Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction: Lung as a target...
Regulation of IL-6 and IL-6...
Role of IL-6 in experimental...
Genetic evidence supporting the...
IL-6 and COPD
Aging, frailty and IL-6
Potential mechanisms mediating...
Conclusions
Acknoledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(9):1281-1290. doi:10.7150/ijbs.4874 This issue Cite
Review
Role of IL-6 in Asthma and Other Inflammatory Pulmonary Diseases
Mercedes Rincon1 ![]() , Charles G. Irvin2
, Charles G. Irvin2
1. Department of Medicine, Immunobiology Division, University of Vermont, Burlington, VT 05405, USA;
2. Pulmonary and Critical Care Medicine Division, University of Vermont, Burlington, VT 05405, USA.
Received 2012-7-14; Accepted 2012-8-14; Published 2012-10-25
Abstract
The incidence and severity of chronic lung diseases is growing and affects between 100 and 150 million people worldwide and is associated with a significant rate of mortality. Unfortunately, the initial cause that triggers most chronic lung diseases remains unknown and current available therapies only ameliorate, but do not cure the disease. Thus, there is a need for identification of new targets and development of novel therapies especially for those most severely affected. IL-6, like other inflammatory cytokines, has been shown to be elevated in different lung diseases, but it was considered a byproduct of ongoing inflammation in the lung. However, recent studies support a dissociation of IL-6 from inflammation in the lung and suggest that this cytokine plays an active role in pathogenesis of asthma and, in all likelihood, COPD. IL-6 may therefore be a germane target for treatment of these and other chronic lung disease. Here, we provide an overview of the studies in mouse models and human patients that provide support for the involvement of IL-6 in lung diseases.
Keywords: IL-6, chronic lung diseases
Introduction: Lung as a target organ for inflammatory diseases
Lung diseases are on the rise and represent a significant source of mortality and morbidity. For example, chronic obstructive pulmonary disease (COPD) is currently the 5th cause of death and is rising such that by 2020 COPD is expected to be the 4th most common cause of death worldwide. Asthma is exceedingly common with a prevalence of about 8% in the U.S. Asthma is the most common chronic disease of children and is a leading cause of missed days of school, and among adults, missed days at work. Other inflammatory lung diseases such as cystic fibrosis (CF) and interstitial lung disease (ILD) are also more frequently encountered and are characterized by poor outcomes, i.e. death and/or poor quality of life. All these lung diseases are characterized by periodic or chronic inflammatory processes and the general use of anti-inflammatory therapeutic interventions such as steroids. While the major focus of this present review will begin with a discussion of the role of IL-6 in asthma we will also discuss the role of this cytokine in other lung diseases.
Allergic asthma is a chronic inflammatory disease of the airways that occurs in response to inhaled allergens such as ragweed pollen, cat dander, house dust mites, and fungi. The development of a CD4 Th2 immune response and its associated cytokines (e.g. IL-4, IL-5, and IL-13) are known to play an important role in the pathogenesis of allergic asthma. IL-4 promotes Ig isotype switching in B cells to produce primarily IgE and IgG1 (1). IL-5 promotes eosinophil survival, differentiation and migration (2) while IL-13 induces mucus metaplasia and airway hyperresponsiveness (3-5). The role of Th17 cells and their signature cytokines IL-17A and IL-17F in allergic asthma is also becoming more apparent. While animal models have provided some conflictive conclusions in part due to the presence of several members of the IL-17 family and a common IL-17R, together with human studies, it appears that IL-17 also contributes to the pathogenesis of asthma, primarily severe and steroid-resistant asthma (6).
Although this Th2/Th17 type of immune response may be influenced by the nature of the antigenic determinants of the allergen and its recognition by CD4 T cells, components of the lung environment (e.g. epithelial cells, smooth muscle cells) can also contribute to the overall immune response by secreting cytokines. In recent years a number of studies have shown that lung epithelial cells produce specific cytokines and that these cytokines can favor Th2 and/or Th17 cell differentiation. Thus, IL-33 is produced by lung epithelial cells and promotes some aspects of a Th2 response such as IL-5 production and eosinophilia (7). Thymic Stromal Lymphopoietin (TSLP) is also produced predominantly by lung epithelial cells and promotes a Th2 response (8). IL-6, a cytokine produced by inflammatory cells is also produced by primary lung epithelial cells in response to a variety of different stimuli including allergens, respiratory virus and exercise (9-12). In addition, a recent study has shown high levels of IL-6 mRNA present in a constitutive manner in mouse primary lung epithelial cells, but not in immune cells resident in the lung (13). IL-6 has emerged as an important regulator of effector CD4 T cell fate (14), promoting IL-4 production during Th2 differentiation, inhibiting Th1 differentiation and, together with TGFβ, promoting Th17 cell differentiation. The pleotropic nature of these immunoregulatory roles suggests that IL-6 could be a potential wide-ranging contributor to asthma as well as other pulmonary diseases where the lung epithelium is damaged.
