Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(10):1398-1407. doi:10.7150/ijbs.5079 This issue Cite
Research Paper
TRAF Family Member-Associated NF-κB Activator (TANK) Induced by RANKL Negatively Regulates Osteoclasts Survival and Function
Mengrui Wu1,2, Yiping Wang1,2, Lianfu Deng3, Wei Chen1 ![]() , Yi-Ping Li1,2
, Yi-Ping Li1,2 ![]()
1. Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA.
2. Institute of Genetics, Life Science College, Zhejiang University, 388 Yuhang Road, Hangzhou 310058, People's Republic of China.
3. Shanghai Key laboratory, Shanghai Institute of Trauma and Orthopaedics, Ruijin Hospital, School of Medicine, Shanghai Jiao-Tong University, 197 Rui Jin Er Road, Shanghai 200025, P.R.China.
Received 2012-8-20; Accepted 2012-10-18; Published 2012-11-6
Abstract
Osteoclasts are the principle bone-resorbing cells. Precise control of balanced osteoclast activity is indispensable for bone homeostasis. Osteoclast activation mediated by RANK-TRAF6 axis has been clearly identified. However, a negative regulation-machinery in osteoclast remains unclear. TRAF family member-associated NF-κB activator (TANK) is induced by about 10 folds during osteoclastogenesis, according to a genome-wide analysis of gene expression before and after osteoclast maturation, and confirmed by western blot and quantitative RT-PCR. Bone marrow macrophages (BMMs) transduced with lentivirus carrying tank-shRNA were induced to form osteoclast in the presence of RANKL and M-CSF. Tank expression was downregulated by 90% by Tank-shRNA, which is confirmed by western blot. Compared with wild-type (WT) cells, osteoclastogenesis of Tank-silenced BMMs was increased, according to tartrate-resistant acid phosphatase (TRAP) stain on day 5 and day 7. Number of bone resorption pits by Tank-silenced osteoclasts was increased by 176% compared with WT cells, as shown by wheat germ agglutinin (WGA) stain and scanning electronic microscope (SEM) analysis. Survival rate of Tank-silenced mature osteoclast is also increased. However, acid production of Tank-knockdown cells was not changed compared with control cells. IκBα phosphorylation is increased in tank-silenced cells, indicating that TANK may negatively regulate NF-κB activity in osteoclast. In conclusion, Tank, whose expression is increased during osteoclastogenesis, inhibits osteoclast formation, activity and survival, by regulating NF-κB activity and c-FLIP expression. Tank enrolls itself in a negative feedback loop in bone resorption. These results may provide means for therapeutic intervention in diseases of excessive bone resorption.
Keywords: TANK, RANKL, NF-κB, Osteoclast.
INTRODUCTION
Osteoclasts are large, multinucleated, TRAP-positive cells which are derived from the monocyte/macrophage lineage[1]. Receptor activator of NF-κB (RANKL) and macrophage-colony stimulating factor (M-CSF), mainly secreted by osteoblasts, are the two major cytokines that drive osteoclastogenesis. Binding with their receptors, RANK and c-Fms, in the membrane of pre-osteoclast, RANKL and M-CSF[2] induces the activation of transcription factors (eg. c-Fos[3], NF-κB[4], Nfatc1[5, 6]) and the expression of osteoclast specific genes (eg. TRAP, calcitonin R[7], DC-STAMP[8]).
There are two pathways for NF-κB signaling: canonical and non-canonical. In the canonical NF-κB pathway, activation of RelA/p65-p50 heterodimer requires the phosphorylation and degaration of IkB by IKK complex. p50 is generated from NF-κB1/p105 fragments. In the noncanonical NF-kB pathway, Nf-kB2/p100 is processed into p52, which dimerizes with RelB to form active complex. An essential role of NF-κB in osteoclast formation was discovered unexpectedly when NF-κB1 and NF-κB2 double-knockout mice were generated, displaying severe osteopetrosis due to the defective osteoclasts [9, 10]. This confirms the importance of NF-κB signaling in controlling bone homeostasis. NF-κB has also been implicated in the induction of c-fos and NFATc1[11].
RANKL, a major NF-κB activator in osteoclastogenesis, binds to its receptor RANK, a member of the tumor necrosis factor (TNF) receptor superfamily. After activation, RANK recruit TRAF family E3 ubiquitin ligases, such as TRAF2, TRAF5, and TRAF6 [12]. TRAF6, in particular, has a vital role in the RANK signaling to NF-κB. TRAF6 Deficiency results in severe osteopetrosis similar to that observed in RANKL- and RANK-deficient mice[13-15]. Overall, it has been suggested that RANKL-RANK interactions and the TRAF6 signaling that follows are important in osteoclast differentiation.
