Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(2):190-199. doi:10.7150/ijbs.5844 This issue Cite
Research Paper
Temporal Profile of Astrocytes and Changes of Oligodendrocyte-Based Myelin Following Middle Cerebral Artery Occlusion in Diabetic and Non-diabetic Rats
Li Jing1, 2, Qingping He1, Jian-Zhong Zhang2 ![]() , P. Andy Li1,2
, P. Andy Li1,2 ![]()
1. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technological Enterprise (BRITE), North Carolina Central University, Durham, North Carolina, USA.
2. Department of Pathology and Ningxia Key Laboratory for Cerebrocranial Diseases, Ningxia Medical University, Yinchuan, Ningxia, China.
Received 2013-1-9; Accepted 2013-1-30; Published 2013-2-11
Abstract
The long-term impacts of cerebral ischemia and diabetic ischemia on astrocytes and oligodendrocytes have not been defined. The objective of this study is to define profile of astrocyte and changes of myelin in diabetic and non-diabetic rats subjected to focal ischemia.
Focal cerebral ischemia of 30-min duration was induced in streptozotocin-induced diabetic and vehicle-injected normoglycemic rats. The brains were harvested for immunohistochemistry of glial fibrillary acidic protein (GFAP) and 2', 3'-cyclic nucleotide 3'-phosphodiesterase (CNPase) at various reperfusion endpoints ranging from 30 min up to 28 days. The results showed that activate astrocytes were observed after 30 min and peaked at 3 h to 1 day after reperfusion in ischemic penumbra, and peaked at 7 days of reperfusion in ischemic core. Diabetes inhibited the activation of astrocytes in ischemic hemisphere. Demyelination occurred after 30 min of reperfusion in ischemic core and peaked at 1 day. Diabetes caused more severe demyelination compared with non-diabetic rats. Remyelination started at 7 days and completed at 14 and 28 days in ischemic region. Diabetes inhibited the remyelination processes. It is concluded that ischemia activates astrocytes and induces demyelination. Diabetes inhibits the activation of astrocytes, exacerbates the demyelination and delays the remyelination processes. These may contribute to the detrimental effects of hyperglycemia on ischemic brain damage.
Keywords: astrocyte, cerebral ischemia, diabetes, hyperglycemia, myelin, oligodendrocyte, remyelination.
Introduction
It is well known that hyperglycemia exacerbates the prognosis of critically ill patients and increases mortality and morbidity of stroke patients [1-3]. Hyperglycemia-exacerbated neuronal damage is associated with an increase in the brain edema and enlargement of infarct volume [2,3]. The underlying cellular and molecular mechanisms by which hyperglycemia aggravates ischemic brain damage may involve tissue acidosis, increased free radical production, DNA oxidation, early mitochondrial damage, activation of cell death pathways, inflammation, compromised capillary patency, and induction of mitochondrial fission [4-7,22].
Astroglial cells are the most abundant cells in the central nervous system (CNS), providing structural, trophic, and metabolic support to neurons and modulating synaptic activity. They are believed to exert a neuroprotective effect in stroke by shielding neurons from oxidative stress [8]. Astrocytes become activated (reactive) in response to many CNS injuries, including brain infection, inflammation, ischemia, trauma, neurodegenerative diseases, and tumor [9]. Reactive astrocytes undergo changes in morphology and in their expression of a wide range of molecules. The hallmarks of astrocyte activation, regardless of its origin, are enhanced expression of glial fibrillary acidic protein (GFAP), enlarged cell body, increased number and length of dendrites. Impairment of astrocytes can critically affect the homeostasis of neuronal cells. Our previous study conducted in a brief period of forebrain ischemia model demonstrated that diabetic hyperglycemia increased damage to astrocytes, which presented as inhibition of activation, decrease in GFAP positive cell number, mitochondrial swelling and nuclear chromatin condensation [10]. However, the impact of diabetes/hyperglycemia on astrocytes after a long period of recovery following a short duration of focal cerebral ischemia has not been reported.
Myelination is a complex process by which an axon becomes insulated by the continuous wrapping of the proteolipid oligodendroglial membrane known as myelin [11-13]. Myelin is a dielectric (electrically insulating) material that forms a layer, the myelin sheath, usually around only the axon of a neuron. Myelin is formed from membranous sheets that are elaborated by Schwann cells in the PNS and oligodendrocytes in the CNS. Myelination is essential for the proper functioning nervous system [12]. Demyelination, loss of myelin sheath around an axon, impairs the conduction of action potential in affected nerves, causing impaired sensation, movement, cognition, or other function aspects depending on which nerves are involved [14]. Remyelination of denuded or regenerating axons in the peri-infarct area has been observed in the CNS [15]. However, the effect of diabetic hyperglycemia on oligodendrocyte-based demyelination and remyelination after cerebral ischemia following a long period of reperfusion has not been studied. The purpose of the present study was to reveal the temporal profile of astrocyte and changes of oligodendrocyte-based myelin in diabetic and non-diabetic rats subjected to 30-min transient cerebral focal ischemia with sequential reperfusion endpoints up to 28 days.
