Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(6):791-801. doi:10.7150/ijbs.4568 This issue Cite
Research Paper
Reduction in Tcf7l2 Expression Decreases Diabetic Susceptibility in Mice
Hyekyung Yang1, Qing Li1, Jong-Hwan Lee2, Yan Shu1 ![]()
1. Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland 21201, USA.
2. Department of Veterinary Anatomy, College of Veterinary Medicine, Konkuk University, Seoul 143-701, Korea.
Received 2012-5-8; Accepted 2012-5-29; Published 2012-6-5
Abstract
Objective: The WNT signaling pathway effector gene TCF7L2 has been associated with an increased risk of type 2 diabetes. However, it remains unclear how this gene affects diabetic pathogenesis. The goal of this study was to investigate the effects of Tcf7l2 haploinsufficiency on metabolic phenotypes in mice.
Experimental Design: Tcf7l2 knockout (Tcf7l-/-) mice were generated. Because of the early mortality of Tcf7l2-/- mice, we characterized the metabolic phenotypes of heterozygous Tcf7l2+/- mice in comparison to the wild-type controls. The mice were fed a normal chow diet or a high fat diet (HFD) for 9 weeks.
Results: The Tcf7l2+/- mice showed significant differences from the wild-type mice with regards to body weight, fasting glucose and insulin levels. Tcf7l2+/- mice displayed improved glucose tolerance. In the liver of Tcf7l2+/- mice fed on the HFD, reduced lipogenesis and hepatic triglyceride levels were observed when compared with those of wild-type mice. Furthermore, the Tcf7l2+/- mice fed on the HFD exhibited decreased peripheral fat deposition. Immunohistochemistry in mouse pancreatic islets showed that endogenous expression of Tcf7l2 was upregulated in the wild-type mice, but not in the Tcf7l2+/- mice, after feeding with the HFD. However, the haploinsufficiency of Tcf7l2 in mouse pancreatic islets resulted in little changes in glucose-stimulated insulin secretion.
Conclusion: These results suggest that decreased expression of Tcf7l2 confers reduction of diabetic susceptibility in mice via regulation on the metabolism of glucose and lipid.
Keywords: Tcf7l2, diabetes, high fat diet, glucose tolerance, gluconeogenesis, hepatic steatosis.
Introduction
Transcription factor 7-like 2 (TCF7L2, formerly called TCF4) is a member of the T-cell-specific high-mobility group (HMG) box-containing family of transcription factors that plays an important role in downstream signals of the canonical morphogenic wingless-type MMTV integration site family (WNT) pathway [1]. The WNT signaling pathway has been well known to be associated with the developmental pathways such as embryogenesis including adipogenesis and pancreatic islet development, and tumorigenesis [2-5]. Activation of this pathway leads to accumulation of β-catenin in the nucleus, which interacts with the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors to regulate the transcription of WNT target genes, many of which are associated with the cell proliferation and cell fate decision [1,6].
Besides the developmental role, several lines of evidence suggest the role of WNT signaling pathway in the etiology of metabolic disorders [7-10]. Particularly, single nucleotide polymorphisms (SNPs) in human TCF7L2 gene are known to be strongly associated with an increased risk of type 2 diabetes through the extensive genome-wide association studies in multiple ethnic populations [11-13]. TCF7L2 variants have been associated with an impaired β-cell function including the impaired insulin secretion and processing, an increased insulin resistance, and hyperglycemia [14-16]. Yet, the mechanisms by which TCF7L2 gene function and its genetic polymorphisms affect the susceptibility to type 2 diabetes remain elusive. There are conflicting reports regarding the role of TCF7L2 expression in increase in diabetes risk versus resistance to diabetes. Several reports have suggested that TCF7L2 expression is upregulated in the pancreatic islet from diabetic subjects and negatively correlated with insulin secretion [16-18]. In other context, however, TCF7L2 has been considered to be necessary in regulation of β-cell survival and function in human pancreatic islets, and a decreased TCF7L2 protein expression has been suggested to be responsible for the reduced islet insulin secretion in response to glucose through impaired GLP-1 signaling [19, 20].
To understand the mechanism underlying how TCF7L2 function change contributes to susceptibility to metabolic derangements and diabetes, we analyzed the metabolic phenotypes of the mice with Tcf7l2 haploinsufficiency in comparison to the wild-type mice. In particular, we sought to demonstrate a role of TCF7L2 in the regulation of glucose and lipid metabolism.