TNF receptor associated factors (TRAF) family member-associated NF-κB activator (TANK)has been shown to bind with TRAF1, TRAF2, TRAF3, TRAF5, and TRAF6 [16-19], to act as a regulator for TRAF-mediated signaling. Controversial roles of tank in regulating NF-κB signaling have been reported in a number of papers. TANK has been described as an NF-κB activator, by interacting with IKK complex[20]. TANK has also been described as an NF-κB inhibitor by competitively interacting with TRAF2[21]. TANK deficient mice develop autoimmune nephritis, with hyperactive NF-κB activity observed in B cells and macrophages[22]. In TANK-/-B cells and macrophages, TRAF6 ubiquitination and activation induced by TLRs is increased compared with WT cells [22]. Because of this controversy, more study must be done to further our understanding of the role that TANK plays in NF-κB signaling.
In this study, we examined the effect of Tank on osteoclasts, by silencing tank expression in pre-osteoclast, using lentivirus carrying Tank-shRNA. We found that TANK knockdown promoted osteoclast differentiation, activation and survival, while having no effect on osteoclast acidification.
MATERIALS AND METHODS
Cell culture and In vitro Analysis of osteoclastogenesis. In vitro osteoclast differentiation and analysis were performed as previously described[23, 24]. Isolated BMMs from C57BL/6 mice were plated at a density of 1X105cells/cm2, and cultured in α-MEM (Gibco) (pH6.9) supplemented with 10% FBS, 100u/ml penicillin-100ug/ml streptomycin, 10ng/ml recombinant RANKL (R&D) and 10ng/ml recombinant M-CSF (R&D) for 5 days or 7 days. Mature osteoclasts were then characterized by staining for TRACP activity using a commercial kit (sigma).
GeneChip analysis. Human osteoclasts were obtained as described[25]. Human peripheral blood mononuclear cells (PBMCs) were cultured for 7 days with human RANKL and human M-CSF. GeneChip data were analyzed using Affymetrics scanner and accompanying gene expression software as described[26, 27]. Three individual samples were used.
Lentivirus production and transduction. Five PLKO.1 vectors encoding short hairpin RNAs (shRNAs) targeting the mRNA of mouse Tank (NM_011529) were purchased from sigma-aldrich, with the sense strand insert sequences of 5'-CCGGCGTACAGAGAATAACAGACAACTCGAGTTGTCTGTTATTCTCTGTACGTTTTTG -3', 5'-CCGGCGGCATCTTAATACACACTTTCTCGAGAAAGTGTGTATTAAGATGCCGTTTTTG -3', 5'- CCGGCCCAGGCTAAAGATGATATAACTCGAGTTATATCATCTTTAGCCTGGGTTTTTG -3' (referred as 'shRNA-1'), 5'-CCGGCCATCCTTTATAGTGATGCTACTCGAGTAGCATCACTATAAAGGATGGTTTTTG -3', 5'-CCGGGCATCACGAAAGGGATAATATCTCGAGATATTATCCCTTTCGTGATGCTTTTTG-3' (referred as 'shRNA-2'). A PLKO.1 vector encoding scrambled shRNA sequence was also purchased as negative control (referred as 'control' in the paper). Lentivirus was produced and transduced as previously described[26, 27]. Lentivirus was produced by co-transfecting three plasmids, PLKO.1, dr8.9 and vsvg, together into HEK-293T cells and harvesting supernatants 48-56hrs after transfection. BMMs were transduced with lentivirus supertanant in the presence of 8ug/ml polybrene (sigma) for 24hrs, on D0 (the first day when BMMs were cultured in medium with both RANKL and M-CSF). Cells were harvested 72 hrs after transfection for tank expression analysis.
Bone resorption pit assays and scanning electron microscopy. Bone resorption pit was analyzed by WGA-lectin stain as previously described[28]. A consistent number of BMM cells were cultured on bovine cortical bone slices in 24-well plates (2X105cells/well). The bone slices were harvested after 6 days. Cells adhering to the bone slices were subsequently removed by sonication in PBS. The slices were then incubated with 20 mg/ml peroxidase-conjugated WGA-lectin (Sigma) for 30-60 min at room temperature and then with DAB Peroxidase Substrate Kit (vector laboratories Inc, SK-4100). Resorption pits will be stained brown. Pictures of resorption pits on bone slides were also taken by a Philips 515 SEM (Department of Materials Science and Engineering, UAB). The assays were performed in triplicate and a representative view from each assay shown. The data were quantified by measuring the percentage of the areas resorbed in three random resorption sites, as determined using the ImageJ analysis software.
Immunofluorescence stain and confocal microscopy. We performed immunofluorescence analysis as outlined previously[23]. Primary antibodies, goat-anti-TANK (C-20) (sc-1997) (1:200) and mouse-anti-Cathepsin K (E-7) antibody (sc-48353) (1:200), were purchased from santa cruz. Secondary antibodies, FITC donkey-anti-goat antibody was purchased from Jackson ImmunoResearch and TR-goat-anti-mouse antibody was purchased from santa cruz Inc. Data was documented using epifluorescence on a Zeiss Axioplan microscope in the Developmental Neurobiology Imaging and Tissue Processing Core at the UAB Intellectual and Disabilities Research Center. Nuclei were visualized with 1 μg/ml DAPI (4',6-diamidino-2-phenylindole; Sigma). The experiments were performed in triplicate.
