114100). We are very grateful to Dr Guo (Harvard University) for improving the text of the manuscript. We also acknowledge three anonymous reviewers for their invaluable comments and suggestions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1726-1780
2. Hebert PDN, Cywinska A, Ball SL, de Waard JR. Biological identification through DNA barcodes. Proc R Soc London Series B Biol Sci. 2003;270:313-321
3. Thao ML, Baumann L, Baumann P. Organization of the mitochondrial genomes of whiteflies, aphids and psyllids (Hemiptera, Sternorrhyncha). BMC Evol Biol. 2004;4:25-37
4. Dowton M, Austin AD. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol. 1999;16:298-309
5. Zhang D, Szymura JM, Hewitt GM. Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol. 1995;40:382-391
6. Nardi F, Carapelli A, Dallai R, Frati F. The mitochondrial genome of the olive fly Bactrocera oleae: two haplotypes from distant geographical locations. Insect Mol Biol. 2003;12:605-611
7. Da Silva NM, de Souza Dias A, da Silva Valente VL, Valiati VH. Characterization of mitochondrial control region, two intergenic spacers and tRNAs of Zaprionus indianus (Diptera: Drosophilidae). Genetica. 2009;137:325-332
8. Wei SJ, Shi M, Sharkey MJ, van Achterberg C, Chen XX. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to holometabolous insects. BMC Genomics. 2010;11:371
9. Wei SJ, Shi M, He JH, Sharkey M, Chen XX. The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome. 2009;52:308-319
10. Yin J, Wang AM, Hong GY, Cao YZ, Wei ZJ. Complete mitochondtial genome of Chilo suppressalis (Walker) (Lepidoptera: Crambidae). Mitochondrial DNA. 2011;22:41-43
11. Li WW, Zhang XY, Fan ZX, Yue BS, Huang FN, King E, Ran JH. Structural characteristics and phylogenetic analysis of the mitochondrial genome of the sugarcane borer, Diatraea saccharalis (Lepidoptera: Crambidae). DNA Cell Biol. 2011;30:3-8
12. Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld MJ, Sun WL, Ba MN, Sanon A, Binso-Dabire CL, Baoua I, Ishiyaku MF, Covas FG, Srinivasan R, Armstrong J, Murdock LL, Pittendrigh BR. Mitochondrial genome sequence and expression profiling for the legume pod borer, Maruca vitrata (Lepidoptera: Crambidae). PLoS ONE. 2011;6:e16444
13. Coates BS, Sumerford DV, Hellmich RL, Lewis LC. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int J Biol Sci. 2005;1:13-18
14. Sheng CF, Wang HT, Sheng SY, Gao LD, Xuan WJ. Pest status and loss assessment of crop damage caused by the rice borers Chilo suppressalis and Tryporyza incertulas in China. Inst Zool. 2003;40:289-294
15. Liu Y, Wang JQ, Feng XD, Jiang XH. Analysis on the occurring of Cnaphalocrocis medinalis in 2007 and forecasting its occuring trends in 2008. China Plant Protection. 2008;28:33-35
16. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947-2948
17. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
18. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
19. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
20. Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, Pande N, Shang Z, Yu N, Gutell RR. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:15
21. Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol. 2006;15:657-686
22. Gong YJ, Shi BC, Kang ZJ, Zhang F, Wei SJ. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol Biol Rep. 2012;39:2893-2900
23. Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci USA. 2000;97:13738-13742
24. Buckley TR, Simon C, Flook PK, Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol Biol. 2000;9:565-580
25. Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573-580
26. Liu YQ, Li YP, Pan MH, Dai FY, Zhu XW, Lu C, Xiang ZH. The complete mitochondrial genome of the Chinese oak silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae). Acta Biochim Biophys Sin. 2008;40:693-703
27. Kim SR, Kim MI, Hong MY, Kim KY, Kang PD, Hwang JS, Han YS, Jin BR, Kim I. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae). Mol Biol Rep. 2009;36:1871-1880
28. Hu XL, Cao GL, Xue RY, Zheng XJ, Zhang X, Duan HR, Gong CL. The complete mitogenome and phylogenetic analysis of Bombyx mandarina strain Qingzhou. Mol Biol Rep. 2010;37:2599-2608
29. Lu C, Liu YQ, Liao XS, Li B, Xiang ZX, Han H, Wang XG. Complete sequence determination and analysis of Bombyx mori mitochondrial genome. J Agri Biotechnol. 2002;10:163-170
30. Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci. 2009;5:351-365
31. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-123
32. Hong MY, Lee EM, Jo YH, Park HC, Kim SR, Hwang JS, Jin BR, Kang PD, Kim KG, Han YS, Kim I. Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene. 2008;413:49-57
33. Lewis DL, Farr CL, Kaguni LS. Drosophila melanogaster mitochon-drial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol. 1995;4:263-278
34. Yang L, Wei ZJ, Hong GY, Jiang ST, Wen LP. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol Biol Rep. 2009;36:1441-1449
35. Yin J, Hong GY, Wang AM, Cao YZ, Wei ZJ. Mitochondrial genome of the cotton bollworm, Helicoverpa armigera (Lepidoptera: Noctuidae), and comparison with other Lepidopterans. Mitochondrial DNA. 2010;21:160-169
36. Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci. 2010;6:172-186
37. Zhu YJ, Zhou GL, Fang R, Ye J, Yi JP. The complete sequence determination and analysis of Lymantria dispar ( Lepidoptera: Lymantriidae) mitochondrial genome. Plant Quarantine. 2010;4:6-11
38. Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the Bag-shelter moth, Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics. 2008;9:1-15
