Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(7):1026-1035. doi:10.7150/ijbs.4703 This issue Cite
Research Paper
Molecular Dynamics Simulation and Bioinformatics Study on Yeast Aquaporin Aqy1 from Pichia pastoris
Yubao Cui1, 2, David A. Bastien2 ![]()
1. Department of Laboratory Medicine, Yancheng Health Vocational & Technical College, Jiangsu Yancheng 224006, P. R. China.
2. Department of Physics, University of Texas at San Antonio, San Antonio, Texas 78249, USA.
Received 2012-6-6; Accepted 2012-7-24; Published 2012-8-9
Abstract
In the present study, an equilibrated system for the Aqy1 tetramer was developed, and molecular biophysics modeling showed that the Aqy1 channel was blocked by Tyr-31 in the N-terminus, which was also supported by the free energy profiles. However, bioinformatics analysis of the amino acid sequence of Aqy1 indicated this Tyr-31 is not conserved across all fungi. Analysis of the equilibrated structure showed that the central pore along the four-fold axis of the tetramers is formed with hydrophobic amino acid residues. In particular, Phe-90, Trp-198, and Phe-202 form the narrowest part of the pore. Therefore, water molecules are not expected to translocate through the central pore, a hypothesis that we confirmed by molecular dynamics simulations. Each monomer of the Aqy1 tetramers forms a channel whose walls consist mostly of hydrophilic residues, transporting through the selectivity filter containing Arg-227, His-212, Phe-92, and Ala-221, and the two conserved Asn-Pro-Ala (NPA) motifs containing asparagines 224 and 112. In summary, not all fungal aquaporins share the same gating mechanism by a tyrosine residue in the N-terminus, and the structural analysis in the present study should aid our understanding of aquaporin structure and its functional implications.
Keywords: Fungi, Aquaporin, Diversity, Molecular Dynamics Simulation, Bioinformatics.
Introduction
Water is indispensable for cells and organisms, and must be able not only to flow into and out of the cells but also pass through subcellular compartments. The presence of biological membranes separating the internal and external environments of a cell led to the hypothesis that proteins embedded in lipid bilayers facilitate the conduction of water across biological membranes. Indeed, water channels, later called aquaporins (AQPs), are transmembrane proteins with a specific three-dimensional structure forming a channel to facilitate the flow of water through cellular membranes [1,2]. Over 450 AQPs have been identified in plants, microorganisms, various animals, and humans, most of which are less than 300 amino acids in length [1,2]. These proteins comprise two highly conserved NPA motifs containing three amino acid residues (asparagine-proline-alanine or Asn-Pro-Ala) and several surrounding amino acids.
Many AQPs in yeast and filamentous fungi have been shown to correlate strongly with freeze tolerance. For example, in S. cerevisiae, two aquaporin-encoding genes, Aqy1 and Aqy2, have been identified and characterized. Deletion of these two genes in a laboratory strain increased the cells' sensitivity to freezing, while over-expression of the genes improved freeze tolerance [3,4,5]. A similar protective effect has been found in C. albicans, S. pombe, and P. pastoris [3,4,5]. It is hypothesized that this effect is associated with a reduced occurrence of intracellular ice crystals. When freezing cells are put in an aqueous medium, extracellular water should freeze faster than intracellular water due to higher osmolarity of the cell contents. AQPs facilitate the rapid flux of water out of the cell, thus less intracellular ice forms in the organism [3,4,5].
Genetic studies of Aqy1 have provided more insight to the channel. Interestingly, Laizé et al. (1999) reported that the C-terminal-truncated Aqy1 variant of S. cerevisiae produced a 3-fold higher water permeability than full-length Aqy1; thus, the C-terminus is important for regulating water flux through the channel [6]. In contrast, deletion of the Aqy1-encoding gene from P. pastoris resulted in a freeze-sensitive phenotype, an effect reversed by reintroduction of the gene or N-terminal-truncated form of Aqy1. Further, overproduction of the N-terminal-truncated Aqy1 in a wild-type strain negatively altered freeze/thaw survival [6]. Therefore, the physiological functions of fungal AQPs, especially the transport of water molecules, may be regulated by various mechanisms.
Recently, high-resolution atomic structures of AQPs—determined by NMR spectroscopy and X-ray diffraction—have offered new insight to the molecular mechanisms of these proteins. All known AQPs form tetramers, and each monomer has a central channel surrounded by six membrane-spanning segments (TM1-6) and five connecting loops (A-E). Interestingly, the first and second halves of AQPs are quite similar, with each half containing three transmembrane domains; the two halves integrate in the membrane in opposite orientations [7, 8]. Additionally, the loops between helices 2 and 3 (H2,3; loop B) and H5 and H6 (loop E), which have a highly conserved NPA motif, meet in the center of the membrane to form the channel [7,8].
