Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
The Src family tyrosine kinases
Discovery of Csk
Function of Csk
Structure of Csk
Regulation of Csk
Csk in diseases
Perspectives
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(10):1385-1397. doi:10.7150/ijbs.5141 This issue Cite
Review
Regulation of the Src Family Kinases by Csk
Masato Okada ![]()
Department of Oncogene Research, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, JAPAN.
Received 2012-8-31; Accepted 2012-10-1; Published 2012-11-1
Abstract
The non-receptor tyrosine kinase Csk serves as an indispensable negative regulator of the Src family tyrosine kinases (SFKs) by specifically phosphorylating the negative regulatory site of SFKs, thereby suppressing their oncogenic potential. Csk is primarily regulated through its SH2 domain, which is required for membrane translocation of Csk via binding to scaffold proteins such as Cbp/PAG1. The binding of scaffolds to the SH2 domain can also upregulate Csk kinase activity. These regulatory features have been elucidated by analyses of Csk structure at the atomic levels. Although Csk itself may not be mutated in human cancers, perturbation of the regulatory system consisting of Csk, Cbp/PAG1, or other scaffolds, and certain tyrosine phosphatases may explain the upregulation of SFKs frequently observed in human cancers. This review focuses on the molecular bases for the function, structure, and regulation of Csk as a unique regulatory tyrosine kinase for SFKs.
Keywords: Csk, Src family, tyrosine kinases
The Src family tyrosine kinases
Tyrosine kinases, which exist only in animals, play important roles in controlling animal-specific cellular functions such as rapid cell-cell communication via the plasma membrane [1, 2]. The most critical feature of tyrosine kinases is the strict regulation of their functions. Due to these enzyme's importance in controlling cellular functions, dysregulation of tyrosine kinases can play a causative role in various diseases, especially cancers. Tyrosine kinases are classified into two major subgroups: receptor and non-receptor. The receptor tyrosine kinases are activated and auto-phosphorylated upon directly receiving extra-cellular cues, creating an intracellular platform for specific adaptors and effectors that activate downstream signaling pathways. The cytoplasmic non-receptor tyrosine kinases are also activated in response to extra-cellular cues via physical and functional interactions with particular transmembrane receptors, and subsequently activate specific downstream pathways to control cell fate.
The Src family tyrosine kinases (SFKs) are the major non-receptor tyrosine kinases expressed in multiple types of animal cells [3]. The SFKs include eight members, c-Src, c-Yes, Fyn, c-Fgr, Lyn, Hck, Lck, Blk. They are activated in concert with the activation of transmembrane receptors, such as EGF receptor, T cell receptor, and integrins. The SFKs regulate a wide range of cellular events, including cell growth, division, differentiation, survival, and programmed death, as well as specialized functions such as immune responses, cell adhesion, cell movement, and endocytosis [4, 5]. Several of the SFKs, c-Src, c-Yes, and c-Fgr, were originally discovered as prototypes of avian retroviral oncogene products (v-Src, v-Yes and v-Fgr) [6-11]. Dysregulation of SFKs has been implicated in the etiology of various human diseases such as cancers, although somatic mutation in the SFK genes has not been detected [12, 13]. There observations underscore the important roles played by SFKs. However, the molecular mechanisms underlying the causes of SFK dysregulation in the diseases remain unclear.
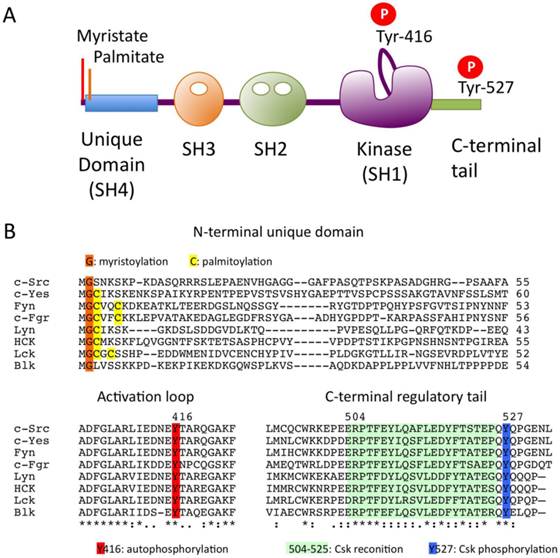
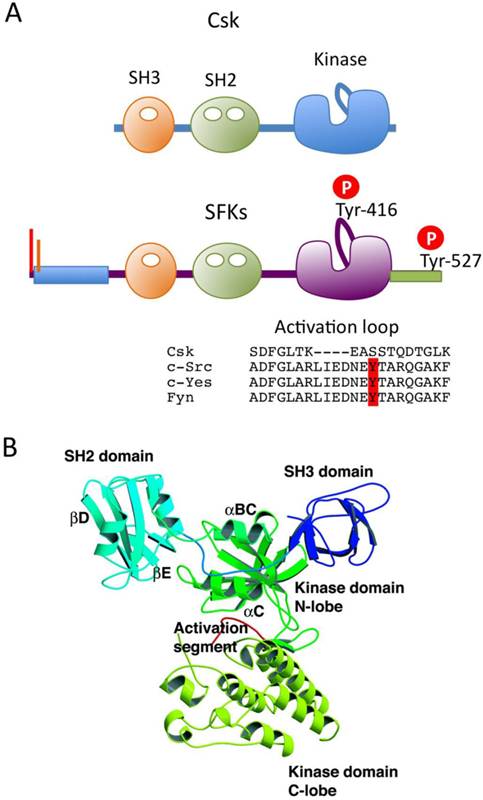
The overall sequences of SFKs, with the exception of the N-terminal ~50 residues, is highly conserved among the family members. The primary structure can be divided into five functional domains (Fig.1A). The N-terminal domain contains signals for lipid modification: myristoylation (in all SFKs) and palmitoylation (in all but c-Src and Blk) signals, both of which are required for membrane association of SFKs (Fig.1B). Palmitoylation has been implicated in the stable localization of SFKs in membrane microdomains called “lipid rafts” [14]. All SFKs share the Src homology 3 (SH3) and SH2 domains, the kinase domain, and the C-terminal regulatory tail. These structural similarities allow for common mechanisms of regulation: the phosphorylation of the conserved tyrosine residues (Tyr-416 and Tyr-527) and the intramolecular interactions among the domains are crucial for the regulation of SFKs (Fig.1B).