39. Hu J, Zhang D, Hao J, Huang D, Cameron S, Zhu C. The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event. Mol Biol Rep. 2010;37:3431-3438
40. Hong GY, Jiang ST, Yu M, Yang Y, Li Feng, Xue FS, Wei ZJ. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochim Biophys Sin. 2009;41:446-455
41. Xia J, Hu J, Zhu GP, Zhu CD, Hao JS. Sequencing and analysis of the complete mitochondrial genome of Calinaga davidis Oberthür (Lepidoptera: Nymphalidae). Acta Entomol Sin. 2011;54:555-565
42. Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol. 2006;15:217-225
43. Kim MJ, Wan XL, Kim KG, Hwang JS, Kim I. Complete nucleotide sequence and organization of the mitogenome of endangered Eumenis autonoe (Lepidoptera: Nymphalidae). Afr J Biotechnol. 2010;9:735-754
44. Kim MI, Baek JY, Kim MJ, Jeong HC, Kim KG, Bae CH, Han YS, Jin BR, Kim I. Complete nucleotide sequence and organization of the mitogenome of the red spotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Mol Cells. 2009;28:347-363
45. Feng X, Liu DF, Wang NX, Zhu CD, Jiang GF. The mitochondrial genome of the butterfly Papilio xuthus (Lepidoptera: Papilionidae) and related phylogenetic analyses. Mol Biol Rep. 2010;37:3877-3888
46. Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene. 2006;373:52-57
47. Zhao JL, Zhang YY, Luo AR, Jiang GF, Cameron SL, Zhu CD. The complete mitochondrial genome of Spilonota lechriaspis Meyrick (Lepidoptera: Tortricidae). Mol Biol Rep. 2011;38:3757-3764
48. Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
49. Ronquist F, Huelsenbeck JP. MrBayes version 3.0: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
50. Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online-a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33:557-559
51. Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173-216
52. Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol. 2006;38:100-116
53. Kristensen NP, Skalski AW. Phylogeny and paleontology. In: (ed.) Kristensen NP. Lepidoptera: Moths and Butterflies, 1 Evolution, Systematics, and Biogeography, Handbook of Zoology Vol IV, Part 35. Berlin and New York: De Gruyter. 1999:7-25
54. Kawahara AY, Mignault AA, Regier JC, Kitching IJ, Mitter C. Phylogeny and biogeography of hawkmoths (Lepidoptera: Sphingidae): evidence from five nuclear genes. PLoS ONE. 2009;4:e5719
55. Meng XF, Shi M, Chen XX. Population genetic structure of Chilo suppressalis (Walker) (Lepidoptera: Crambidae): Strong subdivision in China inferred from microsatellite markers and mtDNA gene sequences. Mol Ecol. 2008;17:2880-2897
56. Bachtrog D. Reduced selection for codon usage bias in Drosophila miranda. J Mol Evol. 2007;64:586-590
57. Fenn JD, Cameron SL, Whiting MF. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol Biol. 2007;16:239-252
58. Junqueira AC, Lessinger AC, Torres TT, da Silva FR, Vettore AL, Arruda P, Azeredo Espin AM. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene. 2004;399:7-15
59. Beckenbach AT, Stewart JB. Insect mitochondrial genomics 3: the complete mitochondrial genome sequences of representatives from two neuropteroid orders: a dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome. 2009;52:31-38
60. Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 1993;133:97-117
61. Masta SE, Boore JL. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol Biol Evol. 2004;21:893-902
62. Xia X. Maximizing transcription efficiency causes codon usage bias. Genetics. 1996;144:1309-1320
63. Lessinger AC, Martins Junqueira AC, Lemos TA, Kemper EL, da Silva FR, Vettore AL, Arruda P, Azeredo-Espin AML. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol Biol. 2000;9:521-529
64. Coleman AW. ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet. 2003;19:370-375
65. Gutell RR, Larsen N, Woese CR. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 1994;58:10-26
66. Yang F, Du YZ, Wang LP, Cao JM, Yu WW. The complete mitochondrial genome of the leafminer Liriomyza sativae (Diptera: Agromyzidae): Great difference in the A+T-rich region compared to Liriomyza trifolii. Gene. 2011;485:7-15
67. Hu L, Liu HY, Cao LM, Shi AM, Yang HL, Cai WZ. The complete mitochondrial genome of the damsel bug Alloeorhynchus Bakeri (Hemiptera: Nabidae). Int J Biol Sci. 2012;8:93-107
68. Negrisolo E, Babbucci M, Patarnello T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics. 2011;12:221
69. Sheffield NC, Song H, Cameron SL, Whiting MF. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol. 2008;25:2499-2509
70. Zhang DX, Hewitt GM. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997;25:99-120
71. Clary DO, Wolstenholme DR. Drosophila mitochondrial DNA: conserved sequences in the A+T rich region and supporting evidence for a secondary structure model of the small RNA. J Mol Evol. 1987;25:116-125
72. Rand DM, Harrison RG. Molecular population genetics of mtDNA size variation in crickets. Genetics. 1989;121:551-569
Author contact
![]() Corresponding author: Yu-Zhou Du, Institute of Applied Entomology, Yangzhou University, 48 Wenhui Road (East), Jiangsu, Yangzhou 225009, China; Tel: 86-514-87971854; Fax: 0086-514-87971854; E-mail: yzduedu.cn; chaihuannacom.
Corresponding author: Yu-Zhou Du, Institute of Applied Entomology, Yangzhou University, 48 Wenhui Road (East), Jiangsu, Yangzhou 225009, China; Tel: 86-514-87971854; Fax: 0086-514-87971854; E-mail: yzduedu.cn; chaihuannacom.
Received 2011-9-21
Accepted 2012-2-24
Published 2012-4-17