Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(4):350-360. doi:10.7150/ijbs.6058 This issue Cite
Research Paper
Genomic Profiling Reveals the Potential Role of TCL1A and MDR1 Deficiency in Chemotherapy-Induced Cardiotoxicity
Timothy A. McCaffrey1,6, Constantine Tziros2, Jannet Lewis2, Richard Katz2, Robert Siegel3, William Weglicki2,4, Jay Kramer4, I. Tong Mak4, Ian Toma1, Liang Chen1, Elizabeth Benas1, Alexander Lowitt1, Shruti Rao1, Linda Witkin2, Yi Lian1, Yinglei Lai5, Zhaoqing Yang1, Sidney W. Fu1,6 ![]()
1. Department of Medicine, Division of Genomic Medicine;
2. Department of Medicine, Division of Cardiology;
3. Department of Medicine, Division of Hematology/Oncology;
4. Department of Biochemistry and Molecular Biology;
5. Department of Statistics and Biostatistics Center;
6. Department of Microbiology, Immunology and Tropical Medicine, The GW Heart and Vascular Institute, and The Dr. Cyrus and Myrtle Katzen Cancer Center, George Washington University School of Medicine and Health Sciences, Washington DC, USA.
Received 2013-2-9; Accepted 2013-2-27; Published 2013-4-22
Abstract
Background: Anthracyclines, such as doxorubicin (Adriamycin), are highly effective chemotherapeutic agents, but are well known to cause myocardial dysfunction and life-threatening congestive heart failure (CHF) in some patients.
Methods: To generate new hypotheses about its etiology, genome-wide transcript analysis was performed on whole blood RNA from women that received doxorubicin-based chemotherapy and either did, or did not develop CHF, as defined by ejection fractions (EF)≤40%. Women with non-ischemic cardiomyopathy unrelated to chemotherapy were compared to breast cancer patients prior to chemo with normal EF to identify heart failure-related transcripts in women not receiving chemotherapy. Byproducts of oxidative stress in plasma were measured in a subset of patients.
Results: The results indicate that patients treated with doxorubicin showed sustained elevations in oxidative byproducts in plasma. At the RNA level, women who exhibited low EFs after chemotherapy had 260 transcripts that differed >2-fold (p<0.05) compared to women who received chemo but maintained normal EFs. Most of these transcripts (201) were not altered in non-chemotherapy patients with low EFs. Pathway analysis of the differentially expressed genes indicated enrichment in apoptosis-related transcripts. Notably, women with chemo-induced low EFs had a 4.8-fold decrease in T-cell leukemia/lymphoma 1A (TCL1A) transcripts. TCL1A is expressed in both cardiac and skeletal muscle, and is a known co-activator for AKT, one of the major pro-survival factors for cardiomyocytes. Further, women who developed low EFs had a 2-fold lower level of ABCB1 transcript, encoding the multidrug resistance protein 1 (MDR1), which is an efflux pump for doxorubicin, potentially leading to higher cardiac levels of drug. In vitro studies confirmed that inhibition of MDR1 by verapamil in rat H9C2 cardiomyocytes increased their susceptibility to doxorubicin-induced toxicity.
Conclusions: It is proposed that chemo-induced cardiomyopathy may be due to a reduction in TCL1A levels, thereby causing increased apoptotic sensitivity, and leading to reduced cardiac MDR1 levels, causing higher cardiac levels of doxorubicin and intracellular free radicals. If so, screening for TCL1A and MDR1 SNPs or expression level in blood, might identify women at greatest risk of chemo-induced heart failure.
Keywords: doxorubicin, adriamycin, heart failure, cardiomyopathy, microarray, expression profiling, free radicals, multidrug resistance protein, MDR1, TCL1A.
Introduction
Anthracyclines, such as doxorubicin (Adriamycin), have been key cancer chemotherapeutic agents for the past 30 years. Despite their demonstrated benefit in cancer chemotherapy, anthracyclines can cause myocardial dysfunction in up to 25% of patients and life threatening heart failure in 1-4% of patients [1]. Cardiotoxicity due to doxorubicin is dose-related, with doses exceeding 500 mg/kg associated with the greatest incidence of left ventricular (LV) dysfunction and development of clinically overt congestive heart failure (CHF), often manifest as a reduction in the left ventricular ejection fraction (EF) [2]. However, patients receiving lower doses of doxorubicin also develop evidence of cardiotoxicity, and a wide individual susceptibility to cardiac damage has been observed.