Materials and Methods
Animals
A total of 77 male Wistar rats (Charles River Laboratories, Wilmington, MA), weighing 310-340 g, were used in the presented experiment. Experimental groups and number of rats in each group were given in Table 1.
Experimental groups and number of rats in each group. Brain sections used for immunohistochemistry staining of GFAP and GNPase were from the same rats.
| ctr | 30m | 3h | 6h | 1d | 3d | 7d | 14d | 28d | |
|---|---|---|---|---|---|---|---|---|---|
| Non-diabetic | 3 | 3 | 6 | 3 | 5 | 3 | 6 | 6 | 4 |
| Diabetic | 4 | 3 | 4 | 4 | 6 | 0 | 10 | 4 | 3 |
All animal use procedures were in strict accordance with the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee at the North Carolina Central University.
STZ-induced diabetes
The rats were fasted overnight, injected intraperitoneally with streptozotocin (STZ, 40 mg/kg, in 0.1 mol/l citrate buffered saline, pH 4.5). Age-matched rats received the same volume of citrate-buffered saline served as normoglycemic controls. Blood glucose levels were measured 2-3 days after STZ injection to verify the successfulness of diabetes induction. Those with glucose>16mM were included in diabetic group. Cerebral ischemia was induced 4 weeks after STZ-induced diabetic and citrate buffer injected non-diabetic animals.
Ischemic model
Both diabetic hyperglycemic and non-diabetic normoglycemic animals were subjected to a 30-min duration of middle cerebral artery occlusion (MCAO) using an intraluminal filament [2]. The choice of a mild MCAO model was based on our previous studies showing that the effect of diabetes on ischemic brain could be clearly distinguished from that of normolgycemic ischemic animals in 30 min MCAO model [2,3]. Rats were revived after reperfusion and neurological defect was examined according to Bederson score [16]. Animals with neurological signs of diminished resistance to lateral push, walking to the left after being pulled backwards by the tail, or with spontaneous contralateral circling were included in the study.
Preparation of Histological Samples
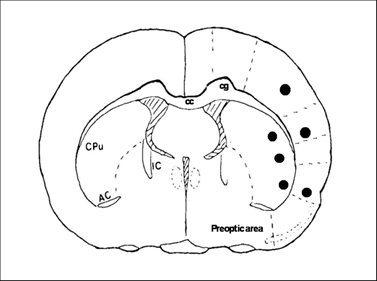
At 30 min, 6 h and 1, 3, 7, 14, 28 days of recirculation, animals were reanesthetized and the thoraxes were exposed. Animals were transcardially perfused with normal saline and followed by ice-cold 4% PBS-buffered paraformaldehyde. The brains were removed and post-fixed in 4% PBS-buffered paraformaldehyde for 48 h. A 2 mm thick block, at a level of -0.3 mm to the bregma, was obtained and embedded in paraffin. This coronal level displays an ideal caudoputamen and cortical areas for detection of MACO damage. A series of consecutive sections (5 µm in thickness) were collected from paraffin-embedded blocks for conventional histologic examination and immunohistochemistry (IHC). Neuronal damage was defined as neurons in triangular shape with nuclear shrinkage. Three microscopic fields at 400X were captured and number of damaged neurons was counted. Fig. 1 shows the sites of image sampling.
Coronal section of a rat brain map at Bregma -3.0 mm illustrates the areas (marked black areas) where images were captured for analysis.
Immunohistochemistry
IHC staining follows the same procedure as having been described previously [7]. In brief, the tissue sections were deparaffinized, rehydrated and washed. Antigen retrival was achieved by heating the sections that were immersed in a sodium citrate buffer (pH 6.0) at 92oC for 40 min in a pre-heated steamer. Nonspecific binding sites were blocked with 10% normal serum in tris-buffered saline containing 0.1% Tween 20 (TBS-T). The sections were incubated overnight with primary antibodies against GFAP at 1:800 (purified mouse monoclonal antibody; Cell Signaling) or CNPase (2', 3'-cyclic nucleotide 3'-phosphodiesterase) at 1:200 (polyclonal antibody, Abcam). The sections were incubated in 0.3% hydrogen peroxide for 15 min at room temperature to quench endogenous hydrogen peroxidase activity. The sections were washed and then incubated with horseradish peroxidase (HRP) conjugated second antibody (1:300, Santa Cruz). Color reaction was achieved by 3,3'-diaminobenzidine (DAB) incubation for 3 min at room temperature. The sections were then rinsed in distilled water and counterstained with haematoxylin. Immunoreactivity of the GFAP was graded as follows: 0 = no reactivity as viewed with no DAB precipitation; 1 = weak reactivity depicted as light brownish staining; 2 = moderate reactivity observed as moderate brownish staining; and 3 = strong reactivity presented as strong dark brownish staining. CNPase images were captured and areas of CNPase positive staining were measured using a computer-assisted imaging system (Nikon Eclipse C1 software). A third person who was blinded to the experimental conditions assessed the IHC sections. Percent of demyelination was measured in brain samples from control, 30 min, 6 h and 1 day ischemic reperfusion groups, while percent of remyelination was presented in brain samples collected from 7, 14, and 28 days of reperfusion. Percent of demyelination = [Total area of ipsilateral caudoputamen - CNPase stained area of ipsilateral caudoputamen] / Total area of ipsilateral caudoputamen. Percent of remyelination = CNPase stained area in caudoputamen/ Total area of ipsilateral caudoputamen.