Regulation of IL-6 and IL-6 receptor in asthma
IL-6 is a small size glycoprotein (21 KDa) produced by cells from the innate immune system (e.g. macrophages, dendritic cells, mast cells, neutrophils), but also by B cells and, to a lesser extent, by some CD4 effector Th cells. In addition, IL-6 is also secreted by non-leukocytes such as endothelial cells, fibroblasts, astrocytes, epithelial cells, and a number of malignant cells (15). Although a large variety of stimuli can induce the production of IL-6 by these cells, a feature common for most of them is their effect promoting cell stress or damage (e.g. UV, irradiation, ROS, microbial products, viruses, or other proinflammatory cytokines). The levels of IL-6 in serum have been found to be elevated in a number of inflammatory diseases (16). As a result, IL-6 has long been considered a general marker of inflammation together with TNFα and IL-1β, two other classical inflammatory cytokines. However, it is becoming evident that IL-6 is not simply a proinflammatory marker, but an active factor that contributes to the pathogenesis of certain inflammatory diseases such as rheumatoid arthritis, and a successful target for some of these diseases (16).
Increased levels of IL-6 in serum have been found in asthmatic patients (11). More importantly, a study examining IL-6 in brochoalveolar lavage fluid (BALF) has shown increased levels of IL-6 in active asthmatic patients compared with the levels in healthy nonsmoker subjects, stable asthmatic and non-asthmatic patients receiving mechanical ventilation (17). In addition, increased levels of IL-6 in BALF from patients with “intrinsic” asthma compared with the levels in patients with allergic asthma have also been reported (18), suggesting that IL-6 may play a role beyond patients with allergic asthma which only accounts for about 50% of all asthmatics. A recent study in mild allergic asthmatic patients with no exuberant airway inflammation has shown increased IL-6 levels in induced sputum compared with the levels in healthy subjects (19). Neither IL-1β nor TNFα levels in induced sputum were increased in this cohort of patients. However, the levels of IL-13 (but not IL-5 or IL-4) were also increased in the allergic asthmatic patients relative to the levels in control subjects. Interestingly, increased IL-13 levels correlate with increased IL-6 levels in the asthmatic patients (19). Although increased levels of IL-6 in the lung of asthmatic patients have been associated with the presence of inflammatory cells (e.g macrophages, neutrophils) that can secrete this cytokine, these studies suggest that the presence of IL-6 in the lung airways may be independent of inflammation. A number of studies have shown an overexpression of IL-6 in bronchial epithelial cells in patients (adult and children) with asthma (9, 10). In addition, the IL-6 gene is constitutively expressed in primary lung epithelial cells from naïve mice and exposure to allergen can trigger the production of this cytokine prior to the recruitment of inflammatory cells (13). Thus, the presence of IL-6 in the airways of asthmatic patients may not be the result of ongoing inflammation, but the result of the “activated” state of pulmonary epithelial cells.
IL-6 binds to IL-6 receptor (IL-6R) that is expressed in leukocytes and hepatocytes. Although IL-6R does not have the ability to trigger signaling, it associates with the gp130 signal transducer glycoprotein that is ubiquitously expressed and is shared by other cytokine receptors (20). The specificity in signaling is determined by the binding of IL-6 to IL-6R and the subsequent association of this complex with gp130. This association triggers the dimerization of gp130, leading to the activation of the specific members of the JAK family of tyrosine kinases (JAK1, JAK2 and Tyk2). Activated JAKs then mediate the phosphorylation and activation of the major transcriptional factor regulated by IL-6, STAT3 (21, 22). In addition, IL-6 also activates the C/EBPβ transcription factor (also known as NF-IL6) through the MAPK pathway (23).
Although IL-6R expression is relatively restricted to immune cells and hepatocytes, IL-6 can regulate the function of other cells through a new pathway that has been defined as “trans-signaling” of IL-6R (24). In addition to the membrane IL-6R, soluble IL-6R (sIL-6R) can be generated and secreted through mRNA alternative splicing, or by shedding of the membrane bound IL-6R mediated by specific metalloproteases (25-27). sIL-6R generated by either mechanism can bind IL-6 and the complex associates with cell surface gp130 in an agonistic manner and transduce signals through gp130. This trans-signaling pathway confers responsiveness for IL-6 to those cells that lack IL-6R. Although macrophages and neutrophils have been considered as the predominant source of sIL-6R (27), both human and mouse CD4 T cells can also produce large amounts of sIL-6R upon activation (28, 29). CD4 T cells could therefore contribute to the development and/or progression of asthma by providing sIL-6R to cells non-responsive to IL-6. In addition to increased IL-6, elevated levels of sIL-6R have also been found in BALF from asthmatic patients after allergen challenge (30), suggesting that IL-6 may have a direct effect on cells other than immune cells. The source of sIL-6R in the airways of these patients remains unknown.
An important question to be addressed is whether the presence of IL-6 in the lung is associated with altered lung function. A prospective cross-sectional study with asthmatic patients has shown that the levels of IL-6 in sputum inversely correlated with FEV1 percentage predictive (31). A similar inverse correlation between IL-6 in sputum and FEV1 was found in a cohort of severe asthmatic patients (32), while in a cohort of allergic asthmatic patients elevation of IL-6 in sputum associates with loss of central airway function (19). In obese asthmatic patients, the increased levels of IL-6 in serum also correlated with impaired lung function (33). Together, these studies indicate that the presence of IL-6 in the lung airways correlates with an impaired lung function in different subsets of asthmatic patients, and suggests that IL-6 is likely to be directly involved in the pathogenesis of asthma and the progressive loss of lung function observed in patients who remain untreated. As such, IL-6 is likely a potential target for new treatments for this important disease.