The Src family kinases (SFKs). (A) Domain organization of SFKs. (B) Amino acid alignments of N-terminal unique domain, activation loop and C-terminal regulatory tail in SFKs.
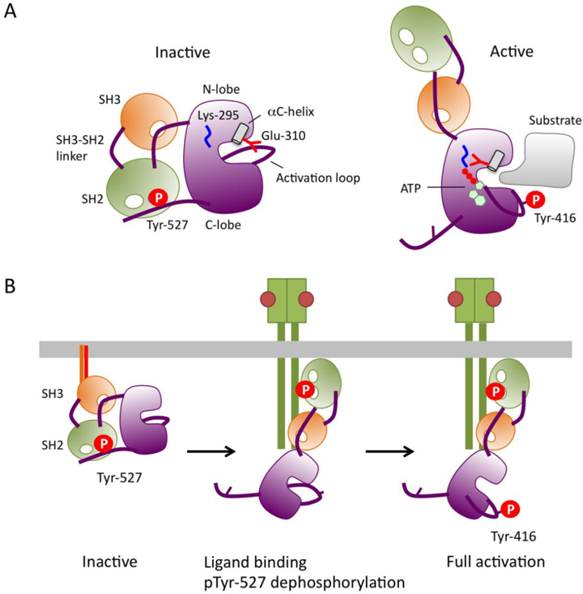
In 1985, Courtneidge first recognized that c-Src is activated by dephosphorylation [15]. Cooper and King subsequently identified the conserved Tyr-527 (in chicken c-Src) in the C-terminal tail as the site of inhibitory phosphorylation [16]. These findings demonstrate that SFK is normally present in its inactive form, in which Tyr-527 is phosphorylated. This is consistent with the fact that the oncogenic v-Src, which lacks the C-terminal tail containing Tyr-527, is constitutively active. By contrast, Tyr-416 in the catalytic domain is highly phosphorylated in v-Src and a c-Src mutant bearing a Tyr-527 to Phe substitution (c-SrcF527) [17, 18], suggesting that these two tyrosine phosphorylation sites are involved in a reciprocal regulatory mechanism. The detailed mechanism of SFK regulation was later resolved at the atomic level by analyses of the crystal structures of the inactive and active forms of c-Src [19-21] (Fig.2A). In resting cells, SFKs are mostly phosphorylated at Tyr-527, and adopt the inactive conformation, which is stabilized by two intramolecular interactions: 1) binding of phosphorylated Tyr-527 to its own SH2 domain, and 2) binding of the SH2-kinase linker to the SH3 domain. These intramolecular interactions affect the configuration of the catalytic pocket. In particular, the hydrogen bond between Glu-310 in the αC-helix and Lys-295, required for Mg-ATP binding, is disrupted in the inactive conformation. The dephosphorylation of Tyr-527 releases the 'lock' by the SH2 domain and causes dramatic conformational change in the kinase domain, resulting in formation of a hydrogen bond between Glu-310 and Lys-295 in the catalytic domain (Fig 2A). This active enzyme can now catalyze the intermolecular auto-phosphorylation of Tyr-416 in the activation loop. This auto-phosphorylation locks the catalytic domain into the active conformation and facilitates access of substrates to the active site [22].
Mechanism of SFK activation. (A) Schematic models of inactive and active forms of c-Src (cited from [21]). (B) A predicted mechanism of SFK activation.
The regulation of SFKs by intramolecular interactions suggests that SFKs can also be regulated by interaction with molecules that compete with the SFK domains. Indeed, SFKs bind to various tyrosine-phosphorylated proteins by recognizing specific phosphopeptide sequences via the SH2 domain [23]. For example, c-Src binds to the phosphorylated forms of Cas, FAK, and paxillin, as well as growth factor receptors such as EGFR, CSF-1 and PDGF, resulting in activation of c-Src [5, 24-26]. Activated c-Src can further phosphorylate these interacting proteins to create new binding sites for other adaptors and effectors, which in turn allows amplification of signals. In addition, the c-Src SH3 domain binds to various signaling proteins that contain proline-rich motifs [27], such as Shc, PI3K, Cas, Tks5, and Arhgef5 [28-30]. Interactions with these proteins disrupt the stabilized inactive conformations of SFKs and promote the phosphorylation of these proteins by SFKs.
Although the molecular mechanisms of SFK activation may vary depending upon cell types and extracellular cues, it is now believed that in general, full activation of SFK is achieved in the following order (Fig.2B): 1) the activated receptors, adaptors, or effectors interact with the SH2/3 domains of inactive SFKs to open up the closed conformation, and harbor SFKs at appropriate intracellular locations such as focal adhesions; and 2) tyrosine phosphatases dephosphorylate the exposed pTyr-527 to stabilize the active conformation; and 3) the activated SFKs undergo intermolecular auto-phosphorylation on Tyr-416 to lock the catalytic pocket into the fully active conformation. The fully activated SFKs then phosphorylate substrate proteins, such as FAK and Cas, many of which can also create binding sites for SFKs; this initiates the positive-feedback loop of SFK activation. When the cell response is terminated, activated SFKs are rapidly degraded via the ubiquitin-proteasome pathway [31] or inactivated by phosphorylation at Tyr-527. Thus, in order to guarantee rapid cell response to extracellular cues, most of SFKs in the resting state should be inactivated by phosphorylation at Tyr-527. Furthermore, this highly secure regulatory system is required in order to strictly control potentially dangerous SFKs that possess inherent oncogenic activity. However, the tyrosine kinase responsible for Tyr-527 phosphorylation was not identified until we discovered Csk (C-terminal Src Kinase). In the next chapter, I will introduce Csk as a genuine suppressor of 'dangerous' SFKs.