Immunohistochemistry stain (IHC). Paraffin sections were prepared from decalcified one-month old mice femur and were used for IHC stain. IHC analysis was performed as outlined previously[23]. Primary antibodies, goat-anti-TANK (C-20) (sc-1997) (1:400) were purchased from santa cruz. VECTASTAIN Elite ABC Kit (Goat IgG) (Vector Labs Inc) and DAB Peroxidase Substrate Kit (SK-4100, Vector Labs Inc) were applied to detect primary antibodies. Then the sections were conterstained by fast green.
Western blotting analysis. Western blotting, performed as previously outlined[24, 29], was visualized and quantified using a Fluor-S Multi-Imager with Multi-Analyst software (Bio-Rad). Goat-anti-TANK (C-20) (sc-1997) and mouse-anti-β-actin (C-4) (sc-47778) anbibodies were purchased from santa cruz Inc. Rabbit-anti-p-IκB (2859) and rabbit-anti-IκB (4812) were purchased from cellsignaling Inc. Horseradish peroxidase (HRP)-goat-anti-mouse and HRP-goat-anti-rabbit antibodies were purchased from Jackson ImmunoResearch.
Quantitative RT-PCR (qRT-PCR). mRNA was prepared as described[27]. Quantitative PCR (qRT-PCR) was performed with Sybr Green reagents (invitrogen, 4309155) and using Step One realtime PCR system (A&B), according to the manufactures instructions. Primers used for PCR were as followings, Bcl-xl 5'-AGGCTGGCGATGAGTTTGAA-3', 5'-CGGCTCTCGGCTGCTGCATT-3'. Cellular FLICE-like inhibitory protein (c-flip) 5'-GCTGCTGTGGTTCTGAACATG-3', 5'-CTTTGACTGTCACGGTATTCCAC-3', IAP-1 5'-GCTTGCAAGTGCTGGATT-3', 5'-CAAGAAGATGAGGATATCTAGCT-3'.
Statistical analysis and data quantification analysis. Experimental data are reported as mean ± SD of triplicate independent samples. Data were analyzed with the two-tailed Student's t-test. P values <0.05 were considered significant. Data quantification analyses were performed using the NIH ImageJ Program as described [23, 24].
TUNEL Stain. TUNEL staining was performed using an in situ cell death detection kit, AP (Roche) according to the manufacturer's instructions. The kit catalyzes an immune complex conjugated with alkaline phosphatase (AP) to the end of single strand DNA breaks. AP activity was detected using a Leukocyte Alkaline Phosphatase Kit (sigma) according to the manufacturer's instructions.
RESULTS
Expression of TANK is induced by RANKL
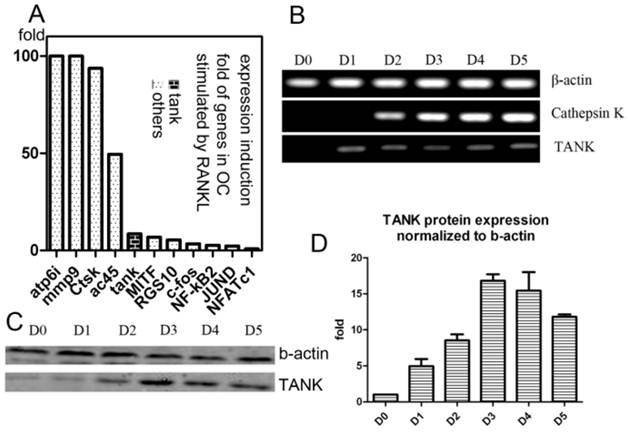
Our lab performed GeneChip to look for genes whose expression were induced by RANKL during osteoclastogenesis, as previously described[26, 27]. We found expression of Tank is induced 10 folds by RANKL, which is significant, compared with other well-known osteoclastogenesis relative signaling proteins, including c-fos, JunD, Nf-kb2, MITF, RGS10 and NFAT2 (Fig. 1A). Then we confirmed Tank expression profile in osteoclast by western blot and semi-quantitative RT-PCR. We performed semi-quantitative RT-PCR using mRNA from cells derived from BMMs under RANKL induction harvested on different time points (Day/D0, D1, D2, D3, D4, D5) (Fig. 1B). We also performed western blot using cells derived from BMMs under RANKL induction harvested on D0-5 (Fig. 1C) and quantified the results (Fig. 1D). TANK protein expression in mature osteoclasts is ~12 folds higher than that in BMMs.
Expression of TANK is induced by RANKL. (A) Induction folds of expression level of TANK and other osteoclast marker genes by RANKL, based on GeneChip data. (B) Time course analysis of TANK transcription level by semi-quantitative RT-PCR in RANKL and M-CSF stimulated bone marrow macrophages (BMMs) on Day(D)0, D1, D2, D3, D4 and D5. (C) Western blot analysis of time-dependent TANK protein expression level in RANKL and M-CSF stimulated bone marrow macrophages (BMMs) on Day(D)0, D1, D2, D3, D4 and D5. (D) Quantification of western blot results in B. Data is expressed as mean ± sd (n=5).