Role of IL-6 in experimental allergic airway inflammation using mouse models
The results described above provide a correlation between IL-6 and asthma in humans, and suggest an active role of IL-6 in determining lung function abnormalities in this disease. However, cause-effect studies in germane animal models provide further proof that IL-6 could both cause disease and be a potential target for this disease. Unfortunately, conflictive results from the mouse model studies diminished the initial interest in IL-6 in asthma and complicated the initiation of potential clinical studies to target this cytokine in lung diseases. The most common mouse model of allergic airway inflammation is the ovalbumin (OVA) model. In this model, mice receive intraperitoneal (i.p.) immunizations of OVA and alum as adjuvant once per week for two weeks and they are challenged one or two weeks after the last immunization with repeated aerosolized doses of OVA (34). Using the OVA model and IL-6 deficient mice (35), initial studies indicated that the lack of IL-6 increases eosinophilia, lung Th2 cytokines (IL-4, IL-5 and IL-13) and eotaxin, as well as airway response to methacholine (36). The same study also reported decreased eosinophilia and Th2 cytokines in transgenic mice overexpressing IL-6 in airway Clara cells (36), although these mice were previously shown to develop spontaneous fibrosis (37). A later study, using the same IL-6 targeted mice in a chronic OVA model of allergic airway inflammation, reported enhanced airway inflammation, yet decreased fibrosis (38). Thus, these early studies suggested that IL-6 could be protective against allergic airway inflammation, but it could promote lung fibrosis.
While the OVA model has been extensively used as a mouse model for allergic airway inflammation, it requires an i.p. immunization to initiate the immune response, bypassing the lung environment (e.g. lung epithelial cells as source of cytokines). In addition, the OVA model uses alum as adjuvant, and it is known that alum trigger the production of IL-1, but it does not trigger the production of IL-6 (39, 40). An alternative model of allergic airway inflammation consists of the oropharyngeal (o.p.) administration of non-infectious extracts from Aspergillus fumigatus, a physiologically significant fungal allergen, once per week for two weeks followed by 2-4 daily o.p. administrations of the same extracts (41, 42). Lung exposure to A. fumigatus extracts rapidly triggers IL-6 production in the airways (13, 40). Repeated administrations cause allergic airway inflammation characterized by an accumulation of eosinophils and Th2 cytokines in the airways, IgE production and airway hyperresponsiveness (41). Using IL-6 null mice (different from the mice used in other studies) (43) in the A. fumigatus model, a more recent study has shown that IL-6 is required for mucus hypersecretion by airway epithelial cells, although is not required for IL-5 and eosinophilia (40). Impaired mucus production in IL-6 null mice correlated with a profound reduction of IL-13, the main inducer of mucus by epithelial cells in the lung. IL-6 promotes IL-13 production by CD4 T cells (40). Interestingly, the association between IL-6 and IL-13 in this mouse model of allergic airway inflammation correlates with the association of these two cytokines in human allergic asthmatic patients (19). Together, these studies therefore suggest that IL-6 could be used as a therapeutic target to decrease airway mucus hypersecretion in asthma and other lung diseases where mucus hyperplasia contributes to the pathogenesis (e.g. cystic fibrosis or chronic bronchitis).
As an alternative for IL-6 deficient mice, other studies have used an IL-6R blockade in wild type mice to address the role of IL-6 in the development of allergic airway inflammation. It has been shown that intranasal administration of a blocking anti-IL-6R antibody in the OVA model decreases Th2 cytokines and eosinophils in the lung (30). More importantly, this IL-6R blockade also ameliorates airway hyperresponsiveness (30). Similar effect was found when gp130-Fc recombinant protein was used as an alternative blockade for IL-6R signaling (30). The attenuating effect of IL-6R blockade on airway inflammation was related to an increased local expansion of Treg cells, and reduced frequency of effector CD4 T cells (30, 44). Although no addressed in these studies, it is possible that the effect of gp130-Fc could be due to an impaired Th17 response, since IL-6 is currently considered a key factor in the balance between Treg cells and Th17 cells. Independently of the mechanism, the results from these studies are supportive of an active role for IL-6 in allergic airway inflammation. Furthermore, these studies represent the first evidence to support that IL-6 might be a suitable target for a new approach to asthma therapy. A blockade for IL-6R (anti-IL6-R neutralizing Ab) is already approved for treatment of rheumatoid arthritis and systemic juvenile arthritis (45).
Genetic evidence supporting the role of IL-6 in asthma
For decades numerous studies have tried to identify genetic links with the susceptibility to asthma, often in unique and highly homogeneous populations. However, most of these studies failed to provide clear and consistent associations that could help to identifying the genetic basis of asthma. This failure has led to questioning the importance of asthma genetics in developing new therapies. However, the rapid growth and development of more comprehensive areas of gene sequencing and genetics (whole genome sequencing) has facilitated studies in very large populations of subjects worldwide. In addition, the current paradigm of replicating results in more than one population is more likely to be useful.
A recent Australian genome-wide association study (GWAS) performed in over 2,000 asthmatics and 4,000 control individuals of European descent from Australia has identified three novel loci associated with asthma (46). Meta-analysis of their results with the results from the GABRIELS consortium identified seven putative novel asthma risk loci. A follow up analysis of these loci in asthmatic patients and controls from other studies (APCAT, Raine, QIMR and NTR) has revealed the association of one SNP (rs4129267) in intron 8 of the IL-6R gene in chromosome 1q21.3 with asthma risk. The presence of the minor T allele in this SNP was previously associated with increased levels of sIL-6R in serum (47-49).
