Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(6):859-869. doi:10.7150/ijbs.3490 This issue Cite
Research Paper
Cdc42-Interacting Protein-4 Promotes TGF-Β1-Induced Epithelial-Mesenchymal Transition and Extracellular Matrix Deposition in Renal Proximal Tubular Epithelial Cells
Shoujun Bai1,3*, Rui Zeng1* ![]() , Qiaodan Zhou1, Wenhui Liao2, Yamin Zhang1, Chuou Xu1, Min Han1, Guangchang Pei1, Lili Liu1, Xiaocheng Liu1, Ying Yao1, Gang Xu1
, Qiaodan Zhou1, Wenhui Liao2, Yamin Zhang1, Chuou Xu1, Min Han1, Guangchang Pei1, Lili Liu1, Xiaocheng Liu1, Ying Yao1, Gang Xu1 ![]()
1. Devision of Nephrology;
2. Department of Geriatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 430030, China.
3. Zhongshan Hospital Qingpu Branch of Fudan University, 1158 East Gongyuan Road, Shanghai, 201700, China.
* Shoujun Bai and Rui Zeng contributed equally to this work.
Received 2011-9-8; Accepted 2012-5-29; Published 2012-6-13
Abstract
Cdc42-interacting protein-4 (CIP4) is an F-BAR (Fer/CIP4 and Bin, amphiphysin, Rvs) family member that regulates membrane deformation and endocytosis, playing a key role in extracellular matrix (ECM) deposition and invasion of cancer cells. These processes are analogous to those observed during the initial epithelial-mesenchymal transition (EMT) of renal tubular epithelial cells. The role of CIP4 in renal tubular EMT and renal tubulointerstitial fibrosis was investigated over the course of the current study, demonstrating that the expression of CIP4 increased in the tubular epithelia of 5/6-nephrectomized rats and TGF-β1 treated HK-2 cells. Endogenous CIP4 evidenced punctate localization throughout the cytosol, with elevated levels observed in the perinuclear region of HK-2 cells. Subsequent to TGF-β1 treatment, CIP4 expression increased, forming clusters at the cell periphery that gradually redistributed into the cytoplasm. Simultaneously, EMT induction in cells was confirmed by the prevalence of morphological changes, loss of E-cadherin, increase in α-SMA expression, and secretion of fibronectin. Overexpression of CIP4 promoted characteristics similar to those commonly observed in EMT, and small interfering RNA (siRNA) molecules capable of CIP4 knockdown were used to demonstrate reversed EMT. Cumulatively, results of the current study suggest that CIP4 promotes TGF-β1-induced EMT in tubular epithelial cells. Through this mechanism, CIP4 is capable of inducing ECM deposition and exacerbating progressive fibrosis in chronic renal failure.
Keywords: Cdc42-interacting protein-4, epithelial-mesenchymal transition, renal proximal tubular epithelial cells, β-catenin, TGF-β1-induced
Introduction
Over the past decade, injured renal tubular epithelial cells have been implicated in increasing kidney myofibroblast populations through the process of epithelial-mesenchymal transition (EMT) [1-3], ultimately leading to inevitable progressive chronic kidney disease (CKD) and renal tubulointerstitial fibrosis [4, 5]. The EMT process consists of several sequential steps [2, 6]: dissolution of tight junctions [7], loss of cell-cell adhesion, loss of apico-basal polarity, reorganization of the actin cytoskeleton, and increases in cell motility. EMT is generally regarded as the method by which renal tubular epithelial cells transform into myofibroblasts [8], paving the way for extracellular matrix (ECM) deposition and ultimately renal fibrosis [1, 2].
Although recent studies using genetic fate mapping techniques in mouse fibrosis models have provided evidence that contradicts former reports of the derivation of myofibroblasts within the tubulointerstitial space from renal tubular EMT [9-11], the results of these studies remain consistent with the notion that an EMT-like process occurs in renal epithelial cells [10, 12]. When primary renal epithelial cells were cultured in vitro in mice and incubated with TGF-β, epithelial cells were found to increase expression of α-SMA while E-cadherin expression decreased, a characteristic indicative of EMT process occurrence [10] that has been referred to as partial EMT [10].
Some researchers have suggested that this partial transition of mesenchymal epithelial cells may regulate interstitial fibrosis development through a paracrine signaling mechanism [10, 11]. Furthermore, Koesters et al. utilizing the inducible expression of TGF-β1 in renal epithelial cells identified overexpression of TGF-β1 in renal tubules with notably increased occurrence of widespread peritubular fibrosis [13], providing strong evidence for the mediation of excessive deposition of ECM by TGF-β1 throughout the process of EMT [6]. Despite the novel nature of this hypothesis and increasing supportive evidence, the molecular mechanism behind this effect has not been fully characterized.
Cdc42-interacting protein-4 (CIP4) belongs to the Bin/amphiphysin/Rvs (BAR) domain protein superfamily. Members of this superfamily are noted for their involvement in membrane remodeling processes that occur in various cellular pathways, such as endocytosis, cytokinesis, T-tubule formation, cell migration, and neuromorphogenesis [14-17]. CIP4 contains a C-terminal SH3 domain that allows it to bind to various proteins involved in cell migration, most notably including dynamin and regulatory proteins [18]. In human renal tumor cells, numerous CIP4 splicing variants have been produced that successfully promote β-catenin tyrosine phosphorylation, leading to renal tumor cell metastasis [19]. Activated CIP4 in pancreatic tumor cells and breast cancer cells has also demonstrated the ability to promote cellular migration and invasion in renal tumor cell lines [20, 21]. Recently, CIP4 has been shown to inhibit ECM degradation by limiting the expression of type-I transmembrane matrix metalloproteases (MMPs) on the cellular surface, suggesting that CIP4 also plays a role in ECM regulation [22]. Based on the previous observation that ECM regulation and invasion of cancer cells are analogous processes to those observed during the renal epithelial-mesenchymal transition (EMT), it is likely that CIP4 may serve as a signaling molecule in the promotion of renal tubular EMT.