Immunofluorescent staining for GFAP and CNPase were also performed using aforementioned antibodies. Following being incubated with anti-GFAP or anti-CNPase antibodies, the sections were incubated with secondary donkey anti-mouse antibody conjugated with Alexa Fluor 561 (1:300, Invitrogen) or donkey anti-rabbit antibody conjugated with Alexa Fluor 488 (1:300, Invitrogen) for 1 h at room temperature. Double labeling of GFAP and CNPase was achieved by incubating the two primary antibodies separately and then mixture of the two fluorescence-conjugated secondary antibodies. The specimens were mounted with Vectashield Hardset Mounting Media (H-1200, Vector) containing 4', 6-diamidino-2-phenylindole (DAPI) and examined using a fluorescence confocal-scanning microscope (Nikon Eclipse C1).
Statistics
Comparison of recirculation endpoints with sham-operated controls within diabetic group or non-diabetic group were made by ANOVA followed by post hoc Scheffe's test. A P value less than 0.05 was considered as statistically significant. Data were presented as means±sd.
Results
Diabetes enhances ischemic brain damage
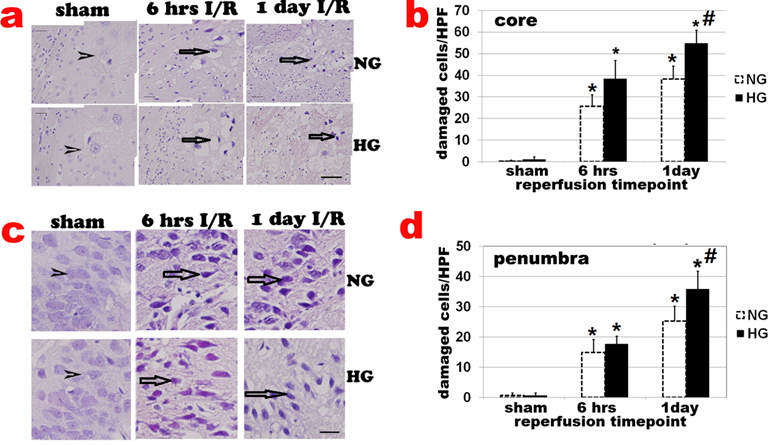
Haematoxylin and eosin (H&E) staining showed no obvious cell morphologic change in sham-operated controls (Fig 2a sham). Neuronal cell death was observed after 6 h of reperfusion and localized mainly in caudoputamen (Fig. 2a & 2b). The number of damaged cell increased after 6 h and peaked at 3-7 days of recovery in normoglycemic animals. After 7 days of reperfusion, glial cells gradually infiltrated into ischemic core and replacing the damaged tissue with glial scar (data not shown). At 28 days, most infarct tissues in the caudoputamen were repaired and indistinguishable from undamaged tissue. Only 1 animal still had small infarct foci. The number of damaged cells in the cortical area (penumbra) also increased after 6 h, 1, and 7 days of reperfusion (Fig 2c & 2d). Comparing to normoglycemic animals, diabetic hyperglycemia increased neuronal death after 6 h, 1 day (Fig. 2a & 2b) and up to 7 day of reperfusion (data not shown).
Histologic changes after MCAO in diabetic and non-diabetic animals. a, Representative H&E stained photomicrographs and enlarged inserts demonstrating changes in the caudoputamen during early reperfusion stages. b, Mean numbers of damaged neuronal cells per high power (400x) microscopic field (HPF) in the caudoputamen. c: Representative photomicrographs showing changes in the cortical area. d: Mean numbers of damaged neurons in the cortex. Arrowheads indicate normal neurons and arrows indicate dead neurons. NG, normoglycemia, HG, diabetic hyperglycemia, I/R, ischemia and reperfusion. *P < 0.01 vs. sham control within same glycemic group and # p<0.05 vs. NG samples at an identical time point. Bar = 50 µm.