Expression of TANK in tissues
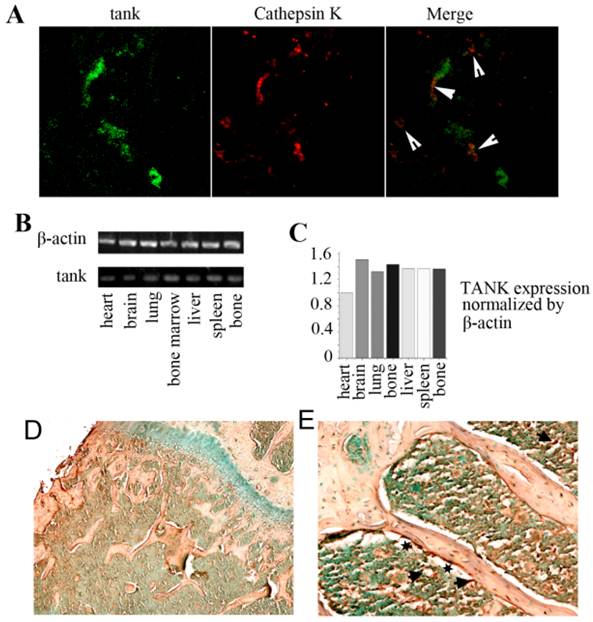
In order to examine the expression of TANK in vitro, we used immunofluorescence staining, IHC stain and RT-PCR analysis. We took frozen femur sections from 10-day-old mice and tested TANK and Cathepsin K expression using immunofluorescence staining (Fig. 2A). Tank (green) and Cathepsin K (red) were partially merged, implicating that tank expression can be detected in osteoclasts in vivo. We then analyzed TANK transcription levels in heart, brain, lung, bone marrow, liver, spleen, and bone tissues using semi-quantitative RT-PCR (Fig. 2B) and quantified the results (Fig. 2C). IHC stain shows that tank is expressing in both bone and bone marrow (marked by arrow), but not in growth plates (Fig. 2D&E). Meanwhile, it seems that higher Tank expression can be found on the surface of trabecular bone (marked by star) (Fig. 2E). Generally, Tank is ubiquitously expressing, including bone marrow.
Expression of TANK in vivo. (A) Immunofluorescence staining of frozen femur sections from 10 day old mice shows that tank expression (green) and Cathepsin K expression (red) are partially merged. (B) Analysis of tank transcription levels in different tissues by semi-quantitative RT-PCR. (C) Quantification of RT-PCR results in B. (D,E) Immunohistochemistry staining of paraffin sections from femur of 1 month old mice shows TANK is expressed in both bone (marked by arrow) and bone marrow (arrow). And high expression can be detected on the surface of trabecular bone (marked by star).
shRNA-1 efficiently depletes Tank expression and promoted osteoclast differentiation by promoting NF-κB activity
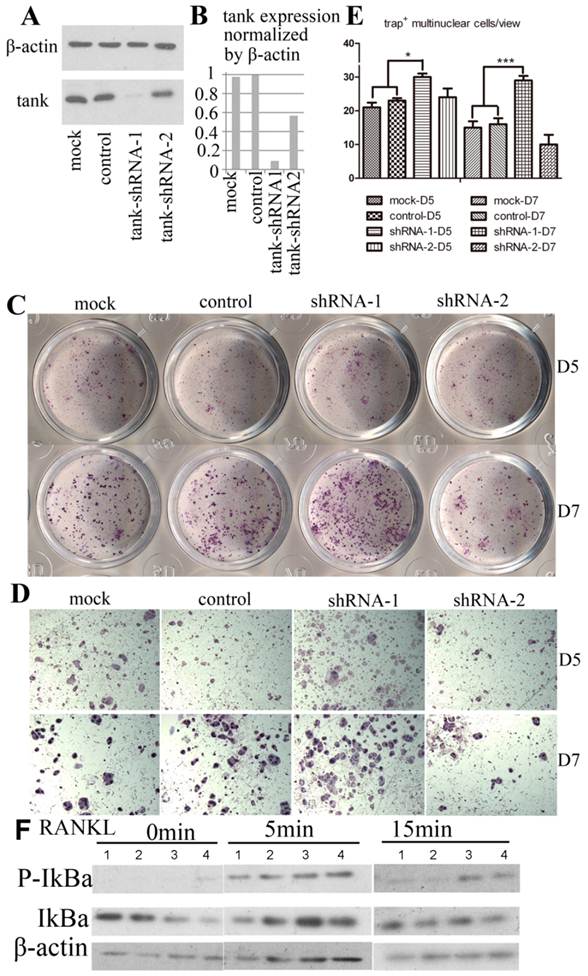
We examine the effect of Tank in osteoclast by loss-of-function experiment. 5 lentivirus encoding shRNA targeting Tank mRNA were produced and transduced into pre-osteoclasts on D1. Western blot analysis was performed with lyses from cells 48hrs post-transfection. We found that Tank expression was reduced by 90% in shRNA-1 group (BMMs which were transduced with lentivirus carrying shRNA-1), 50% in shRNA-2 group (BMMs which were transduced with lentivirus carrying shRNA-1), compared with control group (BMMs which were transduced with lentivirus carrying control shRNA) and mock group (BMMs which were not transduced with lentivirus) (Fig. 3A & B). Mature osteoclasts were stained for TRACP activity on D5 (Fig. 3C,D upper row) and D7 (Fig. 3C,D bottom row). And TRAP+ multi-nuclei cells (≥3 nuclei) number per random view from each of the four groups on days 5 and 7 were quantified (Fig. 3E). We found that TRAP+ multi-nuclei cells were increased to 1.3 folds on D5 and to 1.8 folds on D7 in shRNA-1 group, compared with mock group, implicating promoted osteoclastogenesis after depletion the expression of Tank. NF-κB signaling is essential for osteoclast differentiation [30-32], while the role of Tank in NF-κB signaling is controversial as reported[16, 17, 20, 22]. Thus, we are especially interested in the role of Tank in NF-κB activation in osteoclasts. In the canonical NF-κB pathway, NF-κB activation depends on IκBα phosphorylation and degradation. In our results, IκBα phosphorylation sustained longer in tank depleted cells compared with mock and WT cells (Fig. 3F).