Discovery of Csk
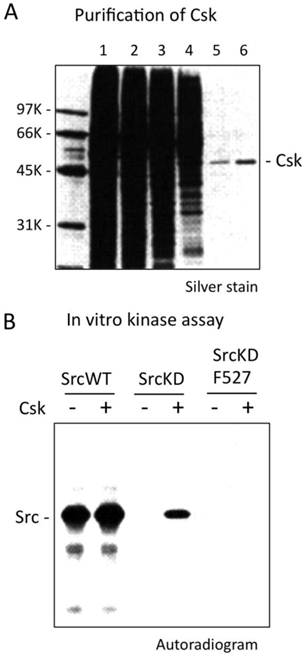
In the late 80's, my research focused on the roles of c-Src in the terminal differentiation of neurons. c-Src+ (neuronal c-Src or n-Src), an alternative splicing variant having 6 amino acid insertion in the SH3 domain, is abundantly expressed specifically in postomitotic neurons [32, 33]. To elucidate the function and regulation of c-Src+, I first attempted to purify the SFK-related tyrosine kinases from neonatal rat brains, a rich source of developing neurons, by means of column chromatography. I successfully isolated c-Src+ and a new tyrosine kinase that was co-purified with c-Src+ [34] (Fig.3A). Because I realized that the new kinase might be involved specifically in the regulation of c-Src+, I challenged it with the purified c-Src+ protein to determine whether it was the missing Tyr-527 kinase. At least in vitro, the answer was “Yes” [35]. Subsequent biochemical analysis confirmed that the kinase (named Csk, from C-terminal Src kinase) [36] specifically phosphorylates Tyr-527 and can inhibit the activity of c-Src (Fig.3B). Next, we cloned rat Csk cDNA and confirmed its specificity for c-Src in a yeast model system [37]. Furthermore, we and another group showed that Csk phosphorylates the conserved negative regulatory sites in other SFKs [36, 38]. Sabe et al. showed that when Csk is overexpressed, it inhibits transformation of fibroblasts by the combined actions of c-Src and the oncoprotein v-Crk [39, 40]. This observation suggests that Csk has the potential to serve as a repressor of c-Src in vivo as well as in vitro.
Discovery of Csk. (A) Sample of each purification step of Csk was analysed by SDS-PAGE and staining with silver. Lane 1, Nonidet P-40 extract; lane 2, DEAE-cellulose column chromatography; lane 3, poly(Glu, Tyr) Sepharose CL-4B: lane 4, Mono Q; lane 5, Sephacryl S200HR; lane 6, Mono S. (B) Phosphorylaion of Src by Csk was determined by incubating immunopurified SrcWT (lanes 1 and 2), SrcKD (lanes 3 and 4) and SrcKDF527 (lanes 5 and 6) with or without Csk. The reaction products were analyzed by SDS-PAGE and autoradiography.
Function of Csk
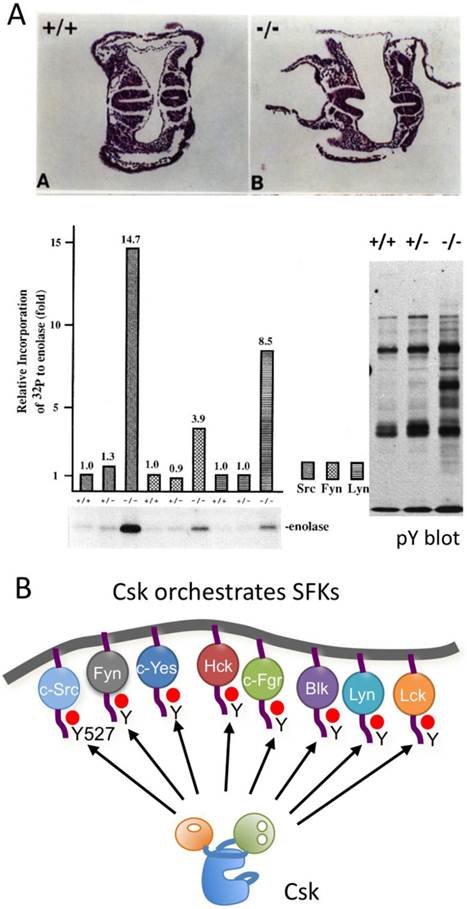
The function of Csk as a negative regulator of SFKs in vivo was demonstrated by the phenotypes of mice lacking Csk [41, 42] (Fig.4A). Homozygous mutant embryos die at neurulation stages with defects in neural tube closure. Mutant embryonal tissues and established Csk-deficient cell lines exhibit increased activity of all SFKs tested, resulting in the accumulation of tyrosine-phosphorylated proteins. These findings strongly suggest that Csk is required to repress SFKs in vivo. Conditional ablation of Csk in specific tissues also causes severe defects associated with constitutive activation of SFKs. Loss of Csk induces dysfunction in acute inflammatory responses [43] and T-cell development [44], hyperplasia of the epidermis [45] and defects in cell adhesion and migration [46]. Even in invertebrates, such as Drosophila and C. elegans, the loss of Csk leads to constitutive activation of SFKs, causing hyperproliferation and defective cytoskeletal function, respectively [47, 48]. These findings indicate that Csk is an indispensable regulator of SFKs (Fig.4B).
Function of Csk. (A: upper) Histological appearances of E8.0 embryos. Transverse sections through the trunk region of wild-type (+/+) and Csk KO (-/-) embryos, respectively. The neural lube began to close in the wild-type embryo, but not in the homozygous mutant embryos. (A: lower left) Activation of SFKs in Csk KO embryos. Src, Fyn and Lyn were immunopurifed from the indicated genotypes of embryos and incubated with acid-treated enolase and [γ-32P]ATP, followed by SDS-PAGE. The Incorporation of 32P to enolase was quantitated. (A: lower wright) Tyrosine-phosphorylated proteins in the indicated genotypes of embryos were analyzed by Western blotting with anti-phosphotyrosine antibody. (B) Csk can phosphorylate the C-terminal regulatory sites of all the members of SFKs to repress their activities.
To further verify the functional connections between Csk and SFKs, we investigated homologs of SFKs and Csk in two primitive animals; a unicellular choanoflagellate M. ovate and a fresh-water sponge E. fluviatilis (Fig.5).
Evolution of SFKs and Csk. Phylogenetic tree of Src, Csk, and related genes by using the maximum likelihood method. The reliability index (1) and bootstrap probability (2) for each branch are indicated before and after the slash, respectively.