Materials and methods
Generation of Tcf7l2 Knockout Mice
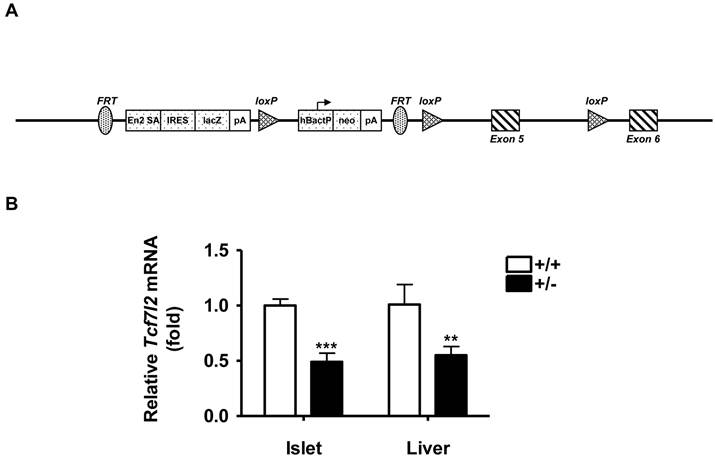
Tcf7l2 knockout mice (Tcf7l2-/-) were generated via the International Knockout Mouse Consortium (http://www.knockoutmouse.org). The procedure involved the insertion of a LoxP site together with a Flippase Recognition Target (FRT) flanked neomycin selection cassette within the intron 4 of mouse Tcf7l2 gene and a single distal LoxP in the intron 5 downstream of exon 5 (Figure 1A). The animals used in the present study were derived from heterozygous Tcf7l2+/- and wild-type C57BL/6 breeding pairs. The genotype of all progeny was confirmed by PCR analysis of DNA extracted from tail biopsies. All procedures were performed in accordance with NIH guidelines for animal experimentation, and all experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the School of Pharmacy, University of Maryland Baltimore. Animals were housed 4-5 mice/cage and maintained under standard laboratory conditions (21 ± 2 ºC, humidity 60 ± 10% and 12 h / 12 h dark / light cycle) with food and water provided ad libitum. All animals used in the present study were male mice with the same genetic background of C57BL/6. Wild-type and Tcf7l2+/- groups were matched for ages in all experiments. Mice were fed a normal chow diet for 11 weeks or a high-fat diet (HFD) from 13 weeks of ages for 9 weeks, which were purchased from Harlan Laboratories. The HFD consisted of 45 kcal% fat, 14.8 kcal% protein and 41 kcal% carbohydrate (4.7 kcal/g), whereas normal chow diet contained 18 kcal% fat, 24% kcal protein, and 58 kcal% carbohydrate (3.1 kcal/g). For each experiment, animals were brought into the experimental room 30 min prior to the experiment in order for them to acclimate to the environment. Body weight was determined once a week at the same time each week.
Generation of Tcf7l2 knockout mice. (A) Targeting construct for mouse Tcf7l2 knockout. There is insertion of a LoxP site and an FRT flanked neomycin selection cassette within intron 4 and a single distal LoxP in intron 5 downstream of exon 5. Detailed information is available from the IKMC web portal (http://www.knockoutmouse.org). (B) qRT-PCR analysis of mouse Tcf7l2 mRNA levels in islets and liver from wild-type (+/+, n = 3) and Tcf7l2 +/- (+/-, n = 3) mice at 11 weeks of age. (**) p < 0.01, (***) p < 0.001 compared with the wild-type mice.
Glucose Tolerance Test and Insulin Meaurement
Glucose tolerance tests were carried out in the mice at 11 weeks of age under a normal chow diet or 22 weeks of age after 9 weeks on the HFD. Mice were fasted overnight, and then intraperitoneally injected with 2 g/kg glucose in saline. Blood glucose levels were analyzed using a glucometer (TRUEresult, HOMEdiagnostics). To measure insulin levels, blood was taken by tail bleeding, collected in microcentrifuge tubes and placed on ice. Following immediate centrifugation at 4°C, serum was separated and stored at -80 °C until analysis. Insulin was determined using a rat/mouse insulin ELISA kit from Mercodia.
Primary Mouse Hepatocyte Isolation and shRNA Construct for Tcf7l2 Knockdown
Primary hepatocytes were isolated from wild-type and Tcf7l2+/- mice using the standard collagenase method [40]. The cells were plated in Williams E medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1% bovine albumin, 0.1 μM dexamethasone, 2 mM l-glutamine, 1 X ITS (100 X Insulin-Transferrin-Selenium; Invitrogen) at a density of 1.5 × 105 cells/cm2 on collagen-coated 6-well plates (BD). After attachment (2-3 hours), hepatocytes were maintained in the completed medium with 0.25 mg/ml Matrigel (BD Biosciences) for 16 hours followed by RNA extraction or regular medium change and lentivirus transduction as described below.
The oligonucleotides encoding a shRNA specific for the Tcf7l2 sequence were subcloned into the shRNA expression vector pGreen-puro containing the H1 promoter (System Bioscience, Mountain View, CA). The sense oligonucleotide is: 5'-GATCCACTCACACCTCTCATCACGTTTCAAGAGAACGTGATGAGAGGTGTGAGTTTTTTG-3', and the antisense: 5'- AATTCAAAAAACTCACACCTCTCATCACGTTCTCTTGAAACGTGATGAGAGGTGTGAGTG-3'. For the scramble shRNA, the sense oligonucleotide is: 5'-GATCCGAGAGTCAGTAAGGATAACATTTCAAGAGAATGTTATCCTTACTGACTCTCTTTTTG-3', and the antisense: 5'-AATTCAAAAAGAGAGTCAGTAAGGATAACATTCTCTTGAAATGTTATCCTTACTGACTCTC-3'. The successful constructs were verified by sequencing. The plasmids were packaged into lentivirus with three other packaging plasmids VSV-G, REV and CPG (Cell Biolabs Inc., San Diego, CA) in HEK-293LTV cells. Mouse primary hepatocytes were transducted by the lentivirus containing Tcf7l2 shRNA or scramble shRNA for 48 hours followed by RNA extraction.
Islet Isolation and Measurement of Glucose-stimulated Insulin Secretion
Islets at 11 weeks of age under a normal chow diet or after 9 weeks on the HFD were aseptically isolated by collagenase digestion of mouse pancreas and the glucose-stimulated insulin secretion was measured in batch incubations as previously described [21].
Histology
Mouse liver at 11 weeks of age under a normal chow diet or after 9 weeks on the HFD was removed and pieces were fixed in 10 % (v/v) neutralized formalin solution (Sigma-Aldrich), embedded in paraffin, sectioned at 5 µm, and stained with haematoxylin and eosin. For Oil Red O staining, frozen liver tissues embedded in O.C.T. compound (Tissue-Tek) were used [22]. Stained liver sections were examined under a light microscope.