The present study examines the potential mediation of TGF-β1-induced EMT by CIP4. To this end, fibrotic renal tissue CIP4 levels in a model of 5/6 nephrectomy were assessed, resulting in the in vitro detection of CIP4 in HK-2 cells treated with TGF-β1. Furthermore, the effect of CIP4 siRNA on TGF-β1-induced EMT was examined in order to provide mechanistic information on these effects.
Materials and Methods
Animal subjects and sampling
All experimental protocols were performed in accordance with the guidelines for the care and use of animals established by the National Institute of Health and the Huazhong University of Science and Technology. Male Sprague-Dawley rats (150-200 g) were obtained from the Tongji Medical Laboratory Animal Center (Wuhan, China). All rats underwent 5/6 nephrectomy according to the previously described method [33]. Rats were subsequently randomized into the sham-operated group (n=10) and the 5/6 nephrectomy group (n=10). All rats were euthanized 12 wk after nephrectomy, and serum was collected for the determination of creatinine and urea nitrogen. Kidneys were rapidly excised after euthanization, and some samples were fixed with 4% paraformaldehyde. Remaining samples were frozen in liquid nitrogen for later examination. Histological examination of renal tissues was conducted using samples stained with Masson's trichrome.
Culture of human proximal tubular cells
A human proximal tubular cell line (HK-2, CRL-2190) provided by the American Type Culture Collection (ATCC) was grown in DMEM/F12 (1:1) (Gibco Industries Inc, USA) culture medium containing 10% calf serum (Gibco Industries Inc, USA). Cells were cultured in an incubator with a 5% CO2 atmosphere at 37ºC. At 70-80% confluency, cells were synchronized with serum-free culture fluid for 24 h and subsequently stimulated with TGF-β1 (10 ng/mL; B&D Systems, MN, USA) for 72 h to obtain the renal tubular EMT. For inhibition study applications, a phosphoinositide 3-kinase inhibitor (PI3K) inhibitor Wortmannin (CST, MA, USA), p38 inhibitor SB 203580 (CST, MA, USA), or negative control (DMSO, Sigma) was incubated with cells for 1 h prior to TGF-β1 treatment.
Gene silencing by small interfering RNA
For small interfering RNA (siRNA) experiments, RNA primers complementary to human CIP4 or SMAD4 were designed and synthesized by the Shanghai Invitrogen Biotechnology Company. HK-2 cells were transfected with the annealed RNA primer pair using Lipofectamine 2000 (Invitrogen Inc, USA) in accordance with the instructions provided by the manufacturer. Cells transfected with scrambled siRNA served as controls. Five hours after transfection, cells were incubated with 10 ng/mL of TGF-β1 for 72 h in order to observe the effects of CIP4 or SMAD4 silencing.
Generation of CIP4 over-expression in HK-2 clones
CIP4 cDNA fragments were cloned into a pcDNA3.1 vector, and the plasmid pcDNA3.1-CIP4 was transfected into HK-2 cells using Lipofectamine 2000 in accordance with instructions provided by the manufacturer. Cells were transfected with an empty pcDNA3.1 vector as a negative control.
RT-PCR
Total RNA was isolated from HK-2 cells with the Trizol reagent (Invitrogen, USA) in accordance with the protocol provided by the manufacturer. Reverse transcription was performed using the standard reagent (Promega, USA) in accordance with the protocols provided by the manufacturer. Primers were synthesized by Invitrogen. The CIP4 primers were: upstream 5′-ACACGGAGTTTGATGAGGAT-3′, and downstream 5′-ATGGTGGAACGATGGTAGAA-3′. The GAPDH primers were: upstream 5′-GGATTTGGTCGTATTGGG-3′, and downstream 5′-GGAAGATGGTGATGGGATT-3′. The conditions for the amplification were denaturation at 94ºC for 5 min, 94 ºC for 45 s, 53 ºC for 45 s, and 72 ºC for 45 s, totaling 32 cycles plus a final extension at 72 ºC for 10 min. A volume of 5μl PCR product was loaded to the 1.5 g/L agarose gel for electrophoresis. Relative amounts of mRNA were normalized by GAPDH. The results were scanned and analyzed using the Kodak science gel image analysis system (Eastman Kodak Co, NY, USA).