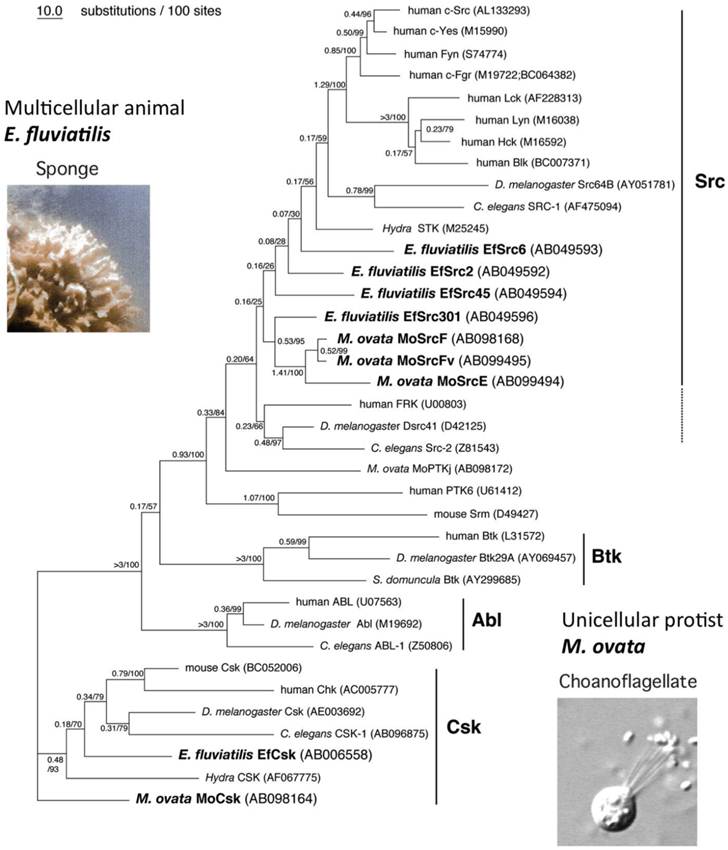
Sequence alignment of invertebrate and vertebrate genes reveal that Csk is highly conserved across the animal kingdom, from the unicellular choanoflagellate to humans, as are the SFKs [49], confirming the intimate relationship between Csk and SFKs. We also found that SFKs form a gene family even in unicellular choanoflagellates, whereas Csk is present as a single gene in all animal phyla: this is consistent with the idea that Csk is the sole conductor of SFKs (Fig.4B). Furthermore, we showed that the SFKs in the unicellular choanoflagellates are more resistant to Csk-mediated inactivation than those of multicellular animals, suggesting that strict regulation of SFKs has been established during the evolution of the multicellular animals [49]. This may mean that the strict 'ON-OFF' regulation of SFKs is prerequisite for the evolution of multicellular organization. The loss of this regulatory system, e.g., upon expression of v-Src, may lead to dysregulation of normal cellular interactions such as that observed in cancers.
A tyrosine kinase homologus to Csk, formerly named Matk (Ctk, Hyl, Chk, or Lsk), has been identified in mammalian hematopoietic cells [50-55]. The results of in vitro analyses suggest that, like Csk, Matk can target SFKs to repress their activity. However, analyses of Matk-deficient mice revealed that loss of Matk did not result in any overt phenotype or affect the activity of SFKs in cells, suggesting that it might not have the same function as Csk [56]. Thus, it is likely that Csk is the major kinase involved in the regulation of SFKs in vivo, and Matk may play specific roles in hematopoietic cells.
As a protein tyrosine kinase, Csk is exceptional in that it exhibits high substrate specificity toward the C-terminal regulatory tyrosine (equivalent to Tyr-527) of SFKs. The surrounding sequence of the SFK regulatory site (QYQ) is highly conserved among SFKs, but biochemical and structural studies reveal that another region (aa 504-525) located distantly from Tyr-527 is rather crucial for specific recognition by Csk [57-59] (Fig.1). In addition to SFKs, several signaling proteins have been reported to serves as substrates of Csk. Those include paxillin [60], P2X3 receptor [61], c-Jun [62] and Lats [63]. However, the physiological relevance of the phosphorylation of these proteins remains unclear.
Although the kinase Csk has been identified as a negative regulator of SFKs, the phosphatases that activate SFKs by acting on Tyr-527 have not yet been identified with certainty. So far, several candidate phosphatases have been reported [64]: those include PTPα [65], PTPε [66], PTPλ [67], CD45 [68] and PTP1B [69], SHP-1 [70], and SHP-2 [71]. However, the substrate specificity of these tyrosine phosphatases is not as strict as that of Csk; most of them can dephosphorylate both Tyr-527 and Tyr-416. These findings imply that specific phosphatase may not be involved in the direct activation of SFKs, and the dephosphorylation of Tyr-527 may be a secondary event that occurs after the ligand-mediated conformational changes earlier in the process of SFK activation.
Structure of Csk
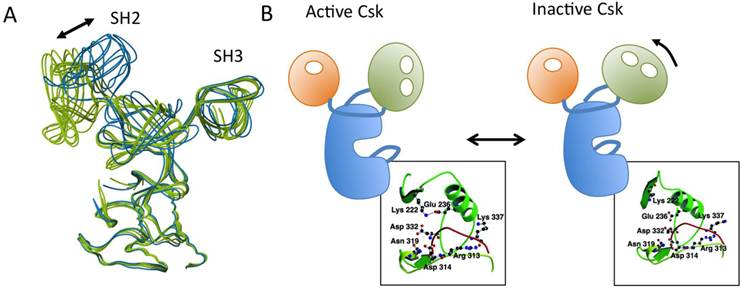
Csk is a non-receptor tyrosine kinase with a molecular mass of 50 kDa. It contains the SH3 and SH2 domains in its N-terminus and a kinase domain in its C-terminus [37]. This arrangement of functional domains within the primary structure is similar to that of SFKs, but Csk lacks the N-terminal fatty acylation sites, the auto-phosphorylation site in the activation loop, and the C-terminal negative regulatory sites, all of which are conserved among SFK proteins and critical for their proper regulation (Fig.6A). The absence of auto-phosphorylation in the activation loop is a distinguishing feature of Csk. The crystal structure of full-length Csk reveals significantly different dispositions of the functional domains relative to those of SFKs [72], indicating that Csk is regulated differently than SFKs (Fig.6B). The most striking feature of the Csk structure is that, unlike the situation in SFKs, the binding pockets of the SH3 and SH2 domains are oriented outward, enabling intermolecular interactions with other molecules. The Csk crystal we analyzed consists of six molecules classified as either active or inactive states according to the coordination states of catalytic residues (specifically, the presence or absence of the salt bridge between Glu236 and Lys222) (Fig.7). In active molecules, the SH2-kinase and SH2-SH3 linkers are tightly bound to the N-terminal lobe of the kinase domain in order to stabilize the active conformation, and there is a direct linkage between the SH2 and the kinase domains. In inactive molecules, the SH2 domains are rotated in a manner that disrupts the linkage to the kinase domain. These observations suggest that the active and inactive conformations of Csk exist in a dynamic equilibrium, and that overall activity is regulated through binding to the SH2 domain of some other factors: such binding would lock the SH2 and kinase domains into the coupled state [72, 73].