Anthracyclines attack tumor cells by intercalating on DNA, inhibiting DNA replication, and thereby generating DNA damage, thus triggering apoptosis. Rapidly growing tumor cells are more susceptible than non-dividing cells, such as cardiomyocytes and neurons, thus conferring a relatively selective anti-tumor effect. However, it is unclear why some women are especially sensitive to the toxic effects of anthracyclines on cardiomyocytes. The mechanism by which anthracyclines produce myocardial damage is thought to involve a marked increase in oxidative stress, culminating in cardiomyocyte apoptosis. There appears to be a subset of individuals who are more susceptible to drug-induced oxidative damage and apoptosis. This susceptibility appears to be exacerbated with the use of trastuzumab, a monoclonal antibody to the HER2 proto-oncogene, a member of epidermal growth factor (EGF) receptor family. The addition of trastuzumab to anthracycline therapy has been shown to improve the chemotherapeutic response, but is also associated with a marked increase in the development of CHF [3], with the incidence of CHF increasing from 8% to 27% when trastuzumab was added [4]. Thus, the identification of women prone to CHF after chemotherapy is key to tipping the balance between effective chemotherapy and potentially life-threatening adverse effects.
A growing literature indicates that changes in gene expression of circulating cells can reflect changes in other organ systems. Published studies have documented mRNA expression changes in blood in disease states such as vasculitis [5], lupus [6], and coronary artery disease [7; 8]. The potential of this methodology in heart failure research has recently been reviewed by Liew [9]. Thus, the present studies were designed to identify mRNA expression markers in patient's blood and to determine whether any of the markers might elucidate possible mechanisms of enhanced susceptibility to anthracycline-induced cardiotoxicity. Minimally, biomarkers of chemo-induced CHF could allow individualization of chemotherapy, and importantly, administration of potentially preventive medications such as angiotensin receptor blockers, anti-oxidants, and beta-blockers.
Methods
Subjects
Patients at The George Washington University Medical Faculty Associates presenting for treatment of cancer using anthracyclines as part of a chemotherapeutic protocol, with or without adjuvant trastuzumab, were enrolled in an Institutional Review Board-approved protocol and gave informed consent.
Inclusion/Exclusion Criteria
All women presenting for anthracycline-based chemotherapy were considered eligible. Patients were excluded if their pre-chemo evaluation indicated abnormal left ventricular systolic function (ejection fraction < 50%), past history of, or active cardiac disease including: myocardial infarction, congestive heart failure, angina pectoris requiring medication, arrhythmia requiring medication, clinically significant valvular disease (i.e., valvular stenosis of any degree, regurgitation greater than mild in severity), or uncontrolled systemic hypertension.
Echocardiography
A standard two-dimensional and Doppler study, including tissue Doppler imaging, was performed at baseline, prior to receiving chemotherapy. Left ventricular systolic function was assessed using biplane Simpson's method for calculation of ventricular volumes and EF. Echocardiograms were repeated after completion of the chemotherapy protocol, or if the patient developed clinical symptoms of heart failure.
Assessment of LV Diastolic Function
Diastolic function was evaluated by pulsed and tissue Doppler for estimation of pulmonary capillary wedge pressure (PCWP) using the formula (PCWP= 1.24 [E/Ea] + 1.9) [10], and color tissue Doppler assessment for diastolic dyssynchrony [11]. Echocardiography and pre-defined parameters were repeated immediately after the final cycle of anthracyclines (typically 8 weeks), 6 months and 12 months. Patients receiving additional chemotherapy with trastuzumab also underwent clinically indicated echocardiograms prior to initiation of therapy, then at 6 months and 12 months.
Markers of Oxidative Stress
Blood samples were obtained prior to chemotherapy, and 24 hours following completion of each infusion cycle. Accumulation of lipid peroxidation products in the blood were assessed semi-quantitatively by the measurement of thiobarbituric-acid reactive substances (TBARS), as described [12]. Briefly, fresh plasma was acidified with trichloroacetic acid to prevent post-isolation changes and then reacted with 2-thiobarbituric acid and butylated hydroxytoluene at 80˚ C for 30 min, and the chromophore developed in the supernatant was measured at 532 nm.