This non-coding SNP is in linkage disequilibrium with the rs2228145 SNP in the coding region where the minor C substitution of A results in a change from Asp358 to Ala. The presence of Ala358 in the IL-6R variant is known to facilitate cleavage of IL-6R and has been associated with increased levels of sIL-6R (50, 51). A recent study has examined the association of Asp358Ala polymorphism with lung function in asthma in subjects of the SARP (Severe Asthma Research Program) study and replicated the results in the CSGA study (Collaborative Study on the Genetics of Asthma). The overall results from these studies indicate that the minor C allele (Ala358) is more frequent within the severe asthma group and it associates with lower percent predicted FEV1, FVC and FEV1/FVC ratio (52). Furthermore, there is an inverse correlation between serum levels of sIL-6R and FEV1 (52). Thus, together these emerging genetic studies provide strong evidences supporting the role of IL-6 in asthma, and it is likely to be a major determinant of progressive loss of lung function in untreated patients.
IL-6 and COPD
COPD is a complex pulmonary disease that is associated with high morbidity and mortality and it is highly prevalent. A number of pathological processes can contribute to the impaired lung function in COPD, including mucus hypersecretion, emphysema, airway obstruction. Like asthma, the involvement of inflammation, especially neutrophils, in COPD is well established. A number of more recent studies are revealing a potential association between COPD and some inflammatory cytokines or products of these cytokines. The immune response has also been implicated in the development or progression of COPD. The first evidence pointing toward a potential role of IL-6 in COPD came from studies describing increased levels of IL-6 in induced sputum of COPD patients and importantly the inverse correlation between IL-6 levels and lung function as determined by the temporal decay in percentage predicted FEV1 (53-55). Furthermore, a recent longitudinal study on inflammatory markers in COPD patients over three years indicates that elevated IL-6 levels in serum, but not TNFα or IL-8 levels, are predictive of increase mortality in COPD (56). Similarly, another study supports the association of elevated IL-6 levels in serum, but not TNFα and IL-8, with poor clinical outcomes in COPD patients (57). The selective association of clinical outcomes with IL-6 but not with other inflammatory cytokines (e.g. TNFα) suggested that IL-6 in the lung is not necessarily a parameter for exacerbated inflammation, but rather an indicative for a potential lung epithelium damage.
Although there are no strong evidences to support that COPD is determined genetically, recent studies suggest an association of the IL-6 gene variants with COPD. Using COPD patients and smokers with no COPD subjects from the NETT (National Emphysema Treatment Trial) and NAS (Normative Aging Study), an association has been shown between the -174G/C SNP in the IL-6 gene and susceptibility to COPD (58). This is the most represented polymorphism of the IL-6 gene in the human population. Another study with COPD patients and healthy smokers identified an association of a different polymorphism in the IL-6 gene with an increased COPD risk (59). In this case the 321G/T polymorphism was located at the 5' flanking region instead of the promoter region.
Thus, together these studies suggested that IL-6 is likely a player in the pathogenesis of COPD by modulating pulmonary function. It is however unclear whether this association is due to inflammation or whether in this case IL-6 may come from “damaged” lung epithelial cells without the need for an exuberant inflammatory response in the lung.
Aging, frailty and IL-6
The age-dependent onset of frailty, loss of lung function, increased prevalence of COPD or ILD and severity of asthma suggests that mechanisms related to the aging process are involved. An emerging concept is that aging is a proinflammatory condition associated to dysfunctional changes in the immune system (60). Indeed it has been suggested that in the case of COPD it may represent an “accelerated aging phenotype” (61) but this accelerated aging mechanism may apply to other lung diseases as well. It is well established that aging results in increased levels of circulating cytokines such as TNFα, IL-1 and IL-6. These cytokines, and especially IL-6, have been associated with negative health outcomes and mortality. The cause for the increased IL-6 levels observed in the elderly is unclear. It is speculated that results of consistent, low grade activation of inflammatory cells. The innate immune system clearly changes with aging; for instance, monocyte-derived dendrite cells that were obtained from older subjects release increased amounts of IL-6 upon stimulation (62). Alternative explanations for this aging associated raise in circulating IL-6 levels include the generation of reactive oxygen species, oxidative stress or persistent herpes or CMV infections observed in elderly (63, 64). Damage caused by oxidative stress in cells other than inflammatory cells (e.g. epithelial cells) can lead to the production of this cytokine. In any event, age related increased IL-6 is strongly correlated to the degree of frailty, morbidity and mortality. Specifically, frailty is associated with respiratory impairment (63, 65) and IL-6 is only an inflammatory mediator included in the PILE score that predicts morbidity in elderly patients with COPD (64). Determining the role and mechanism of IL-6 in causing or modulating frailty, lung disease and adverse outcomes should prove to be a fruitful area of future research.
Potential mechanisms mediating the function of IL-6 in pulmonary diseases
The studies described above lead to the hypothesis that IL-6 is not just the end product of lung pathology, but it is likely to play an active role in the pathogenesis of asthma, COPD, and possibly other lung diseases. The question that remains to be answered is: what are the mechanisms that IL-6 uses to interfere with lung function? Although the answer remains largely unknown, we can speculate that this cytokine probably uses multiple mechanisms and acts on different cell targets. Here, we will summarize some potential mechanisms according to what it is currently known about IL-6.