Diabetes inhibits astrocytes
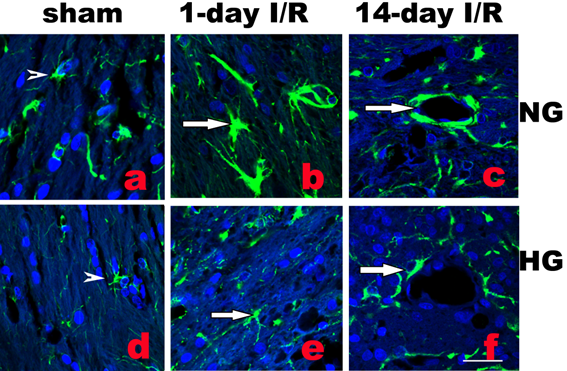
Activation of astrocytes is a universal reaction to brain injury, which seals off the injured tissue and restricts inflammation and neuronal death. Activated astrocytes undergo hypertrophy of their somata and dendritic processes and increase synthesis of GFAP [10]. As shown in Fig. 3b, following normoglycemic ischemia and 1 day reperfusion, astrocytic cell bodies and processes were enlarged compared with the astrocytes in sham operated rats (Fig. 3a). Diabetes inhibited the ischemia-induced astrocyte activation (Fig.3e). Furthermore, at 14 days reperfusion in normoglycemic animals, astrocyte endfoot entirely ensheathed blood vessel wall in infarct area as shown in Fig. 3c. However, in diabetic animals, astrocytic endfoot failed to completely ensheath a vascular wall by attaching to only a small portion of a vascular wall (Fig. 3f).
Double labeling of astrocytes (GFAP, green color) and nuclei (DAPI, blue color) in normoglycemic (NG, a-c) and hyperglycemic (HG, d-f) animals in infarct area. A completely ensheathed by astrocyte end-foot in NG and a partial ensheathed vessel in HG are presented in c and f, respectively. Arrowheads indicate normal astrocytes and arrows indicate activated astrocytes in b and astrocyte end-foot in c and f. Bar = 60 µm.
Temporal profile of astrocyte activation
GFAP immunoreactivity slightly increased in both contralateral and ipsilateral hemispheres after 30 min I/R. Activated astrocytes were first observed in periventricular zones of both hemispheres, followed by an increase in the corpus callosum, cingulum and then in dorsolateral caudoputamen and overlying cortex of ipsilateral hemisphere before the astrocyte infiltration into the ischemic core. In ischemic penumbra region of the normoglycemic animals, number of GFAP positive astrocytes increased after 30 min of reperfusion, accumulated after 3-6 h and peaked at 1-3 days of reperfusion. The GFAP positive cells reduced after 7 days and returned back to non-ischemic control level after 28 days of recovery. The temporal profile of the astrycyte in diabetic animals followed the same pattern with suppressed GFAP immunoreactivity in the penumbra area (Fig 4a & 4b).
Changes of GFAP-positive cells after ischemia and reperfusion (I/R) in normo- and hyperglycemic rats after various reperfusion stages. a, Representative photomicrographs showing GFAP staining in the ischemic core area in normoglycemic (NG) and hyperglycemic (HG) animals. Brown color represents GFAP positive stained astrocytes. Bar =100 µm. b, Temporal profile of GFAP positive staining in the penumbra. c, Temporal profile of GFAP positive staining in the ischemic core, Arrows indicate GFAP positively stained astrocytes. *p<0.05 vs. sham control and #p<0.01 vs. same HG at an identical reperfusion stage.
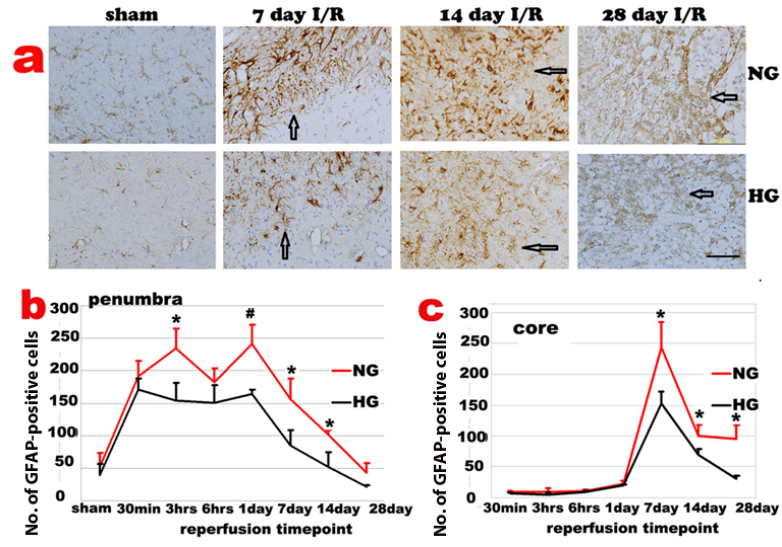
The GFAP positive astrocytes infiltrated into ischemic core at 3 days and peaked at 7 days of reperfusion (Fig 4c). The GFAP positive cells then started to decrease after 14 days but remained to be higher than preischemic value at 14 and 28 days of recovery in normoglycemic animals. Again, astrocyte change in diabetic animals followed the same pattern as the normoglycemic animals, with a blunted peak value at 7 days. Thus, number of GFAP positive cell peaked at 7 days of recovery, decreased at 14 days and completely back to normal at 28 days of recovery in ischemic core (Fig 4c).