TANK knockdown in BMM promotes osteoclast differentiation. (A) Western blot analysis of BMMs (mock), BMMs infected with lentivirus carrying scrambled shRNA (control), BMMs which were infected by lentivirus carrying shRNA targeting tank expression (tank-shRNA-1 and tank-shRNA-2) and treated with M-CSF to confirm the knockdown of TANK. (B) Quantification of TANK protein expression level in osteoclasts with and without viral infection. (C, D) TRAP staining of mature osteoclasts from the mock group, the control-shRNA group, the tank-shRNA-1 group, and the tank-shRNA-2 group on Day 5 (upper row) and on Day 7 (bottom row). (E) Quantification of the number of TRAP+ multiple nucleic (≥3 nuclei) cells per random view on days 5 and 10. (F) Detection of IκBα phosphorylation in BMM induced by RANKL by western blot. 1, mock; 2, control; 3, shRNA-1; 4, shRNA-2. Data is expressed as mean ± sd (n=5).
TANK-depleted osteoclasts have normal acidification but promoted bone resorption
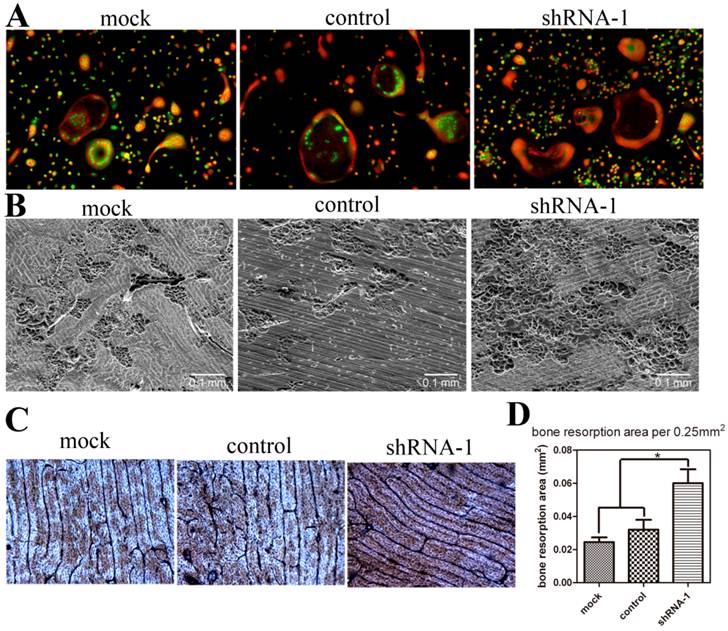
To detect the effect of TANK depletion on osteoclast function, we performed acridine orange stain and bone resorption pit assay with mature osteoclasts. Acridine orange stain shows that acidification doesn't vary between mock, control and tank-knockdown groups (Fig. 4A). Osteoclasts from shRNA-1, mock and control group were cultured on cattle bone slides and the bone slices were subjected to SEM detection (Fig. 4B) and WGA stain (Fig. 4C). Both results show more bone resorption pits in shRNA-1 group. Based on the quantification of the SEM results (Fig. 4D), bone resorption pits were increased to about two folds(p≤0.05) in the tank-shRNA-1 group. These results indicate that TANK knockdown has no effect on osteoclast acidification, but that it promotes bone resorption.
TANK knockdown in BMM doesn't influence osteoclast acidification but promotes bone resorption. (A) Acridine orange staining of osteoclasts from mock group, control-shRNA group, and tank-shRNA-1 group. (B, C) Osteoclasts from mock, control-shRNA, and tank-shRNA-1 groups were cultured on cattle bone slides and analyzed for bone resorption activity through scanning electron microscopy (SEM) (B) and wheat germ agglutinin (WGA) staining (C). (D) Quantification of bone resorption area per view area in B. Data is expressed as mean ± sd (n=15). *p≤0.05.