Western blot analysis
In order to perform Western blot analysis, kidney tissues and cells were extracted with a lysis buffer containing 1% Triton-100, 0.5% Nonidet P-40, 20 mM Tris-HCl, 15 mM NaCl, 1 mmol/L EDTA, 1 mmol/L egtazic acid, 1 mM Na3VO4•10H2O, 2 mM NaF, 2 mM Na2P2O4•10H2O, 10 mM β-glycerophosphate disodium salt (pH 8.0), and a cocktail inhibitor (5 mM phenylmethanesulfonyl fluoride, 5 µg/ml leupeptin, 5 µg/ml pepstatin, and 5 µg/ml aprotinin) for 20 min on ice. Cell debris was removed by centrifugation at 12,000 g for 30 min at 4ºC. Protein concentrations were determined using the Bradford method (BioRad, Hercules, CA). Total protein (100 µg) was subjected to SDS-PAGE. The proteins were then transferred to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). After transfer, the membranes were blocked with 5% nonfat milk in TBS with 0.1% Tween-20 for 1 h at 37 ºC and blotted routinely with CIP4 (Santa Cruz, USA), E-cadherin (BD Biosciences, San Jose, CA, USA), α-SMA (Sigma, USA),Smad4 (Epitomics Inc, CA, USA) and GAPDH (Proteintech Group, Inc) at 4 ºC overnight. The bound antibody complexes were visualized using an enhanced chemiluminescence reaction, and X-ray film was scanned using a ChemiImager 5500 image analysis system (Alpha Innotech, San Leandro, CA, USA). Quantity One software (Bio-Rad) was used to quantify the density of bands.
Immunohistochemistry and immunocytochemistry
For immunohistochemical analysis, paraffin sections were incubated with primary anti-CIP4 antibody (1:100, Santa Cruz, USA) overnight at 4ºC followed by biotinylated goat anti-rabbit IgG (1:500,Peprotech, USA) at 37ºC for 30 min. Antibody reactions were visualized using diaminobenzidine (DAKO, Tokyo, Japan). An irrelevant isotype rabbit Ig was used for the negative control. Microscopic images were captured using a Nikon DXM 1200 digital camera and analyzed with HPIAs-1000 image analysis software (Qianping Image Engineering Co. of the Tongji Medical School, China). For immunocytochemical analysis, HK-2 cells were cultured on sterile glass coverslips in six-well plates. Thereafter, cultures were treated and fixed with iced acetone for 10 min, incubated with 0.1% Triton X-100 for 10 min in order to induce membrane rupture, and incubated with 1% BSA at 37ºC for an additional 30 min for blocking. Cell cultures were then incubated with anti-CIP4 (1:100) overnight at 4ºC, followed by incubation with FITC-goat anti-rabbit IgG at 37ºC for 1 h. Thereafter, the nucleus was stained with DAPI (Sigma, USA) for 5 min and analyzed by laser confocal scanning microscopy (Leica, Heidelberg, Germany).
ELISA assay
Fibronectin (FN) secretion was determined using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Boster Biological Technology, Wuhan, China) in accordance with the instructions provided by the manufacturer, as the previous report [28]. The OD value was detected by an ELISA Reader (Thermo Scientific, Rockford, IL, USA) at a wavelength of 450 nm and calculated in the linear portion of the curve.
Statistical Analysis
All data were expressed as mean ± standard deviation (mean±STD) and processed using the SPSS 12.0 software system (IBM, Chicago, IL). ANOVA was used to compare the mean values. Statistical significance was observed when P was less than 0.05 (P<0.05).
Results
CIP4 expression in renal fibrosis in vivo
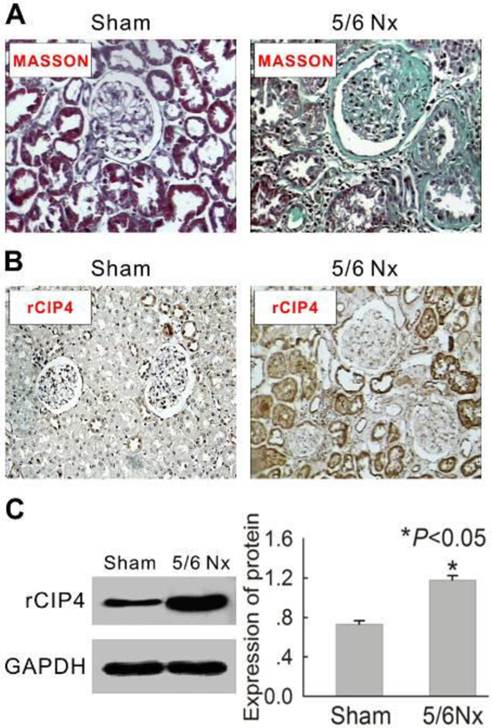
To investigate the distribution of rat CIP4 (rCIP4) in renal tubulointerstitial fibrosis, the classic renal fibrosis model of the 5/6 subtotal nephrectomy was adopted. As shown in Table 1, the serum urea nitrogen and serum creatinine in 5/6-nephrectomized rats were significantly increased compared with levels observed in the sham-operated rats. Light microscopic examination (Masson stain) indicated both glomerulosclerosis and interstitial fibrosis in 5/6-nephrectomized rats (Figure 1A). Immunohistochemical assay further indicated that rCIP4 was extensively expressed in the renal tubules in 5/6-nephrectomized rats, whereas little staining to indicate rCIP4 presence was revealed in the sham-operated rats (Figure 1B). Western blot analysis demonstrated similar results, indicating that rCIP4 protein expression was significantly increased in the kidneys of 5/6-nephrectomized rats compared to levels observed in sham-operated rats (Figure 1C). These results suggest that CIP4 may be involved in renal fibrosis.
Renal function in sham-operated and 5/6-nephrectomized rats
| Groups | n | Bun (mmol/L) | Scr (µmol/L) | BW (g) |
|---|---|---|---|---|
| sham-operated | 10 | 5.78±1.04 | 28.65±6.27 | 378±9 |
| 5/6-nephrectomized | 10 | 37.02±15.9* | 125.43±61.78* | 362±128* |
* P< 0.05, compared with sham-operated rats. Bun, serum urea nitrogen; Scr, serum creatinine; BW, bodyweight.