Structure of Csk. (A) Domain organizations of Csk and SFKs. An amino acid alignment of the activation loop of Csk and SFKs is also shown. (B) Ribon diagram of a representative Csk structure (active molecule) in the crystal.
Regulation of Csk. (A) Superimposed model of the six Csk molecules in an asymmetric unit. Active molecules are shown in green and inactive molecules are in blue. (B) Schematic models of active and inactive Csk structures. (B: boxes) Arrangements of the catalytically important residues in active and inactive Csk. The salt bridge between Lys-222 and Glu-236 is indicated by dotted lines.
The extensive mutagenesis study located the substrate-docking sites on the surface of Csk, providing a structural basis for the high substrate specificity of Csk [58]. The crystal structure of a complex between the kinase domains of Csk and c-Src further revealed that interactions between these kinases position the C-terminal tail of c-Src at the edge of the active site of Csk [59]. Based on this finding, those authors suggested that Csk cannot phosphorylate substrates that lack this docking mechanism, because the conventional substrate binding site is destabilized in Csk by the lack of auto-phosphorylation in the activation loop. Taken together, it is likely that the distinguishing features of Csk structure, i.e., the lack of auto-phosphorylation and the specific orientations of the domains, may determine its exceptionally high substrate specificity.
Regulation of Csk
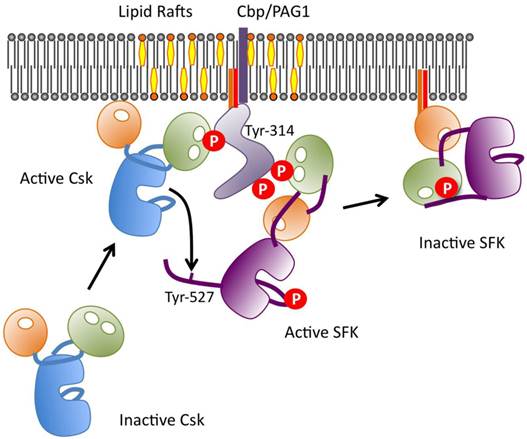
Because Csk lacks a transmembrane domain and fatty acyl modifications, it is predominantly present in cytosol, whereas its substrate SFKs are anchored to the membrane via their N-terminal myristate and palmitate moieties. Therefore, the translocation of Csk to the membrane, where SFKs are activated, is thought to be a critical step of Csk regulation. So far, several scaffolding proteins, e.g., caveolin-1, paxillin, Dab2, VE-cadherin, IGF-1R, IR, LIME, and SIT1, have been identified as membrane anchors of Csk [74-81]. We also searched for intrinsic phosphoproteins that might be involved in the regulation of Csk by binding to the SH2 domain, and successfully isolated a transmembrane adaptor-like molecule Cbp/PAG1 (Csk binding protein/phosphoprotein associated with glycosphingolipid-enriched membrane) from the cholesterol-enriched membrane microdomains “lipid rafts” [82, 83]. Cbp has a single transmembrane domain at its N-terminus and two palmitoyl modification sites just C-terminal to the transmembrane domain, through which Cbp is exclusively localized to lipid rafts. Cbp also contains nine tyrosine residues that can be phosphorylated, four of which are in the typical Src SH2 binding motifs (YEEI/V), and one of which is the Csk SH2 binding site (YSSV). Because Cbp is co-localized with SFKs in lipid rafts, it is a readily available substrate of SFKs, and its five SH2 binding motifs are efficiently phosphorylated. Single molecules of phosphorylated Cbp can simultaneously bind Csk and SFKs, thereby bringing them into close proximity and enabling efficient inactivation of the SFKs by Csk [14, 84, 85] (Fig.8). This negative feedback loop is crucial in preventing tumorigenesis and controlling the cell signaling evoked by activation of growth factor receptors. We also found that Cbp is downregulated by activation of not only SFKs but also other oncogenes such as Ras [84]. It is also downregulated in various cancer cells, potentially through epigenetic silencing [86]; this observation may explain the frequent upregulation of SFK function in cancer cells that lack somatic mutations in SFKs. Other Csk-binding proteins, such as paxillin [75] and caveorin-1 [74], are also phosphorylated by SFKs and participate in the negative regulation of SFKs by recruiting Csk to the sites of SFK activation [87].
Regulation of Csk via Cbp in lipid rafts. A schematic model of the roles of Cbp/Csk and lipid rafts in regulating the function of SFKs. When Cbp is phosphorylated by active SFKs, Csk is recruited to lipid rafts via binding to Cbp at pY314 and phosphorylates Y527 to inactivate the catalytic activity of SFKs. The inactivated SFKs then relocate to non-raft compartments.