The involvement of CD4 T cells in allergic asthma has been supported by numerous studies, but the contribution of CD4 T cells to pathogenesis of severe asthma is more controversial. The regulatory role of IL-6 on CD4 T cell differentiation is the best characterized (14). Although it is not the most effective cytokine relative to others (e.g. IL-2, IL-7, IL-15), IL-6 can provide anti-apoptotic signals to naïve CD4 T cells during activation in vivo and in vitro (66). Nevertheless, IL-6 has emerged as an essential regulator of CD4 T cell differentiation into specific effector CD4 cells. IL-6 is a modulator of the Th1 and Th2 balance (67-70). IL-6 promotes the differentiation of Th2 cells by enhancing endogenous IL-4 production by CD4 T cells (68). IL-4 is a major factor in promoting Ab isotype switching in B cells and the production of IgE, IgG1, IgG2b, and IgG3. As a such, IL-4 was considered as a target for allergic asthma to block IgE production (71). Interestingly, in a fungal antigen-exposure model of allergic airway inflammation, IL-6 deficiency resulted in reduced IgG1 levels, but increased levels of IgE (40). This intriguing phenotype can be explained by the unique effect IL-6 has as an inducer of IL-21 production by CD4 T cells (72). Unlike IL-4, IL-21 promotes B cell plasma differentiation and IgG production but it has a negative effect on IgE production. Similar to IL-6 deficient mice, IL-21 deficient mice also have increased levels of IgE, but reduced levels of IgG (73, 74). Thus, IL-6 or IL-6R blockade might increase the levels of IgE. This is an important caveat to consider if this therapy is utilized in allergic asthmatic patients with already high IgE levels. However, it may not be a problem in the treatment of non-allergic asthmatic patients making this a better targetable population.
Despite its effect on IL-4, IL-6 does not affect IL-5 production by CD4 T cells in vitro or in vivo (40, 68). As a result eosinophilia is not reduced in the absence of IL-6 (36, 40). Thus, it is unlikely that the effect of IL-6 in the asthmatic lung is due to its effect on eosinophils and IL-5. As described above, more recent studies have shown that IL-6 promotes the production of IL-13 (another Th2 cytokine) by CD4 T cells in vitro (40). One of the essential functions of IL-13 in asthma is to promote mucus production by airway epithelial cells (3). Accordingly, IL-13 is now considered a target for treatment of asthma in part through its effects in mucus metaplasia (75-77). Results from Phase II clinical studies using a neutralizing anti-IL-13 Ab in asthmatic patients have been promising and show a benefit in patients who are refractory to corticosteroids. Since the absence of IL-6 in mouse models results in decreased IL-13 production and, consequently, mucus production by airway epithelial cells (40), one significant role for IL-6 to asthma pathogenesis could be augmenting mucus production in the lung airways (Fig. 1). In addition, mucus hypersecretion is also found in COPD and chronic bronchitis, suggesting a potential mechanism by which IL-6 may also contribute to the pathogenesis of COPD. It would be of interest to determine whether the treatment with an IL-6R blockade has a similar effect to IL-13 blockade. IL-13 is also known to promote fibrosis by inducing proliferation of fibroblasts and increasing matrix depositions (78). Thus, some of the observed effects of IL-6 in fibrosis might be mediated through IL-13.
Potential mechanisms by which IL-6 can contribute to lung diseases. IL-6 in the lung can be produced by different sources such as epithelial cells, interstitial fibroblasts, macrophages and other inflammatory cells. The production is induced in response to a variety of stimuli, defined as “insults” because they often cause cell stress or damage. These include allergens, respiratory viruses, excercice, environmental particles, inhaled toxic particles. IL-6 can regulate different aspects of the CD4 T cell mediated response including cytokine production, sIL-6R production, suppressive activity. These mediators in turn contribute to the damage of the lung through their effects on mucus production, matrix deposition, protease release from granulocytes among others. In the presence of sIL-6R, IL-6 can also regulate different functional aspects of non-immune cells such as epithelial cells.
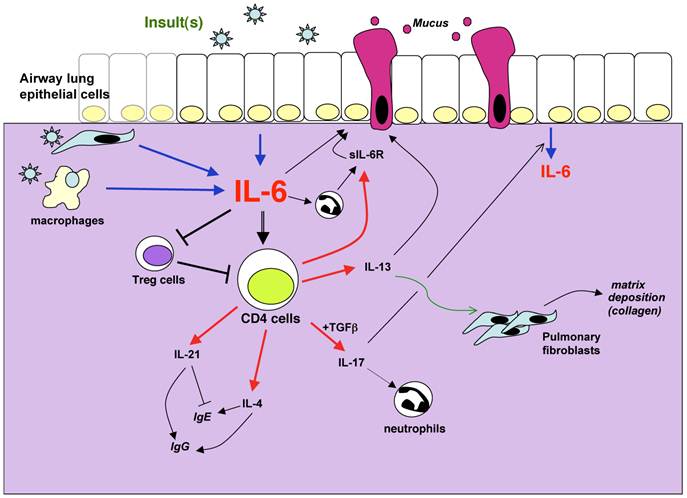
IL-6 has been shown to inhibit Th1 differentiation independently of IL-4. IL-6 interferes with IFNγ production by CD4 T cells through upregulation of SOCSs (SOCS1 or SOCS3) that provide a negative feedback and diminish IFNγ signaling (70). The negative effect of IL-6 in Th1 differentiation could indirectly facilitate the differentiation of other Th subsets, including Th2 and Th17 cells. In addition, during the last five years a number of studies have shown that IL-6 in combination with TGFβ promotes the differentiation of Th17 cells (79-81). Recent studies also indicate that IL-6 promotes Th17 differentiation in the presence of IL-1β and IL-23 (82). Th17 cells can contribute to the development of allergic airway inflammation in mouse models of asthma (83, 84). In human asthma, it is believed that Th17 cells contribute primarily to severe asthma where neutrophils accumulate in the airways (6). In this regard, IL-17 production by CD4 T cells upon induction of allergic airway inflammation was almost abrogated in IL-6 null mice (40). Thus, in human severe asthma IL-6 might be expected to exacerbate lung pathology by its effect on IL-17 and neutrophil accumulation (Fig. 1). In addition, since IL-17 has been shown to trigger the production of IL-6 by lung epithelial cells (85), there may be a positive feedback between these two cytokines (Fig. 1).