Immunohistochemistry
The pancreas from wild-type and Tcf7l2+/- mice at 11 weeks of age under a normal chow diet or after 9 weeks on the HFD were fixed in 10 % (v/v) neutralized formalin solution (Sigma-Aldrich), embedded in paraffin and sectioned (5 µm). For immunohistochemical analyses, sections were treated with 0.03% H2O2 in methanol for 15 min, the slides were then immersed in citrate buffer (0.01 M; pH 6.0) and incubated for 25 min at 90 °C in a steam bath. Slides were washed in 1x Tris Buffered Saline with 0.01% Tween-20 (TBST) and incubated in blocking solution (5% BSA in TBST) for 1 hour at room temperature to block non-specific binding. The primary antibodies were goat anti-TCF-4 (1:200, Santa Cruz) and donkey rabbit anti−insulin antibody (1:200, Santa Cruz). Sections were incubated in an antibody solution overnight at 4 °C. The anti-TCF-4 antibody was visualized with Alexa Fluor® 568 donkey anti-goat IgG (H+L) (Invitrogen); the anti-insulin antibody was detected with Alexa Fluor® 488 goat anti-rabbit IgG (H+L). The tissue was counterstained lightly with DAPI. Digital images of stained sections were captured using a confocal microscope (Nikon).
Measurement of Serum and Hepatic Triglyeride Content
Blood from mice that had been food deprived for overnight at 11 weeks of age under a normal chow diet or after 9 weeks on the HFD was collected by cardiac puncture under deep anesthesia and placed on ice. Following immediate centrifugation at 4°C, serum was separated and stored at -80 °C until analysis for triglycerides. To extract lipids from the liver, each piece of tissues was weighed and homogenized in 4 ml of chloroform: methanol (2:1; v/v) mixture. One milliliter of PBS was then added, and the resulting suspension was mixed vigorously for 15 seconds then centrifuged at 1,500 g for 10 min at room temperature. Five-hundred microliters of the organic phase was transferred into a tube and evaporated under nitrogen gas at room temperature. The residue was resuspended in 200 μl of 10 % Triton X100 in methanol. Serum and hepatic triglyceride levels were determined by an assay kit obtained from Sigma-Aldrich.
Quantitative Real-Time Polymerase Chain Reaction. Total RNA was extracted from the liver, primary hepatocytes, and isolated pancreatic islets of mice using TRIzol (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instruction. Total RNA (2 μg) was reverse transcribed to a complementary DNA with high capability reverse transcript kit (Roche Applied Science, Indianapolis, IN). Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted on an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA) using SYBR Green (Applied Biosystems, Foster City, CA). All primers used in the study were designed by the software Primer3.0 and as follows: Tcf7l2: 5′-CACAGCTCAAAGCATCAGGA-3′ and 5′-CTGCATGTGAAGCTGTCGTT-3′; G6pc: 5′-CATCAATCTCCTCTGGGTGGC-3′ and 5′-TGTTGCTGTAGTAGTCGGTGTCC-3′; Fasn: 5′-AAGGCTGGGCTCTATGGATT-3′ and 5′-GGAGTGAGGCTGGGTTGATA-3′; Acc2: 5′-CCTGTTGCCCAAGAGAGAG-3′ and 5′-ACAGCGGTCAGGTCAAAGTT-3′; Cpt1a: 5′-ATGACGGCTATGGTGTTTCC-3′ and 5′-GGCTTGTCTCAAGTGCTTCC-3′; Acox1: 5′-TTGGAAACCACTGCCACATA-3′ and 5′-GCCAGGACTATCGCATGATT-3′; Srebp1c: 5′-TGATGCTACGGGTACACACC-3′ and 5′-TTGCGATGTCTCCAGAAGTG-3′; Srebp2: 5′-CCAAGGAGAGCCTGTACTGC-3′ and 5′-ACTGCTGGAGAATGGTGAGG-3′; Acc1: 5′-TGGCAGACCACTATGTTCCA-3′ and 5′-GTTCTGGGAGTTTCGGGTTC-3′; Scd1: 5'-CTGACCTGAAAGCCGAGAAG-3' and 5'-GATGAAGCACATCAGCAGGA-3'. All gene expression results were normalized to an internal control with the following primer set: Gapdh: 5′-TCAACGGATTTGGTCGTATTG-3′ and 5′-GCTCCTGGAAGATGGTGATG-3′.
Statistical Analysis
All of the experiments were repeated at least twice. All data were expressed as the mean ± standard deviation (SD). Statistical analyses were performed with the two-tailed Student's t-test. A P value < 0.05 was considered statistically significant.
Results
Generation of Tcf7l2 Knockout Mice
To explore the potential in vivo role of TCF7L2 gene, we generated the mice of which the Tcf7l2 gene was disrupted by insertion of a LoxP site together with an FRT flanked neomycin selection cassette within the intron 4 of mouse Tcf7l2 gene and a single distal LoxP in the intron 5 downstream of exon 5 (Figure 1A), using the “knock-out first strategy” [23]. During the breeding, we could not obtain Tcf7l2 homozygous null mice (Tcf7l2-/-), consistent with previous reports that Tcf7l2-/- mice died within ~ 24 h of birth [24, 25]. We therefore utilized heterozygous Tcf7l2+/- mice for the remaining study. To determine the Tcf7l2 transcript levels, quantitative RT-PCR was performed in isolated islets and livers of mice at 11 weeks of age. As shown in Figure 1B, there were significantly decreases in Tcf7l2 expression up to 55% and 45% in the isolated islets and livers, respectively, of Tcf7l2+/- mice compared with those of wild-type mice. These results indicated that, as expected, the insertion of the cassette within Tcf7l2 locus had disrupted the gene locus and led to decrease in Tcf7l2 mRNA expression in the heterozygotes.