Expression of rat CIP4 (rCIP4) is increased in the kidney tissue of 5/6 nephrectomized rats. (A) Representative histology of kidney sections from sham-operated and 5/6-nephrectomized (5/6 NX) rats are stained with Masson's trichrome stain. Interstitial fibrosis and glomerular sclerosis (blue area) are extensive observed in 5/6 NX rats. The tissue of sham rats was observed to be normal. (Magnification × 200). (B) Representative immunohistochemical detection of rCIP4 in the kidney tissues of sham-operated and 5/6-nephrectomized rats. CIP4 immunostaining (brown area) is strong in the epithelial tubular cells in 5/6 NX rats and weaker in sham rats. (Magnification × 200). (C) Representative Western blot analysis of rCIP4 in the kidney tissues of sham-operated and 5/6-nephrectomized rats. The expression of the rCIP4 protein was elevated in the kidney tissue of 5/6 NX rats. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 4). *P<0.05 compared with sham-operated rats.
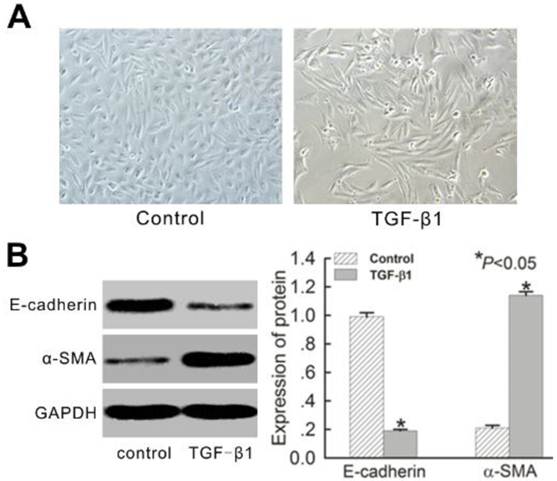
TGF-β1 induced EMT in HK-2 cells and increased expression of CIP4
Normal HK-2 cells presented ovoid or cubic morphologies, forming an epithelial monolayer with evidence of a tight cell-cell junction (Figure 2A). After treatment with 10 ng/mL TGF-β1 for 72 h, cells evidenced notable elongation consistent with the morphology of myofibroblasts, and the intercellular space became enlarged (Figure 2A). E-cadherin and α-SMA marked epithelial cells and fibroblasts, respectively, in the renal interstitium. Normal HK-2 cells showed typical epithelial characteristics of high E-cadherin protein expression and low α-SMA expression. After treatment with TGF-β1 for 72 h, the cells evidenced the general characteristics of myofibroblasts, including a significant reduction in E-cadherin expression and corresponding increase in α-SMA expression (Figure 2B).
TGF-β1-induced EMT in HK-2 cells in vitro for 72 h. (A) Morphological changes of HK-2 cells with or without TGF-β1 treatment. Photographs were attained using an Olympus phase contrast microscope (Magnification × 200). (B) Western blot analysis for E-cadherin and α-SMA protein expression. A representative blot from four independent experiments is shown. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 4). Data is expressed as mean±SEM. *P<0.05 vs. control
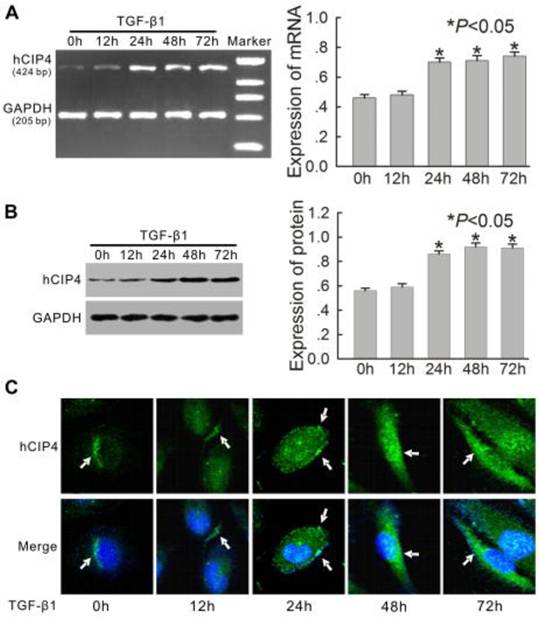
Tubular epithelial cells are the natural in vivo targets of TGF-β1, a compound that plays a critical role in renal fibrosis in the 5/6-nephrectomy rat model [23]. Based on this finding, the expression of human CIP4 (hCIP4) in proximal epithelial cells (HK-2 cells) treated with TGF-β1 in vitro were examined. After stimulation with 10 μg/L of TGF-β1, the mRNA and protein levels of hCIP4 in HK-2 cells increased gradually from 24 h to 72 h (Figure 3A and 3B). Confocal fluorescent microscopy revealed that hCIP4 exhibited punctate localization throughout the cytosol, with the highest levels localized in the perinuclear region of HK-2 cells. After stimulation with TGF-β1, hCIP4 levels increased, and the compound was recruited into cluster formations located adjacent to the cell periphery. Gradually, these clusters were observed to be redistributed into the cytoplasm (Figure 3C). This data suggest that upregulation of hCIP4 is closely related to the development of EMT in HK-2 cells exposed to TGF-β1.