The binding of scaffolds to the SH2 domain of Csk can also activate the enzyme activity of Csk. The occupation of the SH2 domain of Csk by phosphorylated Cbp affects conformation of the catalytic domain, thereby increasing Csk activity toward SFKs [73, 88]. Thus, it is likely that the scaffold proteins positively regulate Csk functions not only by recruiting Csk to the membrane but also by directly activating it. On the other hand, Cloutier and Veillette found that Csk can associate with the protein tyrosine phosphatase PEP/LYP expressed in hemopoietic cells [89, 90]. Their results suggest that the association of Csk with PEP/LYP constitutes a more efficient means of inhibiting signal transduction by SFKs in vivo. However, a more recent report presented results that call into question the postulated function of LYP-Csk interactions [91, 92]. Another regulatory mechanism is suggested by observations that the activity of Csk can be regulated by the oxidation state of the disulfide bond in the SH2 domain, implying that Csk could be regulated by the redox state within the cells [93]. Furthermore, Csk is phosphorylated by PKA at Ser-364, resulting in an increase in kinase activity [94]. However, the physiological relevance of this phosphorylation event has not yet been addressed. Csk is highly expressed in the developing nervous system and lymphoid cells [95], but the mechanisms underlying the regulation of Csk at the expression levels remains to be elucidated
Csk in diseases
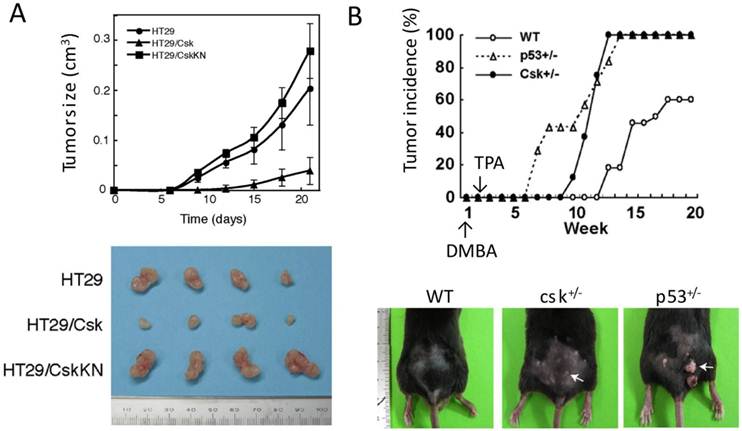
Accumulating evidence suggests that Csk may play a role in tumor suppression by inhibiting the oncogenic activity of SFKs. Indeed, overexpression of Csk can inhibit tumor growth of human colon cancer cells [84]. Heterozygous csk+/- mutant mice exhibit higher sensitivity to chemically induced skin carcinogenesis than wild-type mice (Yagi et al., unpublished observation) (Fig.9). However, there is no convincing evidence that supports the direct involvement of Csk in human cancer. Although Csk is downregulated in some cancers [96], it is likely that Csk is expressed at normal levels in many types of cancer cells [97], and, like the SRC gene [12], there have been no reports of somatic mutation in the csk gene. By contrast, as mentioned above, the expression of Cbp/PAG1 is downregulated in multiple types of cancer cells [84], potentially via an epigenetic mechanism [86]. The downregulation of Cbp/PAG1 may interfere with the translocation of Csk to the membrane, suppressing the inactivation of SFKs. This mechanism may explain the upregulation of SFKs in some cancer cells. Therefore, further analyses of Cbp/PAG1-mediated regulatory system should reveal new opportunities for therapeutic intervention in cancer.
Function of Csk as a tumor suppressor. (A) Effect of Csk overexpression on tumor growth. Human colon cancer cell line HT29, HT29 expressing wild-type Csk (HT29/Csk) and HT29 expressing kinase-negative Csk (HT29/CskKN) were injected s.c. into nude mice. Tumor volume (cm3) obtained from four mice is plotted versus days after inoculation. Excised tumors are shown in lower panel. (B) Tumor incidence in wild-type and heterozygous for Csk (csk+/-) and p53 (p53+/-) after the sequential treatment of skin with dimethyl benzanthracene (DMBA) and phorbol myristate acetate (TPA).
Perspectives
The function of Csk as a regulator of SFKs has been almost completely established, but the question of whether or not Csk has additional functions still remains to be addressed. The greater question of why Csk and SFKs are not mutated in human diseases should also be addressed. More comprehensive search for these mutations in human diseases would give a new clue for understanding the biological significance of the intimate relationship between SFKs and Csk.
Competing Interests
The author has declared that no competing interest exists.
References
1. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140-6
2. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355-65
3. Brown M, Cooper J. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121-49
4. Thomas S, Brugge J. Cellular Functions regulated by Src Family Kinases. Ann Rev Cell Biol. 1997;13:513-609
5. Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC. et al. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677-87
6. Bishop JM, Baker B, Fujita D, McCombe P, Sheiness D, Smith K. et al. Genesis of a virus-transforming gene. National Cancer Institute monograph. 1978:219-23
7. Hunter T. Protein phosphorylated by the RSV transforming function. Cell. 1980;22:647-8
8. Varmus HE, Cohen JC, Hughes SH, Majors J, Bishop JM. The origin and structure of endogenous retroviral DNA. Annals of the New York Academy of Sciences. 1980;354:379-83
9. Toyoshima K, Yoshida M, Kawai S, Kitamura N, Yamamoto T, Segawa K. Oncogene and its production of an avian sarcoma virus Y73. Princess Takamatsu Symp. 1982;12:275-83
10. Takeya T, Hanafusa H. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanism for generating the transforming virus. Cell. 1983;32:881-90
11. Hanafusa H, Halpern CC, Buchhagen DL, Kawai S. Recovery of avian sarcoma virus from tumors induced by transformation-defective mutants. J Exp Med. 1977;146:1735-47
12. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470-80
13. Frame M. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989-98
14. Oneyama C, Iino T, Saito K, Suzuki K, Ogawa A, Okada M. Transforming potential of Src family kinases is limited by the cholesterol-enriched membrane microdomain. Mol Cell Biol. 2009;29:6462-72
15. Courtneidge SA. Activation of the pp60c-src kinase by middle T antigen binding or by dephosphorylation. EMBO J. 1985;4:1471-7
16. Cooper JA, Gould KL, Cartwright CA, Hunter T. Tyr527 is phosphorylated in pp60c-src: implications for regulation. Science. 1986;231:1431-4
17. Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu Rev Cell Biol. 1987;3:31-56
18. Cooper JA, Howell B. The when and how of Src regulation. Cell. 1993;73:1051-4 doi:0092-8674(93)90634-3 [pii]
19. Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J. et al. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13:861-71
20. Xu W, Harrison S, Eck M. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595-602