IL-6 was initially reported to be a cytokine that blocks Treg activity and immunosuppression in vitro (86). Since TGFβ is known to promote the generation of Tregs, it has been proposed that IL-6 could mediate the fine-tuning for the balance between Tregs and Th17 cells (87). The expansion of Treg cells in a mouse model of allergic airway inflammation after blocking IL-6R signaling (30) supports this potential mechanism. The suppressive effect of IL-6 on Treg cells can lead to an amplification of any type of immune response and it could contribute to the exuberant inflammation detected in asthmatic lungs (Fig. 1).
Leukocytes (T cells, macrophages, neutrophils, etc.) are the primary targets of IL-6 in the lung due to the abundant expression of IL-6R in these cells. However, IL-6 can also provide signals to cells lacking IL-6R expression if sIL-6R is available. Thus, in the presence of sIL-6R, IL-6 can directly affect the function of a number of other non-inflammatory cells in the lung (e.g. epithelial cells, endothelial cells, smooth muscle cells, fibroblasts). Although neutrophils and macrophages are an important source of sIL-6R, CD4 T cells upon activation can also secrete large amounts of sIL-6R (28, 29). Thus, CD4 T cells too could contribute to pathogenesis of pulmonary diseases by providing sIL-6R and conferring responsiveness to IL-6 (Fig. 1). Although IL-6 is required for mucus production in the airways likely through its effect on IL-13 production by CD4 T cells, we cannot rule out a direct effect of IL-6 on goblet cells in the lung if sIL-6R is also present. Moreover, IL-6 could modulate the proliferation and/or survival of interstitial fibroblasts and contribute to pulmonary fibrosis without the need of IL-13 as a mediator. Lastly, a potential effect of IL-6 on smooth muscle cells could affect the mechanical function of the lung without affecting inflammation.
Thus, the pleotropic character of IL-6 provides a variety of mechanisms by which this cytokine can contribute to specific aspects of complex lung diseases such asthma. Although most effect may come from its modulatory role of the immune response, the presence of the trans-signaling pathway for IL-6R opens other avenues for the effects of IL-6 on structural components of the lung.
Conclusions
In summary, IL-6 is now emerging as a potentially pivotal player in the pathogenesis of lung diseases. Over the last two decades, more attention and emphasis was given to a number of other cytokines such as IL-4, IL-5, IL-13 and IL-17. In contrast, IL-6 has previously been relegated to be a byproduct of ongoing inflammation in the airways. However, as discussed here, it is evident that IL-6 can play an active role in mediating specific aspects in a number of lung diseases. The success of tocilizumab (the anti-IL-6R blocking Ab) in certain human inflammatory diseases and its recent approval for treatment of rheumatoid arthritis reveals the contribution of IL-6 in disease pathogenesis. In the lung, IL-6 dissociates from other inflammatory markers. IL-6 can be a key modulator of the overall immune response as well as the function of non-immune cells. The recent mouse studies together with the revealing results from the GWAS demonstrating a genetic link of IL-6R and human asthma show that IL-6 (or IL-6R) could be a target for treatment of asthma, COPD or other pulmonary diseases. Future studies will be needed to address the clinical importance of these findings on IL-6 for the treatment of pulmonary diseases.
Acknoledgements
The authors were supported by the National Institute of Health grants PO1 AI045666 and AI094027 (M.R.) and P30 RR031158 (C.I. and M.R.).