Metabolic Phenotypes of Tcf7l2 Heterozygous Mice
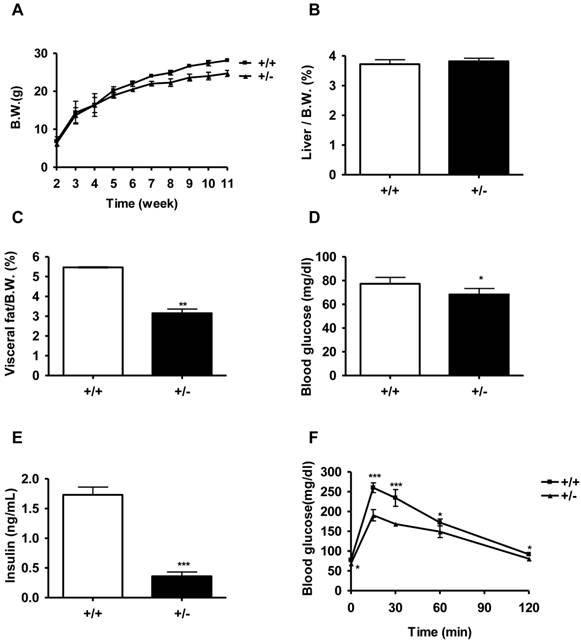
We monitored the body weight of Tcf7l2+/- and wild-type mice beginning from the age of 2 weeks. On a normal chow diet, there was no significant difference in body weight between the two genotypes of mice until 6 weeks of age. From 6 weeks of age, Tcf7l2+/- mice exhibited a significantly less body weight than that observed in the wild-type mice (Figure 2A). At 11 weeks of age, Tcf7l2+/- mice showed 12 % less body weight compared with wild-type mice (P < 0.001). Whereas the liver mass/body weight ratio between Tcf7l2+/- and wild-type mice were not significantly different, the visceral fat mass/body weight ratio decreased in Tcf7l2+/- mice at 11 week old (Figure 2B and 2C). In addition, we observed that the Tcf7l2+/- mice fed with the normal chow diet had reduced levels of fasting glucose and insulin (Figure 2D and 2E), but the level in serum triglyceride was not statistically significant when compared to wild-type mice (Figure 4C). Importantly, compared with the wild-type mice, Tcf7l2+/- mice showed an improved glucose tolerance (Figure 2F). In islets from Tcf7l2+/- mice at 12 weeks old, basal- and glucose-stimulated insulin secretion did not differ between the wild-type and the Tcf7l2+/- mice fed on the normal chow diet (data not shown).
Metabolic characterization of Tcf7l2 +/- mice and wild-type mice on a normal chow diet (NCD). (A) Growth curve for wild-type (■, n = 6) and Tcf7l2 +/- (▲, n = 7) mice from 2 to 11 weeks of age. The Tcf7l2 +/- mice had a significantly less body weight than the wild-type mice since week 6. (B, C) Percentages of liver and visceral fat weight normalized by body weight in wild-type (+/+, n = 6) and Tcf7l2 +/- (+/-, n = 7) mice. (D, E) Fasting blood glucose and serum insulin levels in wild-type (+/+, n = 6) and Tcf7l2 +/- (+/-, n = 7) mice. (F) Intraperitoneal glucose tolerance test (IPGTT) curves of wild-type (■, n = 6) and Tcf7l2 +/- (▲, n = 7) mice. Data are shown as mean ± standard deviation (SD). (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 compared with the wild-type mice.
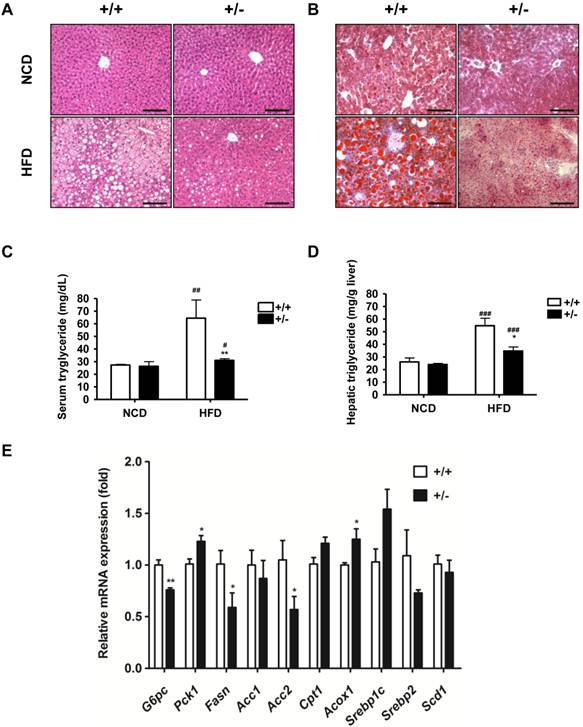
In vivo effects of Tcf7l2 haploinsufficiency on liver histology, triglycerides and hepatic gene expression in mice. The age of mice and feeding duration are described in Materials and Methods. Representative hematoxylin-eosin (A) and Oil Red O (B) stained liver sections in wild-type (+/+) and Tcf7l2 +/- (+/-) mice fed on a NCD and a HFD. Scale bars 100 μm. Tcf7l2 +/- groups showed less hepatic fat deposits than wild-type groups. (C and D) Serum and hepatic triglyceride contents in wild-type (+/+, n = 4) and Tcf7l2 +/- (+/-, n = 4) mice. (E) qRT-PCR analysis of gluconeogenic and lipogenic gene expression in the liver from HFD-fed mice. Data are shown as mean ± SD. (*) p < 0.05, (**) p < 0.01 compared with the wild-type mice. (#) p < 0.05, (##) p < 0.01, (###) p < 0.001 compared with the NCD-fed mice.