TANK depletion promotes osteoclast survival through regulating c-FLIP expression
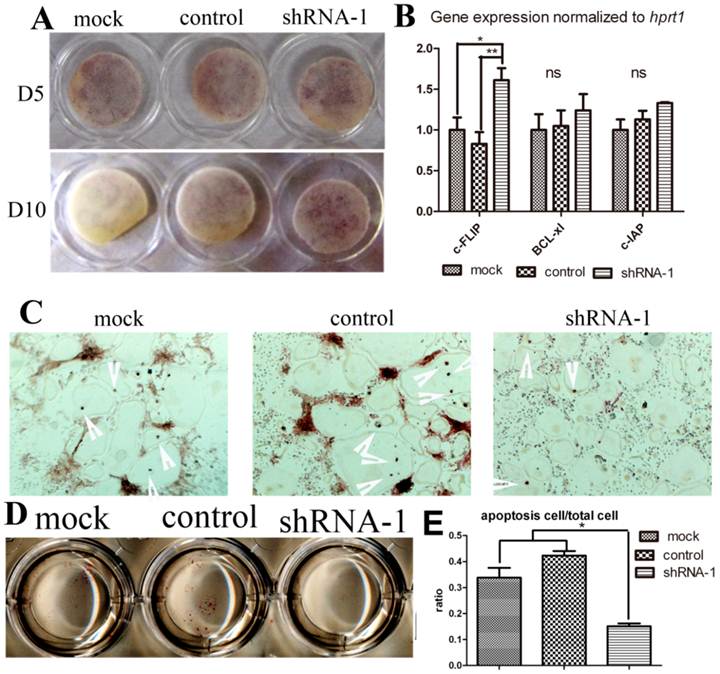
In order to determine whether TANK knock-down influences osteoclast survival, we elongated osteoclast culture to Day 10 and also performed Tunel stain. We continued osteoclast culture until day 10 without medium change after day 5. On day 10, lots of positive trap stain could still be observed in shRNA-1 group, while almost no stain in mock group. (Fig. 5A). This phenomenon hinted us that tank depletion may elongate osteoclast survival. To confirm the effect of Tank in osteoclast apoptosis, we deprived mature osteoclast from serum and cytokine for 8 hours, harvested mRNA for pro-sruvival genes expression detection (Fig. 5B) and examined cell apoptosis by Tunel stain (Fig. 5C & D). Three pro-survival genes (c-FLIP, Bc1-x1, and c-IAP) were examined. Tunel stain showed there was less osteoclast apoptosis observed in the tank-shRNA group when compared to the control-shRNA group, which indicates that TANK depletion inhibits osteoclast apoptosis (Fig. 5C, D & E).
TANK knock-down promotes osteoclast survival through promoting Nf-κB signaling. (A) TRAP staining of osteoclasts from the mock group, the control-shRNA group, and the tank-shRNA group on Day 5 and Day 10. (B) Mature osteoclasts from the mock group, the control-shRNA group, and the tank-shRNA group were cytokine and serum starved for 8hrs. mRNA was derived for analysis of pro-survival genes c-FLIP, Bc1-x1, and c-IAP expression by RT-PCR. (C-D) Mature osteoclasts from the mock group, the control-shRNA group, and the tank-shRNA group were cytokine and serum starved for 8hrs and apoptosis cells (red nuclei) were detected by TUNEL staining. (E) quantification of C&D. Data is expressed as mean ± sd (n=5). *p≤0.05.
DISCUSSION
Bone resorption by osteoclast and ossification by osteoblast are two processes that are responsible for the maintenance of bone homeostasis. Activation and inactivation of osteoclast must be kept in balance by delicate machineries so as to avoid pathological defects such as osteoporosis. In this study, we found that RANKL simultaneously induces osteoclastogenesis and upregulates TANK expression, which induces osteoclast apoptosis and inhibits osteoclast formation and bone resorption, through inhibiting NF-κB signaling and regulating c-FLIP expression, which potentially functions as a negative feedback loop.
Tank was characterized as one of our genes of interest firstly by GeneChip, in which Tank expression is increased 10 folds with RANKL induction. Then we confirmed Tank expression level during osteoclastogenesis by western blot and RT-PCR. We found that TANK expression levels increased over time (Fig. 1). We also found that Tank is expressing in bone marrow tissue by RT-PCR, and Tank expression partially overlapped with Ctsk, a mature osteoclast marker, by immunofluorescence stain (Fig. 2). Expression pattern of Tank in vivo and in vitro implicates the possibility that Tank may have a function in bone resorption.
We found that TANK depletion promotes osteoclast formation (Fig. 3) and bone resorption activity (Fig. 4). Knock-down of Tank promoted the formation of multinuclear osteoclast to 1.3 folds on D5 and to 1.8 folds on D7, and increased bone resorption area to 1.76 folds.