While structural information guides our understanding of the molecular mechanisms of AQPs, experimental studies are needed. Currently, no experimental method offers sufficient spatial and temporal resolution to monitor permeation through AQPs on a molecular level. Recently, molecular dynamics (MD) simulations have served as a surrogate for experimental approaches. These simulations model the progression of a bimolecular system at atomic resolution, revealing the molecular mechanisms underlying the remarkable efficiency and selectivity of these channels. For instance, both human aquaporin-1 (AQP1) and the bacterial glycerol facilitator GlpF act as two-stage filters at NPA motifs and aromatic/arginine (ar/R) constriction regions. MD simulations revealed that AQP selectivity is controlled by a hydrophobic effect and steric restraints [9].
Although AQPs have been identified in many different fungi species, only Aqy1 from P. pastoris has been selected for crystal structure determination. This structure revealed that the channel is closed by the N-terminus, where a unique conformation caps the water channel entrance and thereby closes the channel [10]. In the present study, using the atomic coordinates of Aqy1 from P. pastoris, we conducted MD simulations and free energy calculation to explore its gating phenotype and mechanism. Our results indicate that Tyr-31 in the N-terminus controls the channel entrance, consistent with the observed crystal structure. However, further bioinformatics analysis in this study shows that this Tyr-31 is not a conserved residue in homologous fungal AQPs, so we argue that not all fungal AQPs are gated in the same manner as Aqy1 of P. pastoris.
Materials and methods
Building a structural model of Aqy1
The crystal structure of Aqy1 was obtained from the Protein Data Bank (PDB code 2W1P). The monomeric coordinates were replicated using VMD software [11]. Aqy1 tetramers were generated using the transformation matrices provided with the PDB file. The protein structure file (PSF) and Protein Data Bank (PDB) file for the Aqy1 tetramer, the crystallographic water molecules, and the ions were generated using the AutoPSF plugin of VMD. To solvate the protein, Helmut Grubmüller's SOLVATE program [12] was used to fill any empty space inside the pores, as well as to surround it with water.
Placing Aqy1 tetramer in a membrane
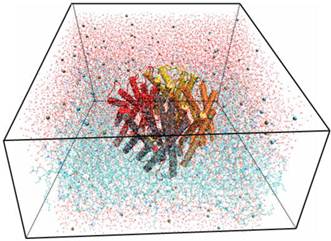
Using the Membrane Builder program provided within VMD, a palmitoyl-oleoyl-phosphatidyl-choline (POPC) lipid bilayer was generated with X length and Y length initially set to 120 Ǻ. The constructed membrane patch and the partially-solvated protein were aligned, moved, and assembled properly. To make room for Aqy1 in the membrane layer and avoid overlap with any lipid molecules, atoms of the “bad lipids” were marked by VMD using the beta field of the PDB files; thus, the majority of the lipids overlapping with Aqy1 were removed. After measuring the water layer using a minmax option, the system was placed in a water box slightly smaller in the XY-plane using VMD's Solvate plugin, since non-equilibrated membranes tend to shrink. Lastly, K+ and Cl- ions with a concentration of 0.4 mmol/L were created for the entire system using VMD's autoionize plugin, which transmutes water molecules into ions. The system consisted of 105,912 atoms total (Fig. 1).
Snapshot for the last frame of the MD simulation of Aqy1 with lipids in lines, water in lines, protein in cartoon and ions in VDW.
Running simulations for Aqy1 tetramer-lipids complex system
Simulations were performed in four phases. In the first phase, melting of lipid tails, lipid tails and bulk water were minimized and equilibrated for 0.74 ns; the protein, crystallographic waters, ions, and lipid head groups were fixed. In the second phase, minimization and equilibration, with protein constrained, the system was minimized and equilibrated for 0.50 ns. Because our system was assembled manually, it has many unnatural atomic positions. A dynamics run would immediately fail from many atoms moving at very a high velocity. Thus, a “minimization” with NAMD software [13] was performed to introduce the system to the nearest local energy minimum in the configuration space, followed by an equilibration with the protein constrained to permit lipids, water, and ions to adapt to the protein in its crystal form. In the third phase, equilibration with protein released, the system was minimized and equilibrated for 0.50 ns. Minimization and equilibration with the protein constrained should cause the lipids to be well packed around the protein, while excluding water from forbidden regions between the protein and lipids. In this phase, we released the harmonic constraints placed on the protein in the second phase and further equilibrated the whole system. In the fourth phase, production runs, simulations were run for 14.24 ns. The protein was equilibrated in previous phases and has been packed well, thus simulations should be run with the XY-plane remaining constant. This approach differed from previous simulations in which the XY-plane fluctuated to permit packing of lipids against the protein. All simulations were performed using the CHARMM22 force field [14,15], the TIP3P water model, and the MD program NAMD2 [16], with periodic boundary conditions at constant temperature of 310K and constant pressure of 1 atm (NpT ensemble). The Langevin dynamics and Langevin piston methods were used to keep temperature and pressure constant. Full electrostatics were employed using the Particle Mesh Ewald (PME) method [16].