21. Young MA, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105:115-26
22. Knighton DR, Xuong NH, Taylor SS, Sowadski JM. Crystallization studies of cAMP-dependent protein kinase. Cocrystals of the catalytic subunit with a 20 amino acid residue peptide inhibitor and MgATP diffract to 3.0 A resolution. J Mol Biol. 1991;220:217-20
23. Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG. et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767-78
24. Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649-55
25. Courtneidge SA, Dhand R, Pilat D, Twamley GM, Waterfield MD, Roussel MF. Activation of Src family kinases by colony stimulating factor-1, and their association with its receptor. EMBO J. 1993;12:943-50
26. Kypta RM, Goldberg Y, Ulug ET, Courtneidge SA. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell. 1990;62:481-92
27. Cheadle C, Ivashchenko Y, South V, Searfoss GH, French S, Howk R. et al. Identification of a Src SH3 domain binding motif by screening a random phage display library. J Biol Chem. 1994;269:24034-9
28. Seals DF, Azucena EF Jr, Pass I, Tesfay L, Gordon R, Woodrow M. et al. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155-65
29. Liu X, Marengere LE, Koch CA, Pawson T. The v-Src SH3 domain binds phosphatidylinositol 3'-kinase. Mol Cell Biol. 1993;13:5225-32
30. Kuroiwa M, Oneyama C, Nada S, Okada M. The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation. J Cell Sci. 2011;124:1726-38
31. Hakak Y, Martin GS. Ubiquitin-dependent degradation of active Src. Curr Biol. 1999;9:1039-42
32. Martinez R, Mathey-Prevot B, Bernards A, Baltimore D. Neuronal pp60c-src contains a six-amino acid insertion relative to its non-neuronal counterpart. Science. 1987;237:411-5
33. Sorge LK, Levy BT, Maness PF. pp60c-src is developmentally regulated in the neural retina. Cell. 1984;36:249-57
34. Okada M, Nakagawa H. Protein tyrosine kinase in rat brain: neonatal rat brain expresses two types of pp60c-src and a novel protein tyrosine kinase. J Biochem. 1988;104:297-305
35. Okada M, Nakagawa H. Identification of a novel protein tyrosine kinase that phosphorylates pp60c-src and regulates its activity in neonatal rat brain. Biochem Biophys Res Commun. 1988;154:796-802
36. Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem. 1991;266:24249-52
37. Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69-72
38. Bergman M, Mustelin T, Oetken C, Partanen J, Flint NA, Amrein KE. et al. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 1992;11:2919-24
39. Sabe H, Knudsen B, Okada M, Nada S, Nakagawa H, Hanafusa H. Molecular cloning and expression of chicken C-terminal Src kinase: lack of stable association with c-Src protein. Proc Natl Acad Sci U S A. 1992;89:2190-4
40. Sabe H, Okada M, Nakagawa H, Hanafusa H. Activation of c-Src in cells bearing v-Crk and its suppression by Csk. Mol Cell Biol. 1992;12:4706-13
41. Nada S, Yagi T, Takeda H, Tokunaga T, Nakagawa H, Ikawa Y. et al. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 1993;73:1125-35
42. Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117-24
43. Thomas RM, Schmedt C, Novelli M, Choi BK, Skok J, Tarakhovsky A. et al. C-terminal SRC kinase controls acute inflammation and granulocyte adhesion. Immunity. 2004;20:181-91
44. Schmedt C, Saijo K, Niidome T, Kuhn R, Aizawa S, Tarakhovsky A. Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature. 1998;394:901-4
45. Yagi R, Waguri S, Sumikawa Y, Nada S, Oneyama C, Itami S. et al. C-terminal Src kinase controls development and maintenance of mouse squamous epithelia. EMBO J. 2007;26:1234-44
46. Nada S, Okada M, Aizawa S, Nakagawa H. Identification of major tyrosine-phosphorylated proteins in Csk-deficient cells. Oncogene. 1994;9:3571-8
47. Read RD, Bach EA, Cagan RL. Drosophila C-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways. Mol Cell Biol. 2004;24:6676-89
48. Takata N, Itoh B, Misaki K, Hirose T, Yonemura S, Okada M. Non-receptor tyrosine kinase CSK-1 controls pharyngeal muscle organization in Caenorhabditis elegans. Genes Cells. 2009;14:381-93
49. Segawa Y, Suga H, Iwabe N, Oneyama C, Akagi T, Miyata T. et al. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc Natl Acad Sci U S A. 2006;103:12021-6
50. Bennett BD, Cowley S, Jiang S, London R, Deng B, Grabarek J. et al. Identification and characterization of a novel tyrosine kinase from megakaryocytes. J Biol Chem. 1994;269:1068-74
51. Sakano S, Iwama A, Inazawa J, Ariyama T, Ohno M, Suda T. Molecular cloning of a novel non-receptor tyrosine kinase, HYL (hematopoietic consensus tyrosine-lacking kinase). Oncogene. 1994;9:1155-61
52. Klages S, Adam D, Class K, Fargnoli J, Bolen JB, Penhallow RC. Ctk: a protein-tyrosine kinase related to Csk that defines an enzyme family. Proc Natl Acad Sci U S A. 1994;91:2597-601
53. McVicar DW, Lal BK, Lloyd A, Kawamura M, Chen YQ, Zhang X. et al. Molecular cloning of lsk, a carboxyl-terminal src kinase (csk) related gene, expressed in leukocytes. Oncogene. 1994;9:2037-44
54. Hamaguchi I, Iwama A, Yamaguchi N, Sakano S, Matsuda Y, Suda T. Characterization of mouse non-receptor tyrosine kinase gene, HYL. Oncogene. 1994;9:3371-4
55. Grgurevich S, Linnekin D, Musso T, Zhang X, Modi W, Varesio L. et al. The Csk-like proteins Lsk, Hyl, and Matk represent the same Csk homologous kinase (Chk) and are regulated by stem cell factor in the megakaryoblastic cell line MO7e. Growth Factors. 1997;14:103-15
56. Hamaguchi I, Yamaguchi N, Suda J, Iwama A, Hirao A, Hashiyama M. et al. Analysis of CSK homologous kinase (CHK/HYL) in hematopoiesis by utilizing gene knockout mice. Biochem Biophys Res Commun. 1996;224:172-9
57. Lee S, Ayrapetov MK, Kemble DJ, Parang K, Sun G. Docking-based substrate recognition by the catalytic domain of a protein tyrosine kinase, C-terminal Src kinase (Csk). J Biol Chem. 2006;281:8183-9
58. Lee S, Lin X, Nam NH, Parang K, Sun G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Natl Acad Sci U S A. 2003;100:14707-12
59. Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134:124-34
60. Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc Natl Acad Sci U S A. 1994;91:3984-8