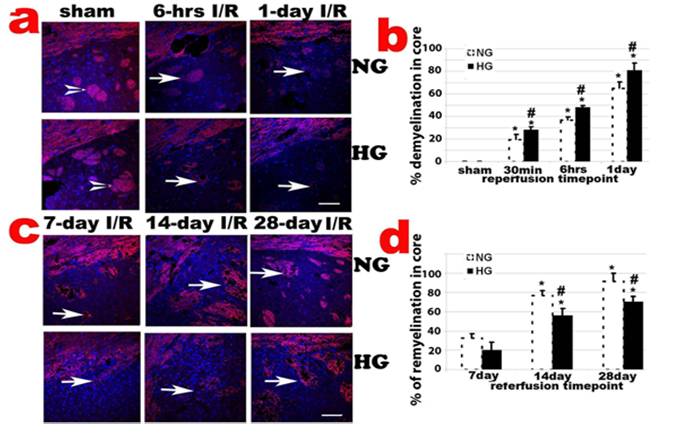
Diabetes enhances ischemia-induced demyelination and inhibites remyelination
CNPase, encoded by the CNP gene, expresses exclusively by oligodendrocytes in the CNS. CNPase is thought to play a critical role in events leading to myelination. Ischemia-caused demyelination was mainly observed in ischemic core area and to a less extend in penumbra. The demyelination, as reflected by significantly decreased or absence of CNPase immunoreactivity, was observed 30 min after and peaked at 1 days of reperfusion in normoglycemic animals (Fig. 5a & 5b). Remyelination, as reflected by the reappearance of positive CNPase staining, started at 7 days of recovery in ischemic core and penumbra areas in normoglycemic animals. The remyelination significantly progressed after 14 days and reached to peak level at 28 day of reperfusion in normoglycemic animals (Fig. 5c & 5d). In fact, at 28 days of reperfusion, oligodendrocyte based myelination reappeared in most ischemic core region. Comparing to normoglycemic animals, hyperglycemia resulted in larger area of demyelination in early reperfusion stage (30 min to 1 day reperfusion), which probably correlates to the severity of the damage in these animals. Hyperglycemia suppressed remyelination process. As shown in Fig. 5d, the area of remyelination is much less in hyperglycemic animals than in normoglycemic animals subjected to ischemia and reperfusion at 7, 14, an 28 days.
CNPase staining in caudoputamen following transient 30-min MCAO. a, Representative photomicrographs of CNPase stained sections in the caudoputamen and the overlaying cortex up to 1 day reperfusion; b, Percentage of demyelination in the caudoputamen. *P < 0.01 vs. sham within same glycemic group and ## p<0.01 HG vs. NG samples at an identical reperfusion endpoint. c, Representative photomicrograph showing CNPase staining in late reperfusion phases (7, 14 and 28 days) in the caudoputamen; d, Percentage of remyelination in the caudoputamen. *P < 0.01 vs. 7 day I/R within same glycemic group and ## p<0.01 HG vs. NG samples at an identical reperfusion endpoint. Bar = 60 µm.
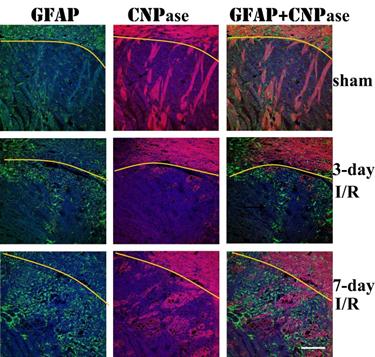
Remyelination correlates with astrocyte infiltration
Double immunolabeling of GFAP and CNPase revealed that astrocyte infiltration into infarct area was concomitant with remyelination in the area. At 3 days of recover, small numbers of GFAP immunoreactive astrocytes infiltrated to ischemic core. The number of infiltrated astrocytes markedly increased at 7 days of recovery. At this time, massive remyelination appeared in the areas where astrocytes had been infiltrated, but not in areas without astrocyte infiltration.