Free energy computation
After simulations, we performed steered molecular dynamics (SMD) to simultaneously pull water through four Aqy1 channels. We pulled 45Å length with start position near -10Å (extra-cellular) to the end near 35 Å (intracellular). Two sets of pulling paths were sampled: each set had five forward paths (from extracellular to intracellular) and five reverse paths (from intracellular to extracellular). The order parameter was chosen as the center-of-mass z-coordinate of one pulled water molecule, and the pulling speed was 0.05 Å/ps. Subsequently, the Brownian dynamics fluctuation-dissipation-theorem (BD-FDT) was used to accurately compute the free-energy profiles for each set of the five forward and five reverse paths [17,18].
Bioinformatics Analysis
The amino acid sequence of Aqy1 was obtained from PDB file 2W1P; sequences for other AQPs were obtained from PDB as follows: Aqp0 from entry 2B6P; Aqp1 from entry 1J4N; Aqp4 from entry 3GD8; and Aqp5 from entry 3D9S. Sequence alignment was conducted with Espript software. Amino acid sequences of other fungal AQPs were obtained by BLASTp search in the NCBI database. The phylogenetic tree was constructed with a neighbor-joining method in Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0.
Results
Equilibrium molecular dynamic simulations
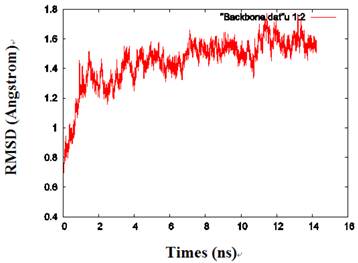
By calculating the root mean square deviation (RMSD) values of the backbone heavy atoms relative to the coordinates of the initial (energy-minimized) structures, the overall changes in the model atomic coordinates were monitored during MD simulations. After an initial fast-rise region ultimately leveling off after about 2ns, the RMSD values changed little and reached a more constant value in the last 1 ns, indicating that the model structure had reached a conformational steady state (Fig. 2).
The RMSD values of the protein backbone in one of the monomers during MD simulations.
The pore along the central four-fold axis
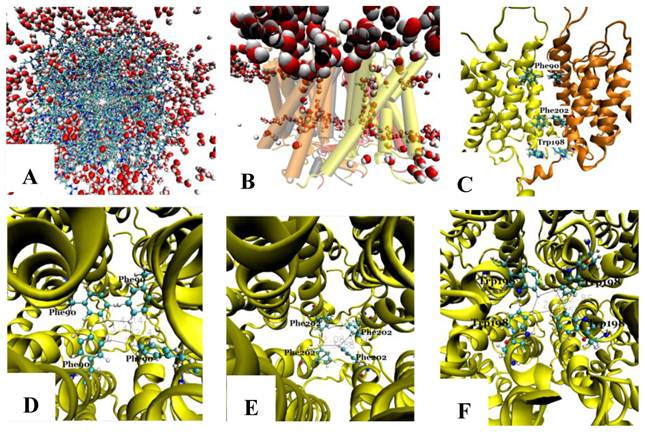
The tetramer was made of four monomers, which formed a pore along the central four-fold axis. During simulations, no water was observed in the pore along the four-fold axis (Fig. 3A and B); however, water flowed into each of the four channels of the Aqy1 tetramer. Cross-sectional analysis of the protein-membrane complex 40 Ǻ in length along the central four-fold axis revealed that the amino acid residues forming the central pore were Pro-82, Ala-83, Ile-86, Met-87, Phe-90, Phe-94, Met-97, Phe-101, Trp-198, Phe-199, Phe-202, Val-203, Ile-206, and Leu-209, all of which are hydrophobic. Phe-90, Trp-198, and Phe-202 formed the narrowest part of the pore, smaller than the diameter of a water molecule (Fig. 3C-F), which would create a hydrophobic block. There although the tetramers were solvated in water, there are not any water molecules in this center hole throughout our simulations as long as 14.24 nanoseconds.