61. D'Arco M, Giniatullin R, Leone V, Carloni P, Birsa N, Nair A. et al. The C-terminal Src inhibitory kinase (Csk)-mediated tyrosine phosphorylation is a novel molecular mechanism to limit P2X3 receptor function in mouse sensory neurons. J Biol Chem. 2009;284:21393-401
62. Zhu F, Choi BY, Ma WY, Zhao Z, Zhang Y, Cho YY. et al. COOH-terminal Src kinase-mediated c-Jun phosphorylation promotes c-Jun degradation and inhibits cell transformation. Cancer Res. 2006;66:5729-36
63. Stewart RA, Li DM, Huang H, Xu T. A genetic screen for modifiers of the lats tumor suppressor gene identifies C-terminal Src kinase as a regulator of cell proliferation in Drosophila. Oncogene. 2003;22:6436-44
64. Roskoski R Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1-14
65. Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature. 1992;359:336-9
66. Granot-Attas S, Elson A. Protein tyrosine phosphatase epsilon activates Yes and Fyn in Neu-induced mammary tumor cells. Exp Cell Res. 2004;294:236-43
67. Fang KS, Sabe H, Saito H, Hanafusa H. Comparative study of three protein-tyrosine phosphatases. Chicken protein-tyrosine phosphatase lambda dephosphorylates c-Src tyrosine 527. J Biol Chem. 1994;269:20194-200
68. Mustelin T, Altman A. Dephosphorylation and activation of the T cell tyrosine kinase pp56lck by the leukocyte common antigen (CD45). Oncogene. 1990;5:809-13
69. Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439-46
70. Somani AK, Bignon JS, Mills GB, Siminovitch KA, Branch DR. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. 1997;272:21113-9
71. Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T. et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Molecular cell. 2004;13:341-55
72. Ogawa A, Takayama Y, Sakai H, Chong KT, Takeuchi S, Nakagawa A. et al. Structure of the carboxyl-terminal Src kinase, Csk. J Biol Chem. 2002;277:14351-4
73. Wong L, Lieser SA, Miyashita O, Miller M, Tasken K, Onuchic JN. et al. Coupled motions in the SH2 and kinase domains of Csk control Src phosphorylation. J Mol Biol. 2005;351:131-43
74. Cao H, Courchesne WE, Mastick CC. A phosphotyrosine-dependent protein interaction screen reveals a role for phosphorylation of caveolin-1 on tyrosine 14: recruitment of C-terminal Src kinase. J Biol Chem. 2002;277:8771-4
75. Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc Natl Acad Sci U S A. 1994;91:3984-8
76. Zhou J, Scholes J, Hsieh JT. Characterization of a novel negative regulator (DOC-2/DAB2) of c-Src in normal prostatic epithelium and cancer. J Biol Chem. 2003;278:6936-41
77. Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J. 2005;24:1686-95
78. Arbet-Engels C, Tartare-Deckert S, Eckhart W. C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J Biol Chem. 1999;274:5422-8
79. Tobe K, Sabe H, Yamamoto T, Yamauchi T, Asai S, Kaburagi Y. et al. Csk enhances insulin-stimulated dephosphorylation of focal adhesion proteins. Mol Cell Biol. 1996;16:4765-72
80. Horejsí V. Transmembrane adaptor proteins in membrane microdomains: important regulators of immunoreceptor signaling. Immunol Lett. 2004;92:43-9
81. Brdicková N, Brdicka T, Angelisová P, Horváth O, Spicka J, Hilgert I. et al. LIME: a new membrane Raft-associated adaptor protein involved in CD4 and CD8 coreceptor signaling. J Exp Med. 2003;198:1453-62
82. Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K. et al. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 2000;404:999-1003
83. Brdicka T, Pavlistova D, Leo A, Bruyns E, Korinek V, Angelisova P. et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J Exp Med. 2000;191:1591-604
84. Oneyama C, Hikita T, Enya K, Dobenecker MW, Saito K, Nada S. et al. The lipid raft-anchored adaptor protein Cbp controls the oncogenic potential of c-Src. Molecular cell. 2008;30:426-36
85. Kanou T, Oneyama C, Kawahara K, Okimura A, Ohta M, Ikeda N. et al. The transmembrane adaptor Cbp/PAG1 controls the malignant potential of human non-small cell lung cancers that have c-src upregulation. Mol Cancer Res. 2010;9:103-14
86. Suzuki K, Oneyama C, Kimura H, Tajima S, Okada M. Down-regulation of the tumor suppressor C-terminal Src kinase (Csk)-binding protein (Cbp)/PAG1 is mediated by epigenetic histone modifications via the mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K) pathway. J Biol Chem. 2011;286:15698-706
87. Howell BW, Cooper JA. Csk suppression of Src involves movement of Csk to sites of Src activity. Mol Cell Biol. 1994;14:5402-11
88. Takeuchi M, Kuramochi S, Fusaki N, Nada S, Kawamura-Tsuzuku J, Matsuda S. et al. Functional and physical interaction of protein-tyrosine kinases Fyn and Csk in the T-cell signaling system. J Biol Chem. 1993;268:27413-9
89. Cloutier JF, Veillette A. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. 1996;15:4909-18
90. Veillette A, Rhee I, Souza CM, Davidson D. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol Rev. 2009;228:312-24
91. Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R. et al. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol. 2012;8:437-46
92. Zhong MC, Veillette A. Immunology: Csk keeps LYP on a leash. Nat Chem Biol. 2012;8:412-3
93. Mills JE, Whitford PC, Shaffer J, Onuchic JN, Adams JA, Jennings PA. A novel disulfide bond in the SH2 Domain of the C-terminal Src kinase controls catalytic activity. J Mol Biol. 2007;365:1460-8
94. Yaqub S, Abrahamsen H, Zimmerman B, Kholod N, Torgersen KM, Mustelin T. et al. Activation of C-terminal Src kinase (Csk) by phosphorylation at serine-364 depends on the Csk-Src homology 3 domain. Biochem J. 2003;372:271-8
95. Inomata M, Takayama Y, Kiyama H, Nada S, Okada M, Nakagawa H. Regulation of Src family kinases in the developing rat brain: correlation with their regulator kinase, Csk. J Biochem. 1994;116:386-92
96. Masaki T, Okada M, Tokuda M, Shiratori Y, Hatase O, Shirai M. et al. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology. 1999;29:379-84
97. Watanabe N, Matsuda S, Kuramochi S, Tsuzuku J, Yamamoto T, Endo K. Expression of C-terminal src kinase in human colorectal cancer cell lines. Jpn J Clin Oncol. 1995;25:5-9
Author contact
![]() Corresponding author: TEL: 81-6-6879-8297 FAX: 81-6-6879-8298, E-mail: okadamosaka-u.ac.jp.
Corresponding author: TEL: 81-6-6879-8297 FAX: 81-6-6879-8298, E-mail: okadamosaka-u.ac.jp.