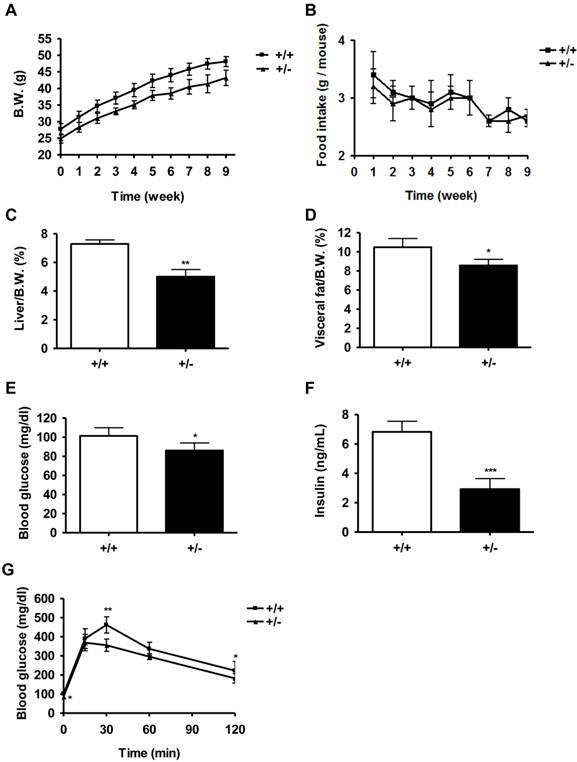
To determine whether Tcf7l2+/- mice were less prone to diabetes than wild-type mice, we fed mice on the HFD for 9 weeks. As shown in Figure 3A, HFD-fed wild-type mice had a significantly greater body weight than Tcf7l2+/- mice, even though the body weight gain in both groups were similar (wild-type : 73.9 ± 5.4 % vs. Tcf7l2+/- : 73.4 ± 8.0 % ) . The food intake was not significantly different between the two genotypes (Figure 3B). The Tcf7l2+/- mice fed with the HFD had significantly less gains of liver and fat mass (Figure 3C and 3D). In addition, although the HFD caused an elevated levels of fasting blood glucose and insulin in both groups, Tcf7l2+/- mice displayed significantly lower levels than those observed in wild-type mice (Figure 3E and 3F). During glucose tolerance tests in the HFD-fed mice, the change of blood glucose levels were also significantly different between wild-type and Tcf7l2+/- mice (Figure 3G). The wild-type mice exhibited an impaired glucose tolerance compared with the Tcf7l2+/-mice. The value of area under the glucose response curves in the Tcf7l2+/- mice was significantly lower than that in the wild-type mice (data not shown). The Tcf7l2+/- mice also showed a lower level of serum triglyceride (P < 0.01, Figure 4C).
Metabolic characterization of Tcf7l2 +/- mice and wild-type mice on a high fat diet (HFD). (A) Growth curve for wild-type (■, n = 7) and Tcf7l2 +/- (▲, n = 6) mice on a HFD from the ages of 13 weeks old for 9 weeks. The Tcf7l2 +/- mice had a significantly less body weight than the wild-type mice during the period of HFD treatment. (B) Food intake in wild-type (■, n = 7) and Tcf7l2 +/- (▲, n = 6) mice on a HFD. (C, D) Percentages of liver and visceral fat weight normalized by body weight in wild-type (+/+, n = 7) and Tcf7l2 +/- (+/-, n = 6). (E, F) Fasting blood glucose and serum insulin levels in wild-type (+/+, n = 7) and Tcf7l2 +/- (+/-, n = 6). (G) Intraperitoneal glucose tolerance test (IPGTT) curves of wild-type (■, n = 7) and Tcf7l2 +/- (▲, n = 6) mice on a HFD. Data are shown as mean ± SD. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 compared with the wild-type mice.
Regulation of Glucose and Lipid Homeostasis
The liver contributes to regulate glucose and lipid homeostasis through gluconeogenesis and lipogenesis [26, 27]. We found that the livers of Tcf7l2+/- mice were less pale and fatty as well as smaller than those of wild-type mice on the HFD. We histologically confirmed the decreased lipid deposition in the liver of Tcf7l2+/- mice on the HFD. In contrast to the wild-type mice with a remarkable hepatic lipid deposit, the Tcf7l2+/- mice had a normal liver histology and no steatosis after 9-week HFD (Figure 4A and 4B). The levels of triglyceride in the liver and serum were consistent with these histological findings (Figure 4C and 4D). We also determined gene expression associated lipid metabolism. While carnitine palmitoyltransferase 1a (CPT1a encoded by CPT1a) and peroxisomal acyl-coenzyme A oxidase 1 (ACOX1 encoded by ACOX1) are thought to be the rate-limiting enzymes of the fatty acid β-oxidation, fatty acid synthase (FAS encoded by FASN) and acetyl-CoA carboxylase 2 (ACC-2 encoded by ACC2) are the genes associated with de novo lipogenesis [28]. In Tcf7l2+/- mice fed with a normal chow diet, there was little or no effect on hepatic expression of Fasn, Acc2 and Cpt1a but significantly decreased Acox1 expression compared with in wild-type mice. Interestingly, Tcf7l2+/- mice fed with a HFD showed increased expressions of Cpt1a and Acox1 as well as reduced expressions of Fasn and Acc2 (Figure 4E), in line with the histological results. However, we did not detect any significant differences in the expression of sterol regulatory element-binding protein gene 1c (Srebp1c), Srebp2, ACC-1, and stearoyl-Coenzyme A desaturase 1 (Scd-1), which are also critical lipogenic genes, between the wild-type mice and the Tcf7l2+/- mice. Since glucose homeostasis was improved in Tcf7l2+/- mice as shown by the IPGTT (Figure 2F and 3F), we also determined hepatic gene expression involved gluconeogenesis (Figure 4E). Glucose-6-phosphatase (G6Pase, which is encoded by G6PC) and phosphoenolpyruvate carboxykinase (PEPCK1, which is encoded by PCK1) are thought to be the rate-limiting enzymes involved in in hepatic gluconeogenesis [6]. On a normal chow diet, Tcf7l2 haploinsufficiency did not significantly affect hepatic expression of these glucose homeostasis genes. However, on the HFD, Tcf7l2+/- mice exhibited a significantly decreased G6pc expression, while it showed slightly increased Pck1 expression (Figure 4E). The increased Pck1 expression seemed contradictory to the data of decreased blood glucose (Figure 3E) and improved glucose intolerance (Figure 3G). To ensure this was not a false result, we knocked down Tcf7l2 expression in the primary hepatocytes isolated from wild-type mice and measured the expression of G6pc and Pck1. The shRNA via lentivirus achieved more than 70% of Tcf7l2 knockdown efficiency in the mouse primary hepatocytes (data not shown). We observed that the Tcf7l2 knockdown caused more than 70% decrease in G6pc expression (p < 0.001), while it resulted in approximately 1-fold increase in Pck1 expression (p < 0.05) (data not shown).