The pore along the central four-fold axis among the four Aqy1 monomers. (A) Top view, the protein is shown in drawing methods of Licorice and waters in VDW; (B) Side view, the protein is shown in drawing methods of Cartoon and Material of Transparent, and water in VDW; (C) Side view, the protein is shown in drawing methods of NewCartoon and the important residues in Licorice. (D) Residue Phe-90 is present in the pore along the central four-fold axis; the protein is shown in drawing method of NewCartoon and residue Phe-90 in drawing method of CPK; (E) Residue Phe-202 is present in the pore along the central four-fold axis; the protein is shown in drawing method of NewCartoon and residue Phe-202 in drawing method of CPK; (F) Residue Phe-198 is present in the pore along the central four-fold axis; the protein is shown in drawing method of NewCartoon and residue Phe-198 in drawing method of CPK.
The conduction channel, selectivity filter, NPA motifs, and extracellular vestibule
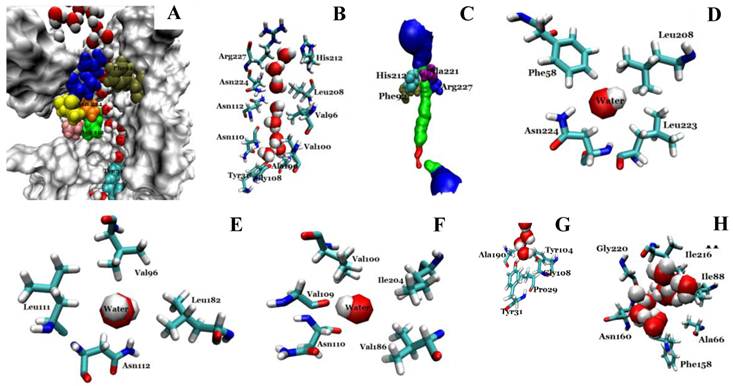
We investigated the structure of Aqy1 tetramers in the protein-lipids complex system of the last frame after simulation and observed eight water molecules in single file in the channel. These molecules were stopped by Tyr-31 (Fig. 4A, B, and G). Due to a selectivity filter containing Arg-227, His-212, Phe-92, and Ala-221 (Fig. 4C), the channel diameter became small, sterically excluding the passage of glycerol, with the narrowest diameter of the channel computed by hole2.0 software (Fig. 5). Asparagines 224 and 112 of the two most conserved NPA motifs form the canonical “fireman's grip-like” structure in the center of the channel (Fig. 4A, B, D, and E). In addition to Asn-224 and Asn-112, other amino acids line the inner surface of the channel, including Leu-208, Leu-223, Phe-58, Val-96, Leu-182, Leu-111, Asn-110, Val-100, Ile-204, Val-109, Val-186, Tyr-31, Gly-108, Gly-108, Ala-190, Tyr-104, and Pro-29, although it is closed by Tyr-31, where the pore diameter measures about 0.8Å (Fig. 4B-G and Fig 5). Before water enters the channel, the molecules remain in the extracellular vestibule, which comprises Gly-220, Ile-216, Asn-160, Ile-88, Ala-66, and Phe-158 (Fig. 4H).
Structural analysis of the conducting channel of one Aqy1 monomer. (A) One channel of the last frame from the MD simulations of the Aqy1 tetramer. There were eight water molecules in single file in the channel. Phe-92 in tan, Asn-112 in green, Pro-113, Ala-114 in pink, Asn-224 in orange, Pro-225 and Ala-226 in yellow, Arg-227 in blue; these residues are shown in drawing methods of VDW, and the other parts of the protein in drawing methods of Surf and white color. (B) The main pathway through the channel. (C) The selectivity filter residues Arg-227, His-212, Phe-92, and Ala-221 shown in drawing method of VDW. (D) One of the NPA motifs' residues Asn-224, which is positioned with Leu-208, Leu-223, and Phe-58, all of which are shown in Licorice drawing method and water in VDW; (E) One of NPA motifs' residues Asn-112, which is positioned with Val-96, Leu-182, Leu-111, all of which are shown in Licorice drawing method and water in VDW; (F) The residue Asn-110, which is positioned with Val-100, Ile-204, Val-109, and Val-186, all of which are shown in Licorice drawing method and water in VDW; (G) The end of the conduction pore, which is stopped by Tyr-31, and Pro-29, Tyr-104, Gly-108, and Ala-190, all of which are shown in Licorice drawing method and water in VDW; (H) The extracellular vestibule of the channel made from Gly-220, Ile-216, Asn-160, Ile-88, Ala-66, and Phe-158, all of which are shown in Licorice drawing method and water in VDW.