Blood RNA Isolation
Blood was collected by venipuncture directly into PaxGene Blood RNA collection tubes (PreAnalytiX). The blood was incubated 2 hours at 25˚ C to complete the cell lysis and RNAse neutralization, moved to -20˚ C overnight, and then stored at -80˚ C until processing. Whole blood RNA was isolated using the PaxGene blood RNA isolation system with on-column DNAse treatment. RNA samples were further purified for microarray analysis using RNAeasy Mini kit (Qiagen). RNA concentration was quantitated by absorbance at 260 nm (Nanodrop), and the RNA integrity was tested by capillary electrophoresis on an Agilent 2100 Bioanalyzer. The typical yield of RNA was ~7 µg/2.5 ml whole blood.
Microarray Data Analysis
Purified RNA (100 ng) was reverse transcribed with SuperScript III using random hexamers and the cDNA was amplified with Ovation RNA Amplification System V2 kit (NuGEN). The cDNA was further purified with DNA Clean & Concentrator-25 (Zymo Research), and then 3.75 µg of cDNA was fragmented and labeled with FL-Ovation cDNA Biotin Module V2 kit (NuGEN) and hybridized to an Affymetrix GeneChip Human Genome U133 Plus 2.0 Array for 18 hours. The hybridization of fluorescently labeled cDNA was quantitated on an Affymetrix GeneChip Scanner 3000 7G.
Statistical Methods
The microarray probeset levels were converted to transcript levels using RMA summarization in GeneSpring GX 10 (Agilent Technologies). To identify transcripts associated with chemo-induced CHF, the 54,000 total transcripts were analyzed by an unpaired t-test (p-value cut-off ≤ 0.05), and further filtered for a fold-change of >1.5 between groups. To understand the possible relationships between these transcripts, the differentially expressed gene (DEG) list was imported into Ingenuity Pathway Analysis (IPA) software. Functional annotation and gene ontology of DEGs was further explored with DAVID [13].
Genotyping Analysis
DNA from subjects was purified from Paxgene preserved whole blood by centrifugation of 500 µl and treating the pellet with RNAse/proteinase K followed by phenol/chloroform extraction and ethanol precipitation [14]. SNPs in TCL1A were determined by TaqMan assay for the rs11849538 SNP on an ABI 7300, with dissociation temperature of 95˚C and annealing temperature of 60˚C. From the 24 patients for which expression data was obtained, sufficient DNA and genotyping results were obtained from 18 of the patients, including all 5 patients in the chemo-induced low EF group.
Cell culture
Rat cardiomyocyte progenitors (H9C2) were obtained from American Type Culture Collection (ATCC) and maintained in DMEM with 10% FBS and antibiotics. The MDR1 inhibitor, verapamil (Sigma), was tested by plating cells at 50,000 cells/well of a 24 well plate and allowing them to adhere overnight. Verapamil was added 2 hours prior to the addition of doxorubicin at specified doses. After 20 hours with doxorubicin, the viable cells were quantitated by their ability to metabolize MTT as described [15]. Briefly, the culture media was replaced with MTT for 4 hours and the metabolic product was quantified by dissolving the cells in 200 µl of DMSO and transferring the media to a 96 well plate, where absorbance at 570 nm was determined in a plate spectrophotometer.
Results
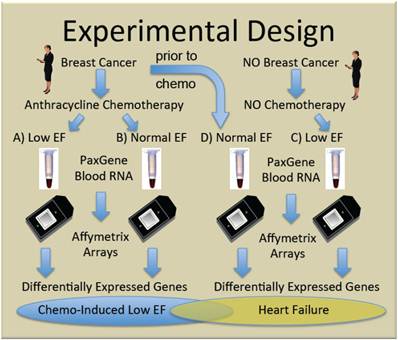
To generate new hypotheses about the etiology of chemo-induced CHF, we performed genome-wide transcript analysis with the Affymetrix U133+2 GeneChip on whole blood RNA from women receiving chemotherapy with anthracyclines who either did (Group A, n=5), or did not (Group B, n=10) develop subsequent cardiac dysfunction, as defined by ejection fractions (EF)≤40%. Women with non-ischemic cardiomyopathy not due to chemotherapy (Group C, n=4) were compared to breast cancer patients prior to chemo with normal EF (Group D, n=5) to identify heart failure-related transcripts independent of chemotherapy. The general experimental design is summarized in Figure 1.