High-fat Diet Regulates Tcf7l2 Expression in a Tissue-specific Manner
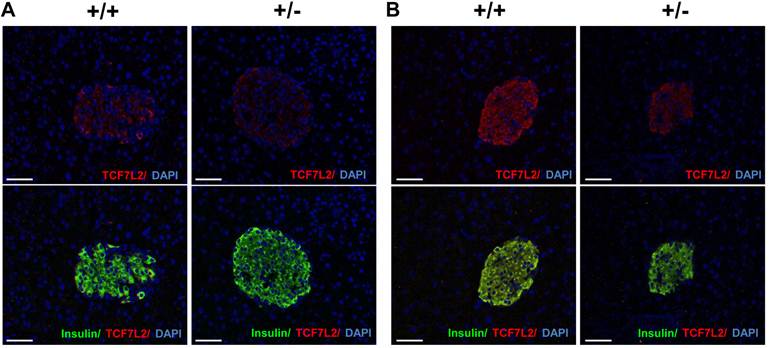
Several literatures have reported TCF7L2 expression in pancreatic islet from type 2 diabetic patients [16-18]. To determine the Tcf7l2 expression in the mice fed with either normal chow diet or HFD, immunohistochemistry was carried out. Triple staining for TCF7L2, insulin and DAPI revealed localization of TCF7L2 in β-cells of pancreatic islet. As shown in Figure 5A, Tcf7l2 was almost undetectable in the sections of pancreatic islet from wild-type and Tcf7l2+/- mice fed with the normal chow diet. In contrast, TCF7L2 expression was upregulated in the wild-type mice, but not much in the Tcf7l2+/- mice fed the HFD (Figure 5B). We also determined the Tcf7l2 expression in the liver from wild-type and Tcf7l2+/- mice. Interestingly, Tcf7l2 expression was down-regulated in the liver when the mice were fed on the HFD (Figure 6).
Expression of Tcf7l2 in mouse pancreatic sections from wild-type (+/+) and Tcf7l2 +/- (+/-) mice fed on a NCD (A) and a HFD (B). Scale bars 50 μm. The Immunohistochemistry was conducted as described in the Materials and Methods.
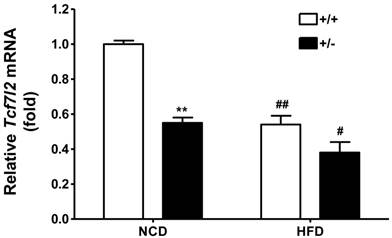
Expression of mouse Tcf7l2 in the liver from wild-type (+/+, n = 3) and Tcf7l2 +/- (+/-, n = 3) mice fed on either NCD or HFD. The age of mice and feeding duration are described in the Materials and Methods. (**) p < 0.01 compared with the wild-type mice. (#) p < 0.05, (##) p < 0.01 compared with the NCD-fed mice.
Discussion
In the present study, we described that Tcf7l2 haploinsufficiency had an effect on glucose- and lipid homeostasis in vivo, suggesting the role of TCF7L2 in metabolic regulation and type 2 diabetic susceptibility. We showed that global reduction of Tcf7l2 expression led to not only a decrease in the level of circulating glucose but also an improved glucose tolerance (Figure 2 and 3). These phenotypes are consistent with those in a recent report [25], where exon 11 of mouse Tcf7l2 was disrupted. Although a detailed profile of gene expression alteration following Tcf7l2 expression change has yet to be characterized to define the effect of Tcf7l2 on hepatic gluconeogenesis, our observation of metabolic phenotypes might be at least partially due to a decreased hepatic glucose production in the Tcf7l2+/- mice. We detected a significant decrease in the expression of G6pc, a rate-limiting enzyme in hepatic gluconeogenesis in the liver from Tcf7l2+/- mice and the wild-type primary hepatocytes of Tcf7l2 knocked-down. Indeed, it has been shown that disruption of WNT signaling by Cre recombinase-mediated deletion of β-catenin reduces the mRNA abundance of G6PC and PCK1 both in vitro and in vivo [6]. However, we surprisingly found a slightly increase in the expression of Pck1, another rate-limiting enzyme in hepatic gluconeogenesis in the liver from Tcf7l2+/- mice and the wild-type primary hepatocytes of Tcf7l2 knocked-down, despite the decreased blood glucose and improved glucose intolerance in the Tcf7l2+/- mice. It is possible that the Pck1 expression increase is due to a feedback mechanism secondary to the more dramatic G6pc suppression by Tcf7l2 knockdown. Tcf7l2 is a transcription factor that may work as either an activator or repressor [41]. It would also be interesting to examine the direct roles of Tcf7l2 on the transcription of G6pc and Pck1 respectively. Lastly, the decreased blood glucose and improved glucose intolerance in the Tcf7l2+/- mice may be reflective of improved glucose utilization due to the global Tcf7l2 knockdown in the whole body. Further in vivo studies, in particular the characterization of glucose disposition and global gene expression using tissue-specific Tcf7l2 knockout mice, are needed to confirm our results and explore the function of hepatic TCF7L2 in pathophysiology of type 2 diabetes.