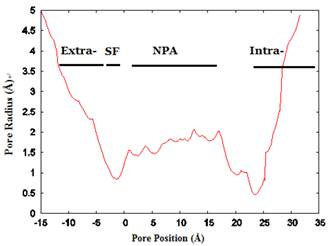
The channel radius versus position. The regions of the channel comprising the extracellular vestibule (Extra-), selectivity filter (SF-), NPA motif region, and intracellular vestibule (Intra-) are labeled. The calculation of pore dimensions was done with the program Hole2.0 software.
Free energy computation
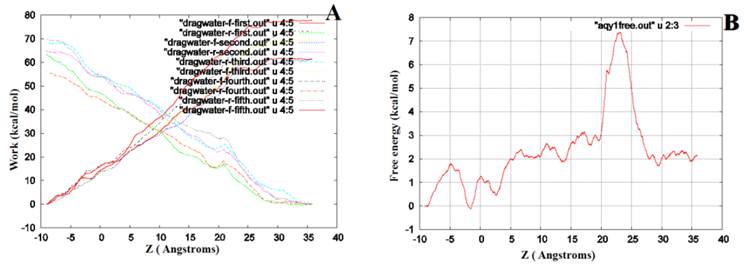
We performed SMD to simultaneously pull water molecules through four channels of Aqy1. The free-energy profile computed by BD-FDT showed an energy barrier near Tyr-31 (Fig. 6), which is consistent with the results of MD simulations.
The free-energy profile of water permeation through the channels of Aqy1. (A) Work done along the forward (dragwater-f) and reverse (dragwater-r) pulling paths. Using the last frame, four water molecules were pulled from the start position at -10Å through Aqy1 channels, one water molecule through each channel; five forward and five reverse paths were performed. (B) The free-energy profile of Aqy1 was computed using the Brownian dynamics fluctuation-dissipation-theorem (BD-FDT) method according to the mmechanical work done in the above pulling paths.
Comparison of Aqy1 with mammalian aquaporins
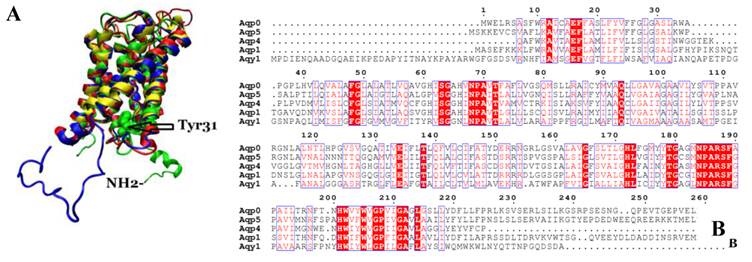
We compared the atomic structure and amino acid sequence of Aqy1 with mammalian aquaporins AQP0, AQP1, AQP4, and AQP5. The amino acid sequences of these aquaporins are quite similar. Interestingly, however, the N-terminus is longer in Aqy1 than in other AQPs (Fig. 7 A and B).
Comparison of Aqy1 with mammalian aquaporins. (A) Comparison of the structure of Aqy1 (in green) monomer with those of Aqp0 (PDB entry 2B6P, in blue), Aqp1 (PDB entry 1J4N, in red), Aqp4 (PDB entry 3GD8, in yellow) and Aqp5 (PDB entry 3D9S, in tan). Each structure is depicted with drawing method of NewCartoon. (B) The amino acid sequence alignment results for Aqy1, Aqp1, Aqp4, and Aqp5 with Espript software.