IκBα phosphorylation sustained longer in Tank shRNA-1 group (Fig. 3). In the NF-κB canonical pathway, IκBα is phosphorylated by IKK complex, ubiquitylated and degraded, so as to release and activate p50-p65 (NF-κB ) complex[35]. NF-κB signaling is essential for osteoclast differentiation. Thus, Tank may negatively regulate osteoclast differentiation by inhibiting NF-κB activity, through inhibiting IκB phosphorylation. Dual functions in regulating NF-κB activity by Tank have been identified[16, 17, 20, 22]. As a negative regulator, Tank has been reported to compete with TRAF2 for the interaction with receptor [21]. Tank has also been reported to inhibit the ubiuititylation of RANK, which is essential for signaling transduction of RANK-TRAF6 axis. Both may serve for the mechanism underlying Tank negatively regulates osteoclasts survival and function [22].
NF-κB signaling is also involved in osteoclast survival. We found that Tank depletion promotes osteoclast survival (Fig. 5). We detected the expression of several pro-survival geneBcl-xl[36], c-FLIP[37, 38] and c-IAP[36]. We found that c-FLIP expression is increased in Tank knock-down group (Fig. 5). c-FLIP is a major resistance factor and critical anti-apoptotic regulator that inhibits tumor necrosis factor-alpha (TNF-alpha), Fas-L, and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis as well as chemotherapy-triggered apoptosis in malignant cells. c-FLIP binds to FADD and/or caspase-8 or -10 in a ligand-dependent and-independent fashion, which in turn prevents death-inducing signaling complex (DISC) formation and subsequent activation of the caspase cascade[39]. Interestingly, c-FLIP expression has been reported to be regulated by Nf-κB[37, 38]. Tank may inhibit osteoclast survival by regulating c-FLIP expression, which may also be due to modified NF-κB activity.
We first reported the negative role of Tank in osteoclast function in ASBMR 2011 annual meeting[33]. Recently, Maruyama K. and colleagues also characterized Tank as a negative regulator of osteoclastogenesis using TANK-/- mice[34]. Severe trabecular bone loss is observed in TANK-/- mice due to increased bone resorption. Osteoclastogenesis is increased in TANK-/- BMMs and decreased in Tank over-expression BMMs[34]. The current manuscript not only confirmed some of these findings reported by Maruyama K et al, but also provided more new information regarding the role of TANK in osteoclast formation and resorption, as well as on NF-κB activation. We showed TANK expression in osteoclasts both in vitro and in vivo. We found more osteoclast remaining on bone slides after extended cultured for five days without medium change and Less apoptosis after cytokine deprivation in tank-deficient mature osteocalsts. We demonstrated that negatively regulated bone resorption without change of osteoclast medicated acidification. Most importantly, we revealed a role of Tank in osteoclast survival by Tunel assay and pro-survival genes expression analysis which was un-reported. These new information and new findings provided further insight into the mechanism underlying Tank negatively regulating bone resorption.
In summary, we found that TANK is ubiquitously expressed in a number of different tissues, including bone marrow. Tank is expressed in BMMs and the expression is induced by RANKL by ~12 times. We efficiently knocked down TANK by 90% (Fig. 3) using TANK-shRNA-1. Tank depletion doesn't affect osteoclast acid production, but promotes osteoclast differentiation, survival and bone resorption, perhaps by activating NF-κB activity and c-FLIP expression.
Abbreviations
RANK: Receptor Activator of NF-κB; RANKL: RANK Ligand; TRAP: Tartrate-Resistant Acid Phosphatase; NF-kB: Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells; TNF: Tumor Necrosis Factor; TRAF: TNF Receptor Associated Factor; TANK: TRAF Family Member-Associated NF-κB Activator; TLF: Toll-Like Receptor; AP-1: Activator Protein-1; BCR: B-Cell Receptor; BMM: Bone Marrow-Derived Macrophage; qRT-PCR: Real-Time Quantitative PCR; ALP: Alkaline Phosphatase; SEM: Scanning Electron Microscopy; WGA: Wheat Germ Agglutinin; TUNEL: Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling.
Acknowledgements
We thank Mr. Zach Nolen for their excellent assistance with the manuscript. This work was supported by NIH grants AR-055307 (Y.P.L.) and R01-AR-44741(Y.P.L.).
Competing Interests
The authors declare no competing financial interests.