Triple immunolabeling of GFAP (green color) and CNPase (purple color) and DAPI (blue color) on ischemic brain sections. Yellow curve indicate the border between caudoputamen and the overlaying cortex. Magnification 200X.
Discussion
Diabetes aggravates cerebral ischemic damage
We have previously shown that diabetes accelerates and enlarges ischemic brain damage in early reperfusion phase [7]. The present study further expended the previous study by showing that the aggravating effects of diabetes on ischemic brain damage persisted to late reperfusion stage. Thus, enhanced neuronal damage was not only observed in caudoputamen at 6 h and 1 day as shown previously, but also at 7 and 14 day of reperfusion in diabetic rats compared to non-diabetic ones. The mechanisms underlying hyperglycemia-enhanced ischemic brain damage may involve tissue acidosis, energy depletion, loss of ionic homeostasis, mitochondrial dysfunction, free radical production, neuroinflammatory processes, damage to the blood-brain barrier (BBB) and neuroglial cells [6,7,17- 21]. Recent study shows that hyperglycemia causes mitochondrial dynamic imbalance and activates autophagy after transient cerebral ischemia [22].
Effects of diabetes on astrocytes
Astrocyte is the most abundant cell type in the CNS. Astrocytes participate in formation of the BBB, maintain electrolyte balance, release neurotrophic factors, limit the expension of tissue injury and repair damaged nerve tissues [8,9]. We observed that astrocytes were first increased in the periventricular zone after 30 min of reperfusion and then appeared in corpus callosum, penumbra, and ischemic core area before infiltration to ischemic core at 3 days of recovery. This may suggest that in response to CNS injury, astroglial cells might be activated at their nesting area of perventricular zone and then migrated along corpus callosum over considerable distances to the injured region, where they join the local pool of reactive astrocytes [23]. We observed that brain infarct area was repaired back to normal level in the brain areas where astrocytes infiltrated, while poor tissue repair was noted in brain areas where astrocytes poorly infiltrated. Thus, it is likely that reactive astrocytes play a key role in the reconstruction of damaged neural tissue after stroke [24]. These results are consistent with the neuroprotective role of astrocytes observed in stroke and other CNS injury models [8,24].
Our results also demonstrated that while astrocyte end-foot entirely ensheathed the wall of some blood vessels under normoglycemic condition, partial or completely absence of astrocyte end-foot around the blood vessel wall was observed in diabetic animals. Recent research suggest that astrocytes serve as tight junctions and as basal lamina of the cerebral endothelial cells, which play the most substantial role in maintaining the BBB [25,26]. Since astrocyte end-foot encircling vascular endothelial cells constitutes the BBB, partial or absence of astrocyte end-foot ensheathed blood vessels cause damages the BBB integrity. In fact, published studies have shown that hyperglycemia increases the BBB permeability [27-29].
The results demonstrated that ischemia resulted in increased number of astrocytes in ischemic penumbra area as early as 30 min after reperfusion in the ipsilateral hemisphere of the normoglycemic animals. The GFAP-positive astrocytes remained increased thereafter, reached to peak level after 1-3 days of reperfusion, and then gradually decreased to control level after 28 days of reperfusion. Preischemic diabetes significantly reduced the number of astrocytes in this region compared with normoglycemic animals. In ischemic core area of the normoglycemic animals, number of astrocytes did not increased significantly until 3 days of reperfusion and reached to peak level after 7 days. Number of astrocytes decreased after 14 days but remained elevated at 28 days of reperfusion. Comparing to normoglycemic animals, the peak of astrocytes in ischemic core reduced and returned to control level after 28 days of reperfusion in diabetic animals. Suppression of actrocytes by presichemic diabetes may contribute to the detrimental effects of hyperglycemia on ischemic brain damage by exacerbating the BBB damage and hindering tissue repair. As discussed elsewhere, astrocytes are especially sensitive to acidosis and die quickly in acidic and hypoxic conditions [30,31]. Diabetic ischemia enhances tissue acidosis during ischemia/hypoxia due to increased anaerobic glucose metabolism [32].
Effects of diabetes on demyelination and remyelination
Oligodendrocytes myelinate neuronal axons in the CNS, which is important for fast conduction of nerve impulses [12]. Demyelination associates with several clinical disorders including multiple sclerosis [33] and virus-induced demyelination [34], characterized by slow or stopped conduction of action potential in certain nerve fibers. Ischemia induces demyelination, as reflected by loss of myelin sheath surround the axons of neurons. Mild demyelination was detected in ischemic core after 30 min of reperfusion in normoglyemic ischemic brain by CNPase IHC. The demyelination persisted and peaked after 1-3 days of reperfusion. Diabetes further enhanced demyelination as reflected by increased area of demyelination and low intensity or absence of CNPase immunoreactivity. The reasons for increased demyelination in hyperglycemic animals are not clear. It may be related to increased brain damage, injury to the BBB that allows the entry of circulating antibodies, in particular demyelinating antibodies such as anti-myelin/oligodendrocyte glycoprotein (MOG) antibodies, into the CNS [27,35], enhanced inflammatory responses [21], and dislocation of iron from their binding sites that triggers formation of reactive oxygen species [29,36-39]. In this context, oligodendrocytes have high iron content, making them especially prone to oxidative stress by reactive oxygen species.