Amino acid sequence homology search, molecular evolution, and alignment
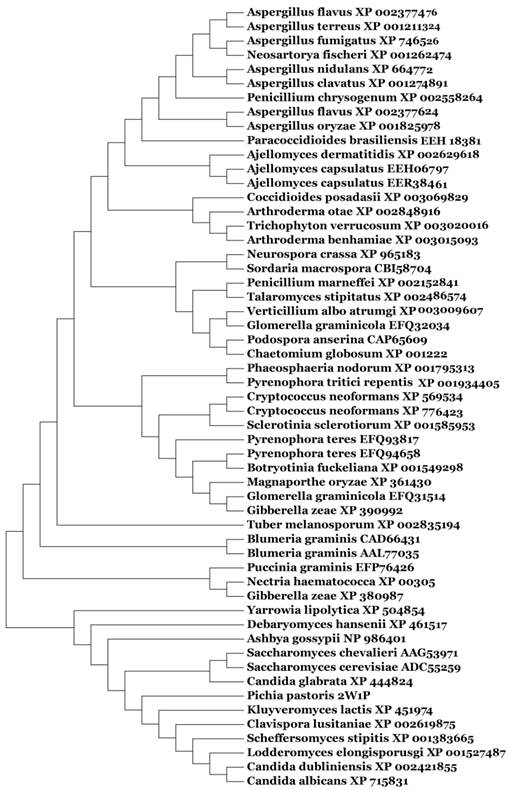
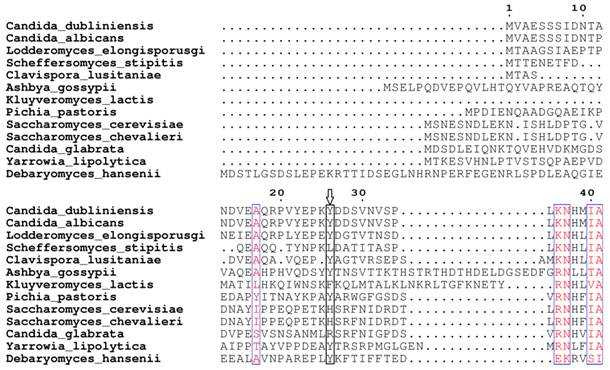
Based on our BLASTp search in the NCBI database, amino acid sequences of similar fungal aquaporins were obtained. We divided these into two groups: one group was clustered with Aqy1, and the other was not (Fig. 8). Amino acid sequence alignments indicated that Tyr-31 in Aqy1 of P. pastoris is not a conserved residue: while present in Candida dubliniensis, C. albicans, Lodderomyces elongisporus, Clavispora lusitaniae, Yarrowia lipolytica, and Debaryomyces hansenii, Tyr-31 was not found in S. cerevisiae, S. chevalieri, Scheffersomyces stipitis, Kluyveromyces lactis, or C. glabrata (Fig. 9). In addition, Pro-29 was present in C. dubliniensis, C. albicans, L. elongisporus and C. lusitaniae; this residue is important to the configuration of the pore in Aqy1 of P. pastoris.
Phylogenetic tree constructed for Aqy1 and homologous amino acid sequences from other fungal aquaporins by Mega 4.0.
Amino acid sequence alignment between Aqy1 and homologous amino acid sequences of other fungal aquaporins. Only N-terminal amino acid sequences are shown here. The arrow indicates Tyr-31 in Aqy1 from Pichia pastoris, which is not conserved in across fungi.
Discussion
Fischer et al. (2009) provided the initial insight to the yeast aquaporin Aqy1 by describing its x-ray structure in P. pastoris [10]. Their work demonstrated that Aqy1 is an N-terminal-gated AQP controlled by Tyr-31. Further, the authors indicated that the hydrogen bond from Try-27 anchors the bundle of Aqy1 to the AQP scaffold, and Pro-29 introduces a kink allowing Try-31 to insert itself and block the water channel [10]. In the present study, we simulated the molecular dynamics of this crystal structure in a solvated lipid bilayer. We confirmed that the Aqy1 channel is blocked by Tyr-31 in the N-terminus, where there is an energy barrier in light on our free energy landscape.
We performed bioinformatics analyses of Aqy1 to gain additional insight to this water channel. We compared the Aqy1 structure with all known mammalian AQPs, including Aqp0, Aqp1, Aqp4 and Aqp5. We observed an extended N-terminus in Aqy1, consistent with the amino acid sequence alignment, suggesting potential differences in the gating mechanisms between fungi and mammalian aquaporins. Additionally, sequence alignment of Aqy1 with other fungal AQPs showed that Tyr-31 of Aqy1 from P. pastoris is not a highly conserved residue, despite the report by Fischer et al. [10] describing this residue as conserved across fungi. Therefore, not all fungal aquaporins share the same gating mechanism by a tyrosine residue in the N-terminus. Indeed, considering that the recombinant protein Aqy1 of S. cerevisiae lacks the C-terminus and mediates a 3-fold higher water permeability than the full-length protein, the channel in that fungus is opened and regulated by the C-terminus [6]. However, the presence of Tyr-31 in C. dubliniensis, C. albicans, L. elongisporus, C. lusitaniae, Y. lipolytica, and D. hansenii and Pro-29 in C. dubliniensis, C. albicans, L. elongisporusgi and C. lusitaniae seem to suggest that the pore in these organisms is blocked by tyrosine. More high-resolution structures are required to identify gating mechanisms in other fungal AQPs.
A physiological role for fungal AQPs in protecting cells against freeze-thaw stress by sustaining low-temperature water permeability has been confirmed in several studies [3,4,5]. Thus, the potential to open and close the N-terminus of Aqy1 may be required for the survival of cells in freezing environments. When the pore is open, a continuous water column would be present within the channel, as demonstrated by the eight water molecules in single-file during our MD simulation. Thus, these simulations confirm the physiological role of aquaporin as a water channel.