Competing Interests
M. Rincon has acted as a consultant for Genentech regarding IL-6 signaling and its role in rheumatoid arthritis.
References
1. Berton MT, Uhr JW, Vitetta ES. Synthesis of germ-line gamma 1 immunoglobulin heavy-chain transcripts in resting B cells: induction by interleukin 4 and inhibition by interferon gamma. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2829-33
2. Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255-81
3. Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A. et al. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol. 2002;27:593-602
4. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL. et al. Interleukin-13: central mediator of allergic asthma. Science (New York, NY. 1998;282:2258-61
5. Walter DM, McIntire JJ, Berry G, McKenzie AN, Donaldson DD, DeKruyff RH. et al. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J Immunol. 2001;167:4668-75
6. Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495-516
7. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC. et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780-90
8. Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829-39
9. Marini M, Vittori E, Hollemborg J, Mattoli S. Expression of the potent inflammatory cytokines, granulocyte-macrophage-colony-stimulating factor and interleukin-6 and interleukin-8, in bronchial epithelial cells of patients with asthma. J Allergy Clin Immunol. 1992;89:1001-9
10. Stadnyk AW. Cytokine production by epithelial cells. Faseb J. 1994;8:1041-7
11. Yokoyama A, Kohno N, Fujino S, Hamada H, Inoue Y, Fujioka S. et al. Circulating interleukin-6 levels in patients with bronchial asthma. Am J Respir Crit Care Med. 1995;151:1354-8
12. Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. 1992;89:958-67
13. Neveu WA, Bernardo E, Allard JL, Nagaleekar V, Wargo MJ, Davis RJ. et al. Fungal allergen b-glucans trigger p38 MAPK-mediated IL-6 translation in lung epithelial cells. Am J Respir Cell Mol Biol. 2011;45:1133-44
14. Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol. 2009;130:27-33
15. Hirano T. Interleukin 6 and its receptor: ten years later. Intern Rev Immunol. 1998;16:249-84
16. Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347-52
17. Tillie-Leblond I, Pugin J, Marquette CH, Lamblin C, Saulnier F, Brichet A. et al. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am J Respir Crit Care Med. 1999;159:487-94
18. Virchow JCJr, Kroegel C, Walker C, Matthys H. Inflammatory determinants of asthma severity: mediator and cellular changes in bronchoalveolar lavage fluid of patients with severe asthma. J Allergy Clin Immunol. 1996;98:S27-33
19. Neveu WA, Allard JL, Raymond DM, Bourassa LM, Burns SM, Bunn JY. et al. Elevation of IL-6 in the allergic asthmatic airway is independent of inflammation but associates with loss of central airway function. Respir Res. 2010;11:28
20. Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science. 1992;258:593-7
21. Akira S. IL-6-regulated transcription factors. Int J Biochem Cell Biol. 1997;29:1401-18
22. Akira S, Nishio Y, Inoue M, Wang X, Wei S, Matsusaka T. et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor3 p91-related transcriptional factor involved in the gp130 stimulated signaling pathway. Cell. 1994;77:63-71
23. Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25-50
24. Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta. 2002;1592:251-63
25. Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227-36
26. Mullberg J, Oberthur W, Lottspeich F, Mehl E, Dittrich E, Graeve L. et al. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J Immunol. 1994;152:4958-68
27. Horiuchi S, Koyanagi Y, Zhou Y, Miyamoto H, Tanaka Y, Waki M. et al. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur J Immunol. 1994;24:1945-8
28. Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J Immunol. 2008;180:7102-6
29. Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R. et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol. 2010;184:2130-9
30. Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ. et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313-25
31. Dixon AE, Raymond DM, Suratt BT, Bourassa LM, Irvin CG. Lower airway disease in asthmatics with and without rhinitis. Lung. 2008;186:361-8
32. Morjaria JB, Babu KS, Vijayanand P, Chauhan AJ, Davies DE, Holgate ST. Sputum IL-6 concentrations in severe asthma and its relationship with FEV1. Thorax. 2011;66:537
33. Dixon AE, Shade DM, Cohen RI, Skloot GS, Holbrook JT, Smith LJ. et al. Effect of obesity on clinical presentation and response to treatment in asthma. J Asthma. 2006;43:553-8
34. Renz H, Smith HR, Henson JE, Ray BS, Irvin CG, Gelfand EW. Aerosolized antigen exposure without adjuvant causes increased IgE production and increased airway responsiveness in the mouse. J Allergy Clin Immunol. 1992;89:1127-38
35. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T. et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339-42
36. Wang J, Homer RJ, Chen Q, Elias JA. Endogenous and exogenous IL-6 inhibit aeroallergen-induced Th2 inflammation. J Immunol. 2000;165:4051-61
37. DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR. et al. Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J Clin Invest. 1994;94:2028-35
38. Qiu Z, Fujimura M, Kurashima K, Nakao S, Mukaida N. Enhanced airway inflammation and decreased subepithelial fibrosis in interleukin 6-deficient mice following chronic exposure to aerosolized antigen. Clin Exp Allergy. 2004;34:1321-8
39. Eisenbarth SC, Colegio OR, O'Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122-6
40. Neveu W, Allard JB, Dienz O, Wargo MJ, Ciliberto G, Whittaker LA. et al. IL-6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol. 2009;183:1732-8
41. Allard JB, Poynter ME, Marr KA, Cohn L, Rincon M, Whittaker LA. Aspergillus fumigatus generates an enhanced Th2-biased immune response in mice with defective cystic fibrosis transmembrane conductance regulator. J Immunol. 2006;177:5186-94
42. Allard JB, Rinaldi L, Wargo MJ, Allen G, Akira S, Uematsu S. et al. Th2 allergic immune response to inhaled fungal antigens is modulated by TLR-4-independent bacterial products. Eur J Immunol. 2009;39:776-88
43. Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H. et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994;13:1189-96
44. Finotto S, Eigenbrod T, Karwot R, Boross I, Doganci A, Ito H. et al. Local blockade of IL-6R signaling induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T cells. Int Immunol. 2007;19:685-93