Remyelination of denuded or regenerating axons in infarct area has been observed in the CNS after focal ischemia [40,41]. In this study, remyelination started at 7 days and persisted to 28 days after reperfusion in normoglycemic animals. Diabetes suppressed the remyelination process as the area of remyelination decreased comparing to normoglycemic animals. This may be correlated to the suppression of astrocytes by diabetes. It has been known for many years that astrocytes secrete promyelinating factors including cytokine leukemia inhibitory factor that promotes the myelinating activity of oligodendrocytes [42]. Therefore, astrocytes have an important role in stimulating oligodendricytes to produce myelin for remyelination [43]. In an attempt to gain a better understanding of the interaction between astrocytes and oligodendrocyte-based remyelination, we performed double immunostanning of GFAP and CNPase. We observed a close association between GFAP and CNPase immunoreactivity in brain injury areas. In areas where astrocyte infiltrated, remyelination followed. Vice versa, in areas where astrocytes were not infiltrated, remyelination was not observed. This observation suggests that astrocytes may promote myelinating activity of oligodendrocytes after brain ischemia. Our finding does not support previous suggestion that the presence of established astrocytes in an area of demyelination has an inhibitory effect on the extent of remyelination [44].
In summary, cerebral ischemia activates astrocytes and causes demyelination. Diabetes inhibits the activation of astrocytes, exacerbates demyelination process and suppresses the remyelination after ischemia and reperfusion. These may contribute to the detrimental effects of hyperglycemia on ischemic brain damage.
Acknowledgements
LJ was supported by National Science Foundation of China (81260184) and Ningxia Medical University Research Fund (2010); JZZ by the National Science Foundation of China (81060105); and PAL by the National Institute of Health (7R01DK075476-06). The BRITE is partially funded by the Golden Leaf Foundation.
Authors Contributions
Conceived and designed the experiments: LJ, JZZ, PAL. Performed the experiments: LJ, QH. Contributed reagents/materials/analysis tools: PAL. Wrote the paper: LJ, JZZ, PAL.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mazighi M, Amarenco P. Hyperglycemia: a predictor of poor prognosis in acute stroke. Diabete Metab. 2001;27:718-20
2. Li PA, Gisselsson L, Keuker J, Vogel J, Smith ML, Kuschinsky W, Siesjo BK. Hyperglycemia-exaggerated ischemic brain damage following 30 min of middle cerebral artery occlusion is not due to capillary obstruction. Brain Res. 1998;804:36-44
3. Gisselsson L, Sm ith ML, Siesjo BK. Hyperglycemia and focal brain ischemia. J Cereb Blood Flow Metab. 1999;19:288-97
4. Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3:296-304
5. Anabela P. R. & Carlos M. P. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212:167-78
6. Ding C, He Q, Li PA. Activation of cell death pathway after a brief period of global ischemia in diabet ic and non-diabetic animals. Exp Neurol. 2004;188:421-29
7. Muranyi M, Fujioka M, He Q, Han A, Yong G, Csiszar K, Li PA. Diabetes activates cell death pathway after transient focal cerebral ischemia. Diabetes. 2003;52:481-86
8. Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468-81
9. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7-35
10. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55:349-55
11. Miron VE, Kuhlmann T, Antel JP. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim Biophys Acta. 2011;1812:184-93
12. Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275-83
13. Gingras M, Beaulieu MM, Gagnon V, Durham HD, Berthod F. In vitro study of axonal migration and myelination of motor neurons in a three-dimensional tissue-engineered model. Glia. 2008;56:354-64
14. Konopaske GT, Dorph-Petersen KA, Sweet RA, Pierri JN, Zhang W, Sampson AR, Lewis DA. Effect of chronic antipsychotic exposure on astrocyte and oligodendrocyte numbers in macaque monkeys. Biol Psychiatry. 2008;63:759-65
15. Waxman SG, Craner MJ, Black JA. Na(+) channel expression along axons in multiple sclerosis and its models. Trends Pharmacol Sci. 2004;25:584-91
16. Baltaci S, Orhan D, Göğüş C, Filiz E, Tulunay O, Göğüş O. Thrombospondin-1, vascular endothelial growth factor expression and microvessel density in renal cell carcinoma and their relationship with multifocality. Eur Urol. 2003;44:76-81