References
1. Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389-406
2. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
3. Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA. et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443-8
4. Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD. et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482-96
5. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901
6. Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y. et al. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277:41147-56
7. Hattersley G, Chambers TJ. Calcitonin receptors as markers for osteoclastic differentiation: correlation between generation of bone-resorptive cells and cells that express calcitonin receptors in mouse bone marrow cultures. Endocrinology. 1989;125:1606-12
8. Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K. et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941-6 doi:10.1084/jem.20040518 [doi] jem.20040518 [pii]
9. Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285-9
10. Soysa NS, Alles N. NF-kappaB functions in osteoclasts. Biochem Biophys Res Commun. 2009;378:1-5
11. Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM. et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282:18245-53
12. Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N. et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345-51
13. Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A. et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015-24
14. Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K. et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353-62
15. Kobayashi T, Walsh PT, Walsh MC, Speirs KM, Chiffoleau E, King CG. et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003;19:353-63
16. Cheng G, Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-kappaB activation. Genes Dev. 1996;10:963-73
17. Chin AI, Shu J, Shan SC, Yao Z, Kehrl JH, Cheng G. TANK potentiates tumor necrosis factor receptor-associated factor-mediated c-Jun N-terminal kinase/stress-activated protein kinase activation through the germinal center kinase pathway. Mol Cell Biol. 1999;19:6665-72
18. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694-704
19. Rothe M, Xiong J, Shu HB, Williamson K, Goddard A, Goeddel DV. I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc Natl Acad Sci USA. 1996;93:8241-6
20. Chariot A, Leonardi A, Muller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem. 2002;277:37029-36
21. Kaye KM, Devergne O, Harada JN, Izumi KM, Yalamanchili R, Kieff E. et al. Tumor necrosis factor receptor associated factor 2 is a mediator of NF-kappa B activation by latent infection membrane protein 1, the Epstein-Barr virus transforming protein. Proc Natl Acad Sci USA. 1996;93:11085-90
22. Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T. et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol. 2009;10:965-72
23. Chen W, Yang S, Abe Y, Li M, Wang Y, Shao J. et al. Novel pycnodysostosis mouse model uncovers cathepsin K function as a potential regulator of osteoclast apoptosis and senescence. Hum Mol Genet. 2007;16:410-23
24. Yang S, Li YP. RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev. 2007;21:1803-16
25. Matsuzaki K, Udagawa N, Takahashi N, Yamaguchi K, Yasuda H, Shima N. et al. Osteoclast differentiation factor (ODF) induces osteoclast-like cell formation in human peripheral blood mononuclear cell cultures. Biochemical and biophysical research communications. 1998;246:199-204
26. Feng S, Deng L, Chen W, Shao J, Xu G, Li YP. Atp6v1c1 is an essential component of the osteoclast proton pump and in F-actin ring formation in osteoclasts. Biochem J. 2009;417:195-203 doi:BJ20081073 [pii] 10.1042/BJ20081073
27. Wu H, Xu G, Li YP. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J Bone Miner Res. 2009;24:871-85
28. Ye S, Fowler TW, Pavlos NJ, Ng PY, Liang K, Feng Y. et al. LIS1 Regulates Osteoclast Formation and Function through Its Interactions with Dynein/Dynactin and Plekhm1. PLoS ONE. 2011;6:e27285
29. Yang S, Wei D, Wang D, Phimphilai M, Krebsbach PH, Franceschi RT. In vitro and in vivo synergistic interactions between the Runx2/Cbfa1 transcription factor and bone morphogenetic protein-2 in stimulating osteoblast differentiation. J Bone Miner Res. 2003;18:705-15
30. Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118:2088-97
31. Ozaki K, Takeda H, Iwahashi H, Kitano S, Hanazawa S. NF-kappaB inhibitors stimulate apoptosis of rabbit mature osteoclasts and inhibit bone resorption by these cells. FEBS Lett. 1997;410:297-300
32. Jimi E, Nakamura I, Ikebe T, Akiyama S, Takahashi N, Suda T. Activation of NF-kappaB is involved in the survival of osteoclasts promoted by interleukin-1. J Biol Chem. 1998;273:8799-805
33. Wu M, Deng L, Wang Y, Chen W, Feng X, Li Y-P. TRAF family member-associated Nf-kappa B activator (Tank), induced by RANKL, Negatively regulates osteoclast survival and function. The Amerian Society for bone and mineral Research. 2011;204:SA0304
34. Maruyama K, Kawagoe T, Kondo T, Akira S, Takeuchi O. TRAF family member-associated NF-kappaB activator (TANK) is a negative regulator of osteoclastogenesis and bone formation. J Biol Chem. 2012;287(34):29114-24
35. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49-62
36. Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D. et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21:5673-83 doi:10.1038/sj.onc.1205664 [doi]
37. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. MolCell Biol. 2001;21:5299-305
38. Benayoun B, Baghdiguian S, Lajmanovich A, Bartoli M, Daniele N, Gicquel E. et al. NF-kB-dependent expression of the antiapoptotic factor c-FLIP is regulated by calpain 3, the protein involved in limb-girdle muscular dystrophy type 2A. The FASEB Journal. 2008;22:1521-9
39. Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers (Basel). 2011Jun;3(2):1639-1671
Author contact
![]() Corresponding author: Yi-Ping Li, Ph.D., Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA; Tel: 205-975-2606, Fax: 205-975-4919, E-mail: ypliedu; Wei Chen, MD, Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA; Tel: 205-975-2607, Fax: 205- 975-4919, E-mail: wechenedu.
Corresponding author: Yi-Ping Li, Ph.D., Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA; Tel: 205-975-2606, Fax: 205-975-4919, E-mail: ypliedu; Wei Chen, MD, Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham AL 35294-2182, USA; Tel: 205-975-2607, Fax: 205- 975-4919, E-mail: wechenedu.