Obesity and type 2 diabetes are highly associated with hepatic steatosis, which is accompanied by not only impaired insulin clearance, but also impaired inhibition of hepatic glucose output [29-31]. In this study, we accelerated hepatic steatosis by feeding a HFD in wild-type and Tcf7l2+/- mice. We observed that HFD-fed wild-type mice exhibited a uniformly pale fatty liver and hepatomegaly. Intriguingly, these phenotypes were abolished by Tcf7l2 haploinsufficiency. There was no significant difference in food intake between the wild-type and the Tcf7l2+/- mice. However, the Tcf7l2+/- mice under a HFD displayed a reduced expression of Fasn and Acc-2, which are associated with de novo lipogenesis, as well as increased expression of Cpt1a and Acox1, the genes for fatty acid oxidation [28]. The effect of Tcf7l2 expression on the transcription of Cpt1a and Acox1 was also observed with primary hepatocytes isolated from the wild-type mice and the Tcf7l2+/- mice (data not shown). These changes in gene expression caused by Tcf7l2 downregulation may contribute to the improvement of diet-induced hepatic steatosis, even though the changes seemed to be mild and need to be confirmed by direct analysis such as indirect calorimetric analysis [32]. In addition, visceral adiposity is associated with increased rates of lipolysis and fat deposition in those vital metabolic organs including the liver [27]. On the HFD, wild-type mice displayed a higher extent of visceral adiposity, which seemed to fail to adequately store excess triglyceride and contribute to the elevated serum triglyceride levels. It should be noted that Tcf7l2 is ubiquitously expressed in different mouse tissues. It is thus likely that the metabolic phenotypes observed in the Tcf7l2+/- mice may also result from a global Tcf7l2 function change in multiple tissues, and the molecular mechanism underlying the effect of Tcf7l2 haploinsufficiency on nutrient metabolism remains to be fully characterized.
Both increased and decreased TCF7L2 expressions in β-cells have been associated with altered insulin secretion and β-cell apoptosis in human islets [16-20]. Recently, Gaulton et al. reported that the region surrounding the diabetic risk allele of the SNP (rs7903146) at TCF7L2 is in a more open chromatin state than is the non-risk C allele in human pancreatic islets [33]. They also showed that the T allele had a greater enhancer activity than the C allele by using an in vitro reporter gene assay. Similar results were reported by Stitzel et al [34]. These studies suggest an effect associated with the risk allele of the TCF7L2 gene, whereby its open chromatin status leads to increased transcription of the gene [33-34]. In our study, TCF7L2 expression was upregulated by feeding the HFD in the pancreatic islets of wild-type mice, but not much in the Tcf7l2+/- mice (Figure 5B). In addition, although islet transcript level of Tcf7l2, as expected, was only about 50% in Tcf7l2+/- mice compared to wild-type mice, there were no apparent defects in insulin secretion. Tcf7l2+/- mice exhibited normal pancreatic histology. We did not detect any difference in glucose-stimulated insulin secretion in the isolated islets between the two genotypes either. Our data suggest that while a minimal Tcf7l2 expression is required for normal insulin secretion in vivo; enhanced expression may interfere with islet function, conferring a metabolic risk. Overall, our in vivo results favor that a global overexpression of TCF7L2 may lead to an increased susceptibility to type 2 diabetes. In consistent with this hypothesis, Savic et al. recently reported that Tcf7l2 over-expressing transgenic mice are likely to develop diabetes and glucose intolerance after a HFD feeding, showing the reciprocal phenotypes compared to the heterozygous Tcf7l2 null mice [25].
Interestingly, the regulation of TCF7L2 expression seems to be varied in different tissues. In our mouse study with the HFD, the expression levels of Tcf7l2 in pancreatic islets was upregulated (Figure 5B), whereas it was down-regulated in the liver (Figure 6). Consistently, Hindle et al. reported that hepatic expression of TCF7L2 is decreased in diabetic patients as their body mass index (BMI) increase [35]. Currently, we do not know if a decreased Tcf7l2 expression in the liver is simply protective as a feedback mechanism during metabolic disregulation. Furthermore, TCF7L2 gene is known to display a complex pattern of spliced variants with several alternative exons and splice sites. These TCF7L2 splice variants are distributed in a tissue-specific manner, which could strongly influence tissue-specific effects of TCF7L2 as a nuclear effector of WNT signaling [36-39]. Although studies have shed a light on the effect of TCF7L2 expression on metabolic homeostasis, further investigation is required to make informative conclusions of the functional consequences of the tissue-specific regulation of TCF7L2 expression, e.g. via utilization of in vivo model targeting Tcf7l2 in a tissue-specific manner. It would also be intriguing to examine the functional consequences of human TCF7L2 genetic polymorphisms in different tissues.
In summary, the current study demonstrated that TCF7L2 plays an important role in regulation of glucose and lipid metabolism, wherein its biological function change may cause alteration in the susceptibility to type 2 diabetes. Our findings may provide insights into the etiology of type 2 diabetes and important information essential for evaluating the role of TCF7L2 in metabolic regulation.