Expression of human CIP4 (hCIP4) is upregulated in the HK-2 cells treated with TGF-β1. (A) Representative RT-PCR analysis for hCIP4 mRNA expression in the HK-2 cells treated by TGF-β1 for 0 h, 12 h, 24 h, 48 h, and 72 h. The expression of hCIP4 mRNA was increased in TGF-β1-treated cells. The histogram shows the average relative amounts of mRNA density corrected for the loading control, GAPDH (n = 4).*P<0.05 compared with the HK-2 cells treated by TGF-β1 for 0 h. (B) Representative Western blot analysis of hCIP4 in the HK-2 cells treated by TGF-β1 for 0 h, 12 h, 24 h, 48 h, and 72 h. The expression of the hCIP4 protein was increased in TGF-β1-treated cells. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 4).*P<0.05 compared with the HK-2 cells treated by TGF-β1 for 0 h. (C) Representative immunofluorescence of hCIP4 localization in a monolayer of control cells and TGF-β1-treated cells. Arrows indicate positions of CIP4 in HK-2 cells. Cell nuclei were enhanced by staining of cell nuclei with DAPI. Scale bars represent 20μm.
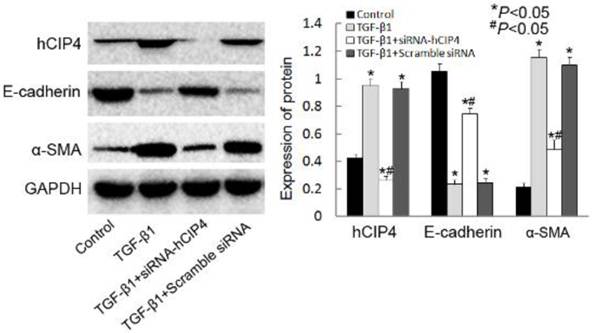
Silencing of CIP4 inhibited TGF-β1-induced EMT
Investigation of the ability of hCIP4 to affect TGF-β1-dependent EMT was conducted by using hCIP4 target siRNA to knockdown hCIP4 expression. As shown in Figure 4, the expression of hCIP4 was dramatically inhibited by hCIP4 siRNAtransfection in TGF-β1 treated cells. TGF-β1 treatment resulted in a decrease of E-cadherin expression and induction of α-SMA expression (Figure 4B). HK-2 cells transfected with CIP4-siRNA, however, demonstrated resistance to TGF-β1-induced EMT. As E-cadherin protein levels increased, a dramatic decrease in α-SMA expression was clearly evident (Figure 4B).These results have been further confirmed by another hCIP4 siRNA (Supplemental Material).
Knockdown of human CIP4 represses TGF-β1-induced EMT. Representative Western blot of hCIP4, E-cadherin, and α-SMA in control cells, TGF-β1-treated cells, TGF-β1-treated hCIP4-siRNA cells, and TGF-β1-treated scrambled siRNA cells. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 4). *P<0.05 compared with control cells, #P<0.05 compared with TGF-β1-treated cells.
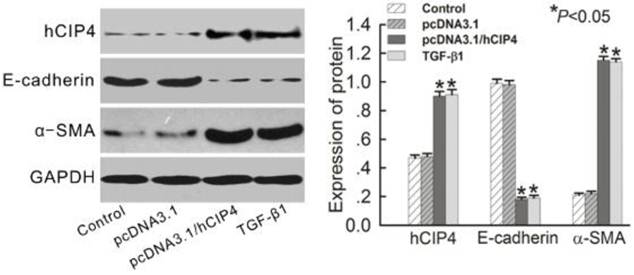
CIP4 induced EMT in CIP4-transfected HK-2 cells
Since hCIP4 expression was increased by TGF-β1 in EMT, the effect of overexpression of hCIP4 on the progress of EMT was investigated. Plasmid pcDNA3.1-hCIP4 was transfected into HK-2 cells, and overexpression of hCIP4 was confirmed by Western blot analysis (Figure 5). No difference was observed between protein expression in control cells and cells transfected with pcDNA3.1/Zeo empty vector (Figure 5). Surprisingly, E-cadherin was downregulated in hCIP4-transfected cells compared to control cells and empty vector-transfected cells (Figure 5). In contrast, α-SMA was upregulated in hCIP4-transfected cells (Figure 5). These results indicated that overexpression of CIP4 is involved in the process of TGF-β1-induced tubular EMT.
Over-expression of human CIP4 promotes EMT. Representative Western blots of hCIP4, E-cadherin, and α-SMA in control cells, pcDNA3.1 empty vector-transfected cells, pcDNA3.1-CIP4-transfected cells, and TGF-β1-treated cells. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 4). *P<0.05 compared with control cells.