As observed in other AQPs [19], two sites interact strongly with water in the conduction channel: the ar/R constriction and the NPA motif. In Aqy1, the ar/R constriction, i.e., selectivity filter, is formed by four residues, Arg-227, His-212, Phe-92, and Ala-221. These reflect similar sites in Aqp1 (Arg-195, His-180, Phe-56, and Cys-189) and GlpF (Arg-205, Phe-200, Gly-191, and Trp-48). Histidine is typically found in water-specific channels, and, together with the highly conserved arginine, provides a hydrophilic edge in juxtaposition to an aromatic residue. The hydrophobic Phe-92 side chain orients the water molecules to enforce strong hydrogen bonds to Arg-227 and His-212. The NPA motifs in the middle of the channel contain both asparagines lying on one side and hydrophobic side chains of Leu-208, Leu-223 and Phe-58, Val-96, Leu-182, Leu-111 on the other. Farther down the channel, toward the intracellular side, water molecules interact with Asn-110 and the carbonyl groups of residues Val-100, Ile-204, Val-109, and Val-186 in the pore. At the end of the conduction pore, water molecules interact with Tyr-31 and Pro-29 and Tyr-104, Gly-108, and Ala-190.
According to our MD simulations, before entering into the Aqy1 channel, some water molecules present in the extracellular vestibule comprised of Gly-220, Ile-216, Asn-160, Ile-88, Ala-66, and Phe-158. Members of the water channel superfamily include aquaporins and aquaglyceroporins. In aquaglyceroporins, the vestibule is regarded as a place for glycerol recruitment and desolvation of solutes for transport through the channel, a hypothesis confirmed by the crystal structures of GlpF and PfAQP (malarial aquaglyceroporins), with glycerol molecules located in the entry vestibule [20, 21, 22]. However, in the 1.8Å structure of the water-specific hAQP4, three glycerol molecules were included in the extracellular vestibule but no glycerol was included in the selectivity filter [19].
In natural membranes, every AQP studied to date forms a tetramer, and each monomer has its own channel with independent function. The tetramer is held together by extensive interactions between helices and loops of the four monomers, with a hole in the center of the molecule. The physiological function of the four-fold axis has been the most intriguing question in the structure of aquaporin. Some suggest this axis functions as an ion or gas channel [8], while others believe it insulates against all solutes and water [19]. Upon simulation, we observed no water in the four-fold axis of Aqy1. Cross-section analyses suggest that there are many hydrophobic residues that may help insulate to prevent flux of water through these holes, especially Phe-90, Trp-198, and Phe-202, which are smaller than other residues.
Accurate computation of free-energy is of essential importance in quantitative studies of biological systems because most biophysical/chemical processes are driven by the free-energy gradients. Methods to calculate free-energy differences between two states (conformations) A and B fall into two classes, equilibrium methods and, more recently developed, non-equilibrium methods. Equilibrium approaches, such as the thermodynamic integration (TI) method, require more computing resources because they are based on “full” sampling of the equilibrium ensembles involved in a given biophysical process. Non-equilibrium approaches exploit the stochastic dynamics of the system driven with some applied forces and aim to map out the free-energy landscape in terms of the potential of mean force (PMF) [23]. The free-energy difference between States A and B is extracted from the measurements of work along the transition paths connecting the two states. Recently, a non-equilibrium PMF type of approach has been developed on the basis of the Brownian dynamics fluctuation-dissipation theorems (BD-FDT), extracting the equilibrium free-energy differences from irreversible (non-equilibrium) work measurements in steered molecular dynamics (SMD) simulations [17, 18]. In the present study, we computed the free energy profiles of water through Aqy1 by BD-FDT, and the result is consistent with the previous report [10] using equilibrium statistical distribution, which is accurate in the limit of finitely long simulations. Using BD-FDT, a non-equilibrium PMF type of approach, we can run shorter simulations and still obtain accurate results.
Our findings provide additional insight to the potential mechanics of Aqy1 and similar aquaporins and may offer important information upon which to develop experimental approaches to understanding these channels.
Acknowledgements
The authors acknowledge helpful discussions with Prof. Liaoyuan Chen, Dr. Guodong Hu and Dr. Yubo Zhang, Department of Physics, University of Texas at San Antonio. They acknowledge support from the NIH (Grant No. SC3 GM084834) and the Texas Advanced Computing Center.