45. Tanaka T, Narazaki M, Kishimoto T. Anti-interleukin-6 receptor antibody, tocilizumab, for the treatment of autoimmune diseases. FEBS Lett. 2011;585:3699-709
46. Ferreira MA, Matheson MC, Duffy DL, Marks GB, Hui J, Le Souef P. et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. 2011;378:1006-14
47. Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I. et al. A genome-wide association study identifies protein quantitative trait loci (pQTLs). PLoS Genet. 2008;4:e1000072
48. Rafiq S, Frayling TM, Murray A, Hurst A, Stevens K, Weedon MN. et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8:552-9
49. Reich D, Patterson N, Ramesh V, De Jager PL, McDonald GJ, Tandon A. et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716-26
50. Galicia JC, Tai H, Komatsu Y, Shimada Y, Akazawa K, Yoshie H. Polymorphisms in the IL-6 receptor (IL-6R) gene: strong evidence that serum levels of soluble IL-6R are genetically influenced. Genes Immun. 2004;5:513-6
51. Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. Faseb J. 2001;15:43-58
52. Hawkins GA, Robinson MB, Hastie AT, Li X, Li H, Moore WC. et al. The IL6R variation Asp(358)Ala is a potential modifier of lung function in subjects with asthma. J Allergy Clin Immunol. 2012Aug;130(2):510-5
53. Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TM, Hurst JR. et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128:1995-2004
54. Grubek-Jaworska H, Paplinska M, Hermanowicz-Salamon J, Bialek-Gosk K, Dabrowska M, Grabczak E. et al. IL-6 and IL-13 in Induced Sputum of COPD and Asthma Patients: Correlation with Respiratory Tests. Respiration. 2012;84(2):101-7
55. Eickmeier O, Huebner M, Herrmann E, Zissler U, Rosewich M, Baer PC. et al. Sputum biomarker profiles in cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) and association between pulmonary function. Cytokine. 2010;50:152-7
56. Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P. et al. Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1065-72
57. Agusti A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE. et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7:e37483
58. He JQ, Foreman MG, Shumansky K, Zhang X, Akhabir L, Sin DD. et al. Associations of IL6 polymorphisms with lung function decline and COPD. Thorax. 2009;64:698-704
59. Yanbaeva DG, Dentener MA, Spruit MA, Houwing-Duistermaat JJ, Kotz D, Passos VL. et al. IL6 and CRP haplotypes are associated with COPD risk and systemic inflammation: a case-control study. BMC Med Genet. 2009;10:23
60. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F. et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92-105
61. Barnes PJ. Future treatments for chronic obstructive pulmonary disease and its comorbidities. Proc Am Thorac Soc. 2008;5:857-64
62. Agrawal A, Tay J, Ton S, Agrawal S, Gupta S. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J Immunol. 2009;182:1138-45
63. Sharma G, Hanania NA, Shim YM. The aging immune system and its relationship to the development of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:573-80
64. Mehrotra N, Freire AX, Bauer DC, Harris TB, Newman AB, Kritchevsky SB. et al. Predictors of mortality in elderly subjects with obstructive airway disease: the PILE score. Ann Epidemiol. 2010;20:223-32
65. Vaz Fragoso CA, Enright PL, McAvay G, Van Ness PH, Gill TM. Frailty and respiratory impairment in older persons. Am J Med. 2012;125:79-86
66. Teague TK, Marrack P, Kappler JW, Vella AT. IL-6 rescues resting mouse T cells from apoptosis. J Immunol. 1997;158:5791-6
67. Rincón M, Anguita J, Nakamura T, Fikrig E, Flavell RA. IL-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997;185:461-9
68. Diehl S, Chow CW, Weiss L, Palmetshofer A, Twardzik T, Rounds L. et al. Induction of NFATc2 expression by interleukin 6 promotes T helper type 2 differentiation. J Exp Med. 2002;196:39-49
69. Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39:531-6
70. Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E. et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13:805-15
71. Barnes PJ. Cytokine-directed therapies for asthma. J Allergy Clin Immunol. 2001;108:S72-6
72. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R. et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. 2009;206:69-78
73. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A. et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630-4
74. Shang XZ, Ma KY, Radewonuk J, Li J, Song XY, Griswold DE. et al. IgE isotype switch and IgE production are enhanced in IL-21-deficient but not IFN-gamma-deficient mice in a Th2-biased response. Cell Immunol. 2006;241:66-74
75. Hodsman GP, Ashman C, Cahn A, De Boever E, Locantore N, Serone A. et al. A Phase 1, Randomized, Placebo-Controlled, Dose-Escalation Study of an Anti-IL-13 Monoclonal Antibody in Healthy Subjects and Mild Asthmatics. Br J Clin Pharmacol. 2012
76. Oh CK, Faggioni R, Jin F, Roskos LK, Wang B, Birrell C. et al. An open-label, single-dose bioavailability study of the pharmacokinetics of CAT-354 after subcutaneous and intravenous administration in healthy males. Br J Clin Pharmacol. 2010;69:645-55
77. Singh D, Kane B, Molfino NA, Faggioni R, Roskos L, Woodcock A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10:3
78. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J. et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779-88
79. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-8
80. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179-89
81. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231-4
82. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE. et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967-71
83. Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V. et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715-25
84. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173-7
85. van den Berg A, Kuiper M, Snoek M, Timens W, Postma DS, Jansen HM. et al. Interleukin-17 induces hyperresponsive interleukin-8 and interleukin-6 production to tumor necrosis factor-alpha in structural lung cells. Am J Respir Cell Mol Biol. 2005;33:97-104
86. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033-6
87. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830-5
Author contact
![]() Corresponding author: Mercedes Rincon, Ph.D. Professor, Department of Medicine/Immunobiology, Given C331, University of Vermont, 89 Beaumont Ave., Burlington, VT 05405. Phone: 802-656.0937 Fax: 802-656-3854 mrinconedu.
Corresponding author: Mercedes Rincon, Ph.D. Professor, Department of Medicine/Immunobiology, Given C331, University of Vermont, 89 Beaumont Ave., Burlington, VT 05405. Phone: 802-656.0937 Fax: 802-656-3854 mrinconedu.