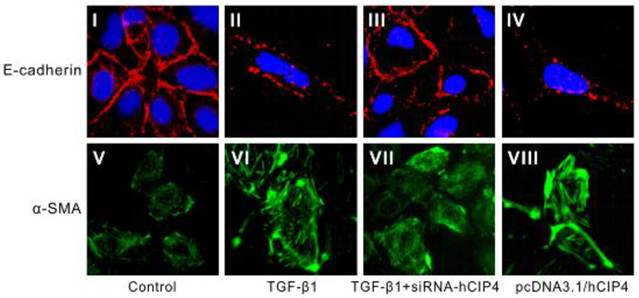
CIP4 involved in TGF-β1-induced EMT was confirmed by morphology
Confocal fluorescent microscopy indicated a continuous distribution of E-cadherin near the perimeter of control cells (Figure 6 I) but a discontinuous distribution of E-cadherin near the perimeter of TGF-β1-treated cells (Figure 6 II). Furthermore, α-SMA was present exclusively in the cytosol of TGF-β1-treated cells (Figure 6 VI), and little endogenous expression in control cells was observed (Figure 6 V), suggesting that these cells underwent EMT in response to TGF-β1. After treatment with hCIP4-siRNA, E-cadherin re-localized to regions surrounding the cellular junctions (Figure 6 III), a reduction in α-SMA expression compared with that observed in TGF-β1-treated cells was observed (Figure 6 VI and 6 VI). In contrast, the hCIP4-transfected cells indicated characteristics common to myofibroblasts, including E-cadherin distribution alteration from a linear pattern to a zipper-like pattern (Figure 6 I and 6 IV) and the appearance of α-SMA (Figure 6 V and 6 VIII). Collectively, this data reconfirms that CIP4 is involved in TGF-β1-induced EMT.
Localization of E-cadherin and α-SMA in variant cells. Representative confocal microscopy of E-cadherin and α-SMA in monolayers of control cells, 72 h TGF-β-treated cells, TGF-β1-treated hCIP4-siRNA cells, and pcDNA3.1/hCIP4 transfected cells. HK-2 cells were immunostained for E-cadherin (I, II, III, and IV) and α-SMA (V, VI, VII, and VIII) and visualized by confocal fluorescent microscopy. Cell nuclei were enhanced by staining of cell nuclei with DAPI for E-cadherin. Scale bars represent 30μm. (Magnification × 400)
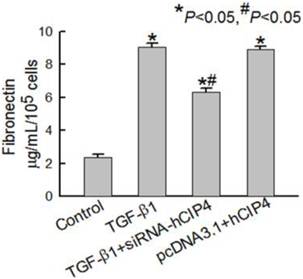
CIP4 promoted fibronectin secretion in HK-2 cells during TGF-β1-induced EMT
Based on the observation that accumulated interstitial matrix components were a consequence of EMT, fibronectin secretion by HK-2 cells was measured in the culture supernatant. Treatment of HK-2 cells for 72 h with TGF-β1 resulted in a significant increase in fibronectin secretion compared to that observed in control cells (Figure 7). HK-2 cells transfected with CIP4-specific siRNA were resistant to TGF-β1-induced fibronectin secretion (Figure 7). When HK-2 cells were transfected with CIP4, a significant increase in fibronectin in the culture supernatant was observed (Figure 7). These results indicate that CIP4 has the potential to regulate ECM secretion in HK-2 cells during TGF-β1-induced EMT.
Fibronectin secretion in variant cells. Representative fibronectin secretion in the culture media of control cells, TGF-β1-treated cells and, TGF-β1-treated CIP4-siRNA cells (n = 4). *P<0.05 compared with control cells, #P<0.05 compared with TGF-β1-treated cells
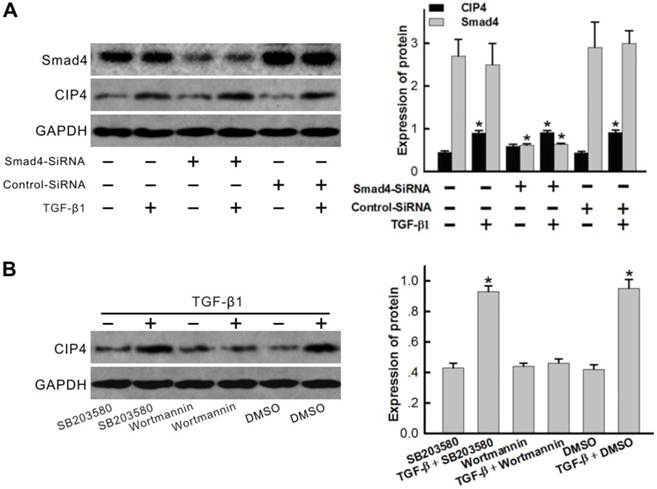
TGF-β1 upregulation of CIP4 expression through a PI3K/Akt dependent pathway
As SMAD pathways are central mediators of TGF-β1 signaling pathways, SMAD4 specific siRNAs were designed in order to examine the role of the SMAD pathway as an essential element of hCIP4 upregulation. As seen in Figure 8A, TGF-β1 exhibited the ability to induce hCIP4 expression; however, SMAD4 silencing did not block TGF-β1-induced hCIP4 upregulation. HK2 cells were subsequently pretreated with Wortmannin (PI3K/AKT inhibitor) or SB203580 (p38 inhibitor). Western blot analysis revealed that the increased CIP4 level in response to TGF-β1 is efficiently inhibited by Wortmannin; however, SB203580 did not evidence the ability to inhibit the increase in CIP4 level (Figure 8B). These results suggest that the PI3K/AKT pathway is critical for TGF-β1-induced hCIP4 upregulation.
TGF-β1 upregulated CIP4 expression in HK-2 cells through a PI3K/Akt dependent pathway, not by SMAD activation. (A) Cells were transfected with SMAD4-specific siRNA or Scrambled siRNA with TGF-β1 treatment for 72 h, and Western blot analyses were performed to examine CIP4 and Smad4 expression. *P<0.05 compared with control cells. (B) Western blot analysis for CIP4 expression in HK2 cells treated with TGF-β1 and/or (SB203580, Wortmannin) for 72 h. DMSO treated cells were used as a control. The histogram shows the average volume density corrected for the loading control, GAPDH (n = 3). *P<0.05 compared with DMSO treated cells.