Abbreviations
MD, molecular dynamics; SMD, steered MD; PDB, Protein Data Bank; RMSD, root mean-squared deviation; POPC, palmitoyl-oleoyl-phosphatidyl-choline; PSF, protein structure file; NPA, Asn-Pro-Ala; PME, Particle Mesh Ewald; BD-FDT, Brownian dynamics fluctuation-dissipation-theorem; MEGA, Molecular Evolutionary Genetics Analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Agre P, King LS, Yasui M. et al. Aquaporin water channels--from atomic structure to clinical medicine. J Physiol. 2002;542:3-16
2. Benga G. Water channel proteins (later called aquaporins) and relatives: past, present, and future. IUBMB Life. 2009;61:112-133
3. Tanghe A, Van Dijck P, Dumortier F. et al. Aquaporin expression correlates with freeze tolerance in baker's yeast, and overexpression improves freeze tolerance in industrial strains. Appl Environ Microbiol. 2002;68:5981-5989
4. Pettersson N, Fillipsson C, Becit E. et al. Aquaporins in yeasts and filamentous. Biol Cell. 2005;97:487-500
5. Tanghe A, Kayingo G, Prior BA. et al. Heterologous auaporin (AQY2-1) expression strongly enhances freeze tolerance of Schizosaccharomyces pombe. J Mol Microbiol Biotechnol. 2005;9:52-56
6. Laizé V, Gobin R, Rousselet G. et al. Molecular and functional study of Aqy1 from Saccharomyces cerevisiae: role of the C-terminal domain. Biochem Biophys Res Commun. 1999;257:139-144
7. Walz T, Fujiyoshi Y, Engel A. The AQP structure and functional implications. Handb Exp Pharmacol. 2009;190:31-56
8. Gonen T, Walz T. The structure of aquaporins. Quarterly reviews of biophysics. 2006;39:361-396
9. Hub JS, Grubmuller H, de Groot BL. Dynamics and energetics of permeation through aquaporins. What do we learn from molecular dynamics simulations? Handb Exp Pharmacol. 2009;190:57-76
10. Fischer G, Kosinska-Eriksson U, Aponte-Santamaría C. et al. Crystal structure of a yeast aquaporin at 1.15Å reveals a novel gating mechanism. Plos Biology. 2009;7:e1000130
11. Humphrey W, Dalke A, Schulten K. VMD-Visual Molecular Dynamics. J Mol Graph. 1996;14:33-38
12. Grubmüller H. SOLVATE. 1.0 ed. Munich: Theoretical Biophysics Group, Institute for Medical Optics, Ludwig-Maximilians University. 1996
13. Kalé L, Skeel R, Bhandarkar M. et al. NAMD2: Greater scalability for parallel molecular dynamics. J Comp Phys. 1999;151:283-312
14. MacKerell Jr AD, Bashford D, Bellott M. et al. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB Journal. 1992;6:A143
15. MacKerell Jr AD, Bashford D, Bellott M. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3568-3616
16. Essmann U, Perera L, Berkowitz ML. et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577-8593
17. Chen LY. Free-energy landscape of glycerol permeation through aquaglyceroporin GlpF determined from steered molecular dynamics simulations. Biophysical Chemistry. 2010;151:178-180
18. Chen LY, Bastien DA, Espejel HA. Determination of equilibrium free-energy from non-equilibrium work measurements. Phys Chem Chem Phys. 2010;12:6579-6582
19. Ho JD, Yeh R, Sandstrom A, Chorny l. et al. Crystal structure of human aquaporin 4 at 1.8Å and its mechanism of conductance. Proc Natl Acad Sci USA. 2009;106:7437-7442
20. Hub JS, de Groot BL. Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Natl Acad Sci USA. 2008;105:1198-1203
21. de Groot BL, H Grubmüller. Water permeation across biological membranes: mechanism and dynamics of Aquaporin-1 and GlpF. Science. 2001;294:2353-2357
22. Newby ZE, O′Connell 3rd, Robbes-Colmenares Y. et al. Crystal structure of the aquaglyceroporin PfAQP from the malarial parasite Plasmodium falciparum. Nat Struct Mol Biol. 2008;15:619-625
23. Hummer G, Szabo A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci USA. 2001;98:3658-3661
Author contact
![]() Corresponding author: Dr Yubao Cui, Email: ybcui1975com. Address to: Department of Laboratory Medicine, Yancheng Health Vocational & Technical College, Jiefangnan Road 263, Yancheng 224006, Jiangsu Province, P. R. China.
Corresponding author: Dr Yubao Cui, Email: ybcui1975com. Address to: Department of Laboratory Medicine, Yancheng Health Vocational & Technical College, Jiefangnan Road 263, Yancheng 224006, Jiangsu Province, P. R. China.