Acknowledgements
This research was supported by intramural start-up funds from the School of Pharmacy, University of Maryland Baltimore.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jin T. The WNT signaling pathway and diabetes mellitus. Diabetologia. 2008;51:1771-1780
2. Christodoulides C, Lagathu C, Sethi JK, Vidal-Puig A. Adipogenesis and WNT signaling. Trends Endocrinol Metab. 2009;20:16-24
3. Heller RS, Dichmann DS, Jensen J, Miller C, Wong G, Madsen OD. et al. Expression patterns of Wnts, Frizzleds, sFRPs, and misexpression in transgenic mice suggesting a role for Wnts in pancreas and foregut pattern formation. Dev Dyn. 2002;225:260-270
4. Welters HJ, Kulkarni RN. Wnt signaling: relevance to β-cell biology and diabetes. Trends Endocrinol Metab. 2009;19:349-355
5. Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:4914-4919
6. Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O. et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011;4:ra6
7. Kanazawa A, Tsukada S, Sekine A, Tsunoda T, Takahashi A, Kashiwagi A. et al. Association of the gene encoding wingless-type mammary tumor virus integration-site family member 5B (WNT5B) with type 2 diabetes. Am J Hum Genet. 2004;75:832-843
8. Christodoulides C, Scarda A, Granzotto M, Milan G, Dalla Nora E, Keogh J. et al. WNT10B mutations in human obesity. Diabetologia. 2006;49:678-684
9. Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C. et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278-1282
10. Salpea KD, Gable DR, Cooper JA, Stephens JW, Hurel SJ, Ireland HA. et al. The effect of WNT5B IVS3C>G on the susceptibility to type 2 diabetes in UK Caucasian subjects. Nutr Metab Cardiovasc Dis. 2009;19:140-145
11. Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J. et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;8:320-323
12. Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PI, Shuldiner AR. et al. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Eng J Med. 2006;355:241-250
13. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL. et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341-1345
14. Bonetti S, Trombetta M, Malerba G, Boselli L, Trabetti E, Muggeo M. et al. Variants and haplotypes of TCF7L2 are associated with β-cell function in patients with newly diagnosed type 2 diabetes: the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 1. J Clin Endocrinol Metab. 2011;96:E389-E393
15. Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D. et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60:2624-2634
16. Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P. et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155-2163
17. Elbein SC, Chu WS, Das SK, Yao-Borengasser A, Hasstedt SJ, Wang H. et al. Transcription factor 7-like 2 polymorphisms and type 2 diabetes, glucose homeostasis traits and gene expression in US participants of European and African descent. Diabetologia. 2007;50:1621-1630
18. Lee SH, Demeterco C, Geron I, Abrahamsson A, Levine F, Itkin-Ansari P. Islet specific Wnt activation in human type II diabetes. Exp Diabetes Res. 2008;2008:728763
19. Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57:645-653
20. Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet. 2009;18:2388-2399
21. El Muayed M, Billings LK, Raja MR, Zhang X, Park PJ, Newman MV. et al. Acute cytokine-mediated downregulation of the zinc transporter ZnT8 alters pancreatic beta-cell function. J Endocrinol. 2010;206:159-169
22. Lillie RD, Ashburn LL. Supersaturated solutions of fat stains in dilute isopropanol for demonstration of acute fatty degeneration not shown by Herxheimer's technique. Arch Pathol. 1943;36:432-440
23. Testa G, Schaft J, van der Hoeven F, Glaser S, Anastassiadis K, Zhang Y. et al. A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis. 2004;38:151-158
24. Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ. et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379-383
25. Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. 2011;21:1417-1425
26. Wahren J, Ekberg K. Splanchnic regulation of glucose production. Annu Rev Nutr. 2007;27:329-345
27. Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881-887
28. Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC. et al. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474:506-510
29. Rijzewijk LJ, van der Meer RW, Lubberink M, Lamb HJ, Romijn JA, de Roos A. et al. Liver fat content in type 2 diabetes: Relationship with hepatic perfusion and substrate metabolism. Diabetes. 2010;59:2747-2754
30. Seppälä-Lindroos A, Vehkavaara S, Häkkinen AM, Goto T, Westerbacka J, Sovijärvi A. et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023-3028
31. Kotronen A, Vehkavaara S, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H. Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab. 2007;293:E1709-E1715
32. Haramizu S, Nagasawa A, Ota N, Hase T, Tokimitsu I, Murase T. Different contribution of muscle and liver lipid metabolism to endurance capacity and obesity susceptibility of mice. J Appl Physiol. 2009;106:871-879
33. Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP. et al. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42:255-259
34. Stitzel ML, Sethupathy P, Pearson DS, Chines PS, Song L, Erdos MR. et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443-455
35. Hindle AK, Brody F, Tevar R, Kluk B, Hill S, McCaffrey T. et al. TCF7L2 expression in diabetic patients undergoing bariatric surgery. Surg Endosc. 2009;23:700-704
36. Prokunina-Olsson L, Welch C, Hansson O, Adhikari N, Scott LJ, Usher N. et al. Tissue-specific alternative splicing of TCF7L2. Hum Mol Genet. 2009;18:3795-3804
37. Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S. et al. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 2010;38:1964-1981
38. Mondal AK, Das SK, Baldini G, Chu WS, Sharma NK, Hackney OG. et al. Genotype and tissue-specific effects on alternative splicing of the transcription factor 7-like 2 gene in humans. J Clin Endocrinol Metab. 2010;95:1450-1457
39. Le Bacquer O, Shu L, Marchand M, Neve B, Paroni F, Kerr Conte J. et al. TCF7L2 splice variants have distinct effects on beta-cell turnover and function. Hum Mol Genet. 2011;20:1906-1915
40. Moldeus P, Hogberg J, Orrenius S. Isolation and use of liver cells. Methods Enzymol. 1978;52:60-71
41. Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochim Biophys Acta. 1999;1424:M23-37
Author contact
![]() Corresponding author: Yan Shu, Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 Penn Street, HSF2 Room 555, Baltimore, MD 21201. Tel: 410-706-7358 Fax: 410-706-5017. e-mail: yshuumaryland.edu.
Corresponding author: Yan Shu, Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 Penn Street, HSF2 Room 555, Baltimore, MD 21201. Tel: 410-706-7358 Fax: 410-706-5017. e-mail: yshuumaryland.edu.