Discussion
The expression of CIP4 is increased in renal fibrotic conditions both in vivo and in vitro after exposure to TGF-β1 in the current study. TGF-β1 is known for its ability to induce and participant in the development and progression of renal tubulointerstitial fibrosis through EMT [8, 24-26]. In 5/6-nephrectomized rats, an increased in the expression of rCIP4 in the renal tubules was identified, suggesting that upregulation of CIP4 is involved in the EMT processes that occur as a prominent feature of renal fibrosis. In addition to the upregulation of rCIP4 in vivo, overexpression of hCIP4 was demonstrated to promote the development of EMT in HK-2 cells exposed to TGF-β1. Conversely, silencing of hCIP4 appeared to reverse TGF-β1-induced EMT by increasing the expression of E-cadherin, an epithelial marker, and reducing the expression of mesenchymal markers, such as α-SMA. Furthermore, by using an hCIP4-transfected cell culture system, overexpression of hCIP4 was shown to directly induce EMT in HK-2 cells. To more accurately determine the location of CIP4 and provide superior assessment of the role of CIP4 expression in HK-2 cells, immunofluorescence laser scanning confocal microscopy was used to show that hCIP4 exhibited punctate localization throughout the cytosol. Notably, the highest levels were shown to be within the perinuclear region of HK-2 cells. After stimulation with TGF-β1, hCIP4 increased and formed visible clusters adjacent to the cell periphery that gradually were redistributed into the cytoplasm. This data suggests that upregulation of CIP4 plays an important role in EMT occurrence in proximal kidney tubules epithelial cells.
CIP4 serves as a scaffolding protein for Cdc42, a member of the F-BAR family. Activation of Cdc42 has been demonstrated to be directly related to cell scattering/migration through a mechanism that involves the disruption of cell-cell junctions and the rearrangement of the actin cytoskeleton [27, 28]. Cytoskeletal rearrangement is required for the migration and invasion of cells [29]. In tumor cell lines, this effect may be evidenced by the acquisition of a migratory phenotype atypical of epithelial-type cells, a phenotype that correlates with the formation of specialized structures such as lamellipodia and invadopodia [20]. Endogenous CIP4 localizes to the invadopodia of migrating cells to promote cancer cell motility and invasion [20]. In epithelial tissue, CIP4 has been identified to regulate the early E-cadherin trafficking/endocytosis [30], which is the key process of EMT in renal tubular epithelial cell [8]. In this study, hCIP4 was observed to gather in clusters at the cell periphery of HK-2 cells during early stages of TGF-β1 stimulation, analogous to similar processes in invasive breast tumor cells [22]. The compound then was translocated into the cytoplasm, thus initiating cellular loss of apico-basal polarity and epithelial cell phenotypic characteristics. These findings suggest the re-distribution of CIP4 induced by TGF-β1 may be closely related with the process of EMT in vivo. In addition, TGF-β1 exhibited the ability to upregulate CIP4 expression via the PI3K/Akt dependent pathway; however, no SMAD or p38 activation was observed.
The results leave little doubt that renal tubular epithelial cells can undergo EMT in vitro in response to TGFβ-1 [31]. Recent experimental data provided by new techniques for genetic fate mapping has provided evidence against the derivation of myofibroblasts within the tubulointerstitial space from EMT [10], conflicting with earlier conclusions [32]. Even when new evidence is taken into account, the concept of renal tubular epithelial cells undergoing various degrees of change to become local producers of extracellular matrix collagens through the EMT process as part of a normal response to injury is worthy of note [31]. Thus, detection of the extracellular matrix components in culture supernatant of HK-2 cells was an important step in determination of the mechanism, resulting in indications of fibronectin secretion, a consequence of EMT, in the TGFβ-1 treated HK-2 cell culture supernatant. Furthermore, this result was able to be inhibited by CIP4 knockdown, indicating that CIP4 may play a regulatory role in ECM secretion of renal tubular epithelial cells. Through paracrine signaling, it may also have the ability to promote the progression of renal tubular interstitial fibrosis.
CIP4 was observed to increase in conditions of renal interstitial fibrosis, specifically EMT, in the animal rat model of 5/6 subtotal nephrectomy applied in the current study. The overexpression of CIP4 further served to promote EMT in the course of TGF-β1 treatment by disrupting the epithelial structure and secretion of ECM components in HK-2 cells. Cumulatively, the results of the current study demonstrate the importance of CIP4 as a downstream target of the TGF-β1 signal in the development and progression of renal fibrosis. These results suggest that this is a novel therapeutic target for the treatment of renal fibrosis.
Supplementary Material
Fig.S1.