17. Song EC, Chu K, Jeong SW, Jung KH, Kim SH, Kim M, Yoon BW. Hyperglycemia exacerbates brain edema and perihematomal cell death after intracerebral hemorrhage. Stroke. 2003;34:2215-20
18. Mehta SL, Lin Y, Chen W, Yu F, Cao L, He Q, Chan PH, Li PA. Manganese Superoxide Dismutase Deficiency Exacerbates Ischemic Brain Damage Under Hyperglycemic Conditions by Altering Autophagy. Transl Stroke Res. 2011;2:42-50
19. Zhang JZ, Jing L, Ma Y, Guo FY, Chang Y, Li PA. Monosialotetrahexosy-1-ganglioside attenuates diabetes-enhanced brain damage after transient forebrain ischemia and suppresses ohosphorylation of ERK1/2 in the rat brain. Brain Res. 2010;16:200-8
20. Muranyi M, Li PA. Hyperglycemia increases superoxide production in the CA1 pyramidal neurons after global cerebral ischemia. Neurosci Lett. 2006;393:119-22
21. Ding C, He Q, Li PA. Diabetes increases expression of ICAM after a brief period of cerebral ischemia. J Neuroimmunol. 2005;161:61-67
22. Kumari S, Anderson L, Farmer S, Mehta SL, LI PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjecte to cerebral ischemia and reperfusioin. Transl Stroke Res. 2012;3:296-304
23. Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisén J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25-34
24. Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137-49
25. Lee J, Baird A, Eliceiri BP. In vivo measurement of glioma-induced vascular permeability. Methods Mol Biol. 2011;763:417-22
26. Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307-17
27. Dietrich WD, Alonso O, Busto R. Moderate hyperglycemia worsens acute blood-brain barrier in jury after forebrain ischemia in rats. Stroke. 1993;24:111-16
28. Paljarvi L, Rehncrona S, Soderfeldt B, Olsson Y, Kalimo H. Brain lactic acidosis and ischemic cell damage: quantitative ultrastructural changes in capillaries of rat cerebral cortex. Acta Neuropathol (Berl). 1983;60:232-40
29. Chan PH, Schmidley JW, Fishman RA, Longar SM. Brain injury, edema, and vascular permeability changes induced by oxygen derived free radicals. Neurology. 1984;34:315-20
30. Giffard RG, Monyer H, Choi DW. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990;530:138-41
31. Bondarenko A. Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134-42
32. Li PA, Shamloo M, Katsura K, Smith ML, Siesjo BK. Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiol Dis. 1995;2:97-108
33. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122:1180-8
34. Shishido-Hara Y. Progressive multifocal leukoencephalopathy and promyelocytic leukemia nuclear bodies: a review of clinical, neuropathological, and virological aspects of JC virus-induced demyelinating disease. Acta Neuropathol. 2010;120:403-17
35. Jaśkiewicz E. Epitopes on myelin proteins recognized by autoantibodies present in multiple sclerosis patients. Postepy Hig Med Dosw. 2004;58:472-82
36. Rehncrona S, Hauge HN, Siesjo BK. Enhancement of iron-catalyzed free radical formation by acidosis in brain homogenates: differences in effect by lactic acid and CO2. J Cereb Blood Flow Metab. 1989;9:65-70
37. Bralet J, Schreiber L, Bouvier C. Effect of acidosis and anoxia on iron delocalization from brain homogenates. Biochem pharma. 1992;43:979-83
38. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Brück W, Parisi JE, Scheithauer BW, Giannini C, Weigand SD, Mandrekar J, Ransohoff RM. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188-97
39. Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. Molecular Biology, Epidemiology, and Pathogenesis of Progressive Multifocal Leukoencephalopathy, the JC Virus-Induced Demyelinating Disease of the Human Brain. Clin Microbiol Rev. 2012;25:471-506
40. Irving EA, Bentley DL, Parsons AA. Assessment of white matter injury following prolonged focal cerebral ischaemia in the rat. Acta Neuropathol. 2001;102:627-35
41. Chida Y, Kokubo Y, Sato S, Kuge A, Takemura S, Kondo R, Kayama T. The alterations of oligodendrocyte, myelin in corpus callosum, and cognitive dysfunction following chronic cerebral ischemia in rats. Brain Res. 2011;1414:22-31
42. Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823-32
43. Schulz K, Kroner A, David S. Iron efflux from astrocytes plays a role in remyelination. J Neurosci. 2012;32:4841-7
44. Blakemore WF, Gilson JM, Crang AJ. The presence of astrocytes in areas of demyelination influences remyelination following transplantation of oligodendrocyte progenitors. Exp Neurol. 2003;184:955-63
Author contact
![]() Corresponding author: Dr. P. Andy Li, Email: pliedu or Dr. Jian-zhong Zhang, zhangjzedu.cn.
Corresponding author: Dr. P. Andy Li, Email: pliedu or Dr. Jian-zhong Zhang, zhangjzedu.cn.