Acknowledgements
The study was supported by the National Natural Science Foundation of China (grant no. 30871172 and no. 30971372), the Science Fund for Young Scholars (grant no. 30800525 and no. 30800383), and the Doctoral Fund for Youth Scholars of Ministry of Education of China (grant no. 200804871042).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12:25-29
2. Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1-12
3. Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212-222
4. Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290-301
5. Rodríguez-Iturbe B, Johnson RJ, Herrera-Acosta J. Tubulointerstitial damage and progression of renal failure. Kidney Int. 2005(Suppl 99):S82-86
6. Hills CE, Squires PE. TGF-beta 1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. American Journal of Nephrology. 2010;31(1):68-74
7. Lee DB, Huang E, Ward HJ. Tight junction biology and kidney dysfunction. Am J Physiol Renal Physiol. 2006;290:F20-34
8. Zheng G, Lyons JG, Tan TK. et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol. 2009;175:580-591
9. Humphreys BD, Lin SL, Kobayashi A. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85-97
10. Duffield JS, Humphreys BD. Origin of new cells in the adult kidney: results from genetic labeling techniques. Kidney International. 2011;79:494-501
11. Bielesz B, Sirin Y, Si H. et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040-4054
12. Ivica Grgic, Duffield JS, Benjamin D. Humphreys, The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol. 2012;27(2):183-93
13. Koesters R, Kaissling B, Lehir M. et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632-643
14. Holbert S, Dedeoglu A, Humbert S, et al.Cdc42-interacting protein4 binds to huntingtin. neuropathologic and biological evidence for a role in Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2712-2717
15. Koduru S, Kumar L, Massaad MJ. et al. Cdc42 interacting protein 4 (CIP4) is essential for integrin-dependent T-cell trafficking. Proc Natl Acad Sci USA. 2010;107:16252-16256
16. Takano K, Toyooka K, Suetsugu S. EFC/F-BAR proteins and the N-WASP-WIP complex induce membrane curvature-dependent actin polymerization. EMBO J. 2008;27:2817-2828
17. Toguchi M, Richnau N, Ruusala A, Aspenström P. Members of the CIP4 family of proteins participate in the regulation of platelet-derived growth factor receptor-beta-dependent actin reorganization and migration. Biol Cell. 2010;102:215-230
18. Chitu V, Stanley ER. Pombe Cdc15 homology (PCH) proteins: coordinators of membrane-cytoskeletal interactions. Trends Cell Biol. 2007;17:145-156
19. Tsuji E, Tsuji Y, Fujiwara T. et al. Splicing variant of Cdc42 interacting protein-4 disrupts beta-catenin-mediated cell-cell adhesion: expression and function in renal cell carcinoma. Biochem and Biophys Res Commun. 2006;339(4):1083-1088
20. Pichot CS, Arvanitis C, Hartig SM. et al. Cdc42-Interacting Protein 4 Promotes Breast Cancer Cell Invasion and Formation of Invadopodia through Activation of N-WASp. Cancer Res. 2010;70:8347-8356
21. Roignot J, Taïeb D, Suliman M. et al. CIP4 is a new ArgBP2 interacting protein that modulates the ArgBP2 mediated control of WAVE1 phosphorylation and cancer cell migration. Cancer Lett. 2010;288:116-123
22. Hu J, Mukhopadhyay A, Truesdell P, Chander H, Mukhopadhyay UK, Mak AS, Craig AW. Cdc42-interacting protein 4 is a Src substrate that regulates invadopodia and invasiveness of breast tumors by promoting MT1-MMP endocytosis. J Cell Sci. 2011;124:1739-1751
23. Hou CC, Wang W, Huang XR. et al. Ultrasound- microbubble mediated gene tran sfer of inducible Smad7 blocks transforming growth factor-β signaling and fibrosis in rat remnant kidney. Am J Pathol. 2005;166:761-771
24. Gore-Hyer E, Shegogue D, Markiewicz M. et al. TGF-beta and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am J Physiol Renal Physiol. 2002;283:F707-716
25. Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764-5774
26. Zeng R, Han M, Luo Y, Li C, Pei G, Liao W, Bai S, Ge S, Liu X, Xu G. Role of Sema4C in TGF-{beta}1-induced mitogen- activated protein kinase activation and epithelial- mesenchymal transition in renal tubular epithelial cells. Nephrol Dial Transplant. 2011;26(4):1149-1156
27. Patel S, Takagi KI, Suzuki J. et al. RhoGTPase activation is a key step in renal epithelial mesenchymal transdifferentiation. J Am Soc Nephrol. 2005;16:1977-1984
28. Royal I, Lamarche-Vane N, Lamorte L. et al. Activation of Cdc42, Rac PAK, and Rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol Biol Cell. 2000;11:1709-1725
29. Vignjevic D, Montagnac G. Reorganization of the dendritic actin network during cancer cell migration and invasion. Semin Cancer Biol. 2008;18:12-22
30. Leibfried A, Fricke R, Morgan MJ, Bogdan S, Bellaiche Y. Drosophila Cip4 and WASp define a branch of the Cdc42-Par6-aPKC pathway regulating E-cadherin endocytosis. Curr Biol. 2008;18(21):1639-48
31. Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Path. 2011;92:143-150
32. Iwano M, Plieth D, Danoff TM. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341-350
33. Zeng R, Yao Y, Han M. et al. Biliverdin reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J Am Soc Nephrol. 2008;19:380-387
Author contact
![]() Corresponding author: Prof. Rui Zeng or Prof. Gang Xu. Devision of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Wuhan, Hubei, 430030, China. Tel: +0086-027-83663397, E-mail: zengruitjmu.edu.cn or E-mail: xugangtjmu.edu.cn
Corresponding author: Prof. Rui Zeng or Prof. Gang Xu. Devision of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Wuhan, Hubei, 430030, China. Tel: +0086-027-83663397, E-mail: zengruitjmu.edu.cn or E-mail: xugangtjmu.edu.cn