Experimental design. Women with, and without breast cancer gave informed consent and were enrolled in the study. Some women with breast cancer given doxorubicin-containing chemotherapy developed clinically significant low ejection fractions (EF) (Group A) and were compared to similar women who did not develop low EF (Group B). To identify transcripts correlating with CHF, independent of chemotherapy, women with breast cancer and normal ejection fractions, were sampled prior to chemotherapy (Group D), and compared with women who developed low EFs but never received chemotherapy (Group C).
Clinical Parameters
Demographic and baseline cardiovascular parameters are shown in Table 1. While increasing age has been reported to be associated with reduced LV performance after chemotherapy [16], there was no significant difference in age between women who did or did not develop HF after chemo in our cohort (p>0.05). There were more women in Group D with hypertension and diabetes, but with the small group size, the difference was not statistically significant.
Classification and Baseline Values of Patients.
| Group | A | B | C | D | |
|---|---|---|---|---|---|
| Name | BrCa-Chemo-Low EF | BrCa-Chemo-Norm EF | No Ca-NoChemo-Low EF | BrCa-preChemo-Norm EF | |
| N= | 5 | 10 | 4 | 5 | |
| Breast Cancer | Yes | Yes | No | Yes | |
| Chemotherapy | Yes | Yes | No | No | |
| Low Ejection Fraction | Yes | No | Yes | No | |
| Age (mean) | 59.2 | 58.3 | 53.4 | 48.1 | NS |
| Age (median, range) | 56(51-80) | 60(42-76) | 51(47-63) | 53(43-58) | |
| Ejection Fraction (EF) | 24.8 | 60.9 | 26.6 | 68.0 | p<.001 |
| EF (median, range) | 20(15-40) | 58(52-69) | 24(22-35) | 66(55-86) | |
| Hypertensive | 0% | 0% | 0% | 40% | NS |
| Diabetic | 0% | 0% | 0% | 20% | NS |
A comparison of the two-dimensional echo images of a patient with normal LV systolic function versus a patient with chemo-induced dilated cardiomyopathy is shown in Additional file 1: Supplementary Figure 1. In the patient with normal LV function, the heart size is normal in diastole and decrease normally in systole, reflecting a normal EF. In contrast, with chemo-induced cardiomyopathy, the heart is dilated and shows little change in size with systolic contraction, and thus, in these patients, EF is typically significantly reduced.
Gene Expression Profiling of Whole Blood RNA
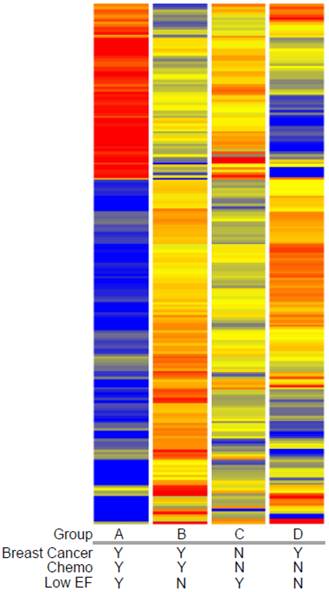
Expression profiling of blood indicates that women who developed a low EF after chemotherapy had 260 transcripts that differed >2-fold, with a t-test probability of p<0.05, compared to women receiving chemo who maintained normal EFs (A vs. B). Most of these transcripts (201) were not shared by women who developed low EFs independent of chemotherapy (C vs. D). As shown in Figure 2, the 201 transcripts that differentiate the chemo-induced low EF women (A) from women similarly treated, but retaining normal EF (B), tend to be more down-regulated (blue) than up-regulated (red). However, these differentially expressed transcripts are not similarly regulated in women with low EFs who never received chemo (C). However, 59 transcripts were identified that were differentially expressed in both chemo-dependent (A vs. B) and chemo-independent low EFs (C vs. D), suggesting these transcripts report some general cause or effect of low EFs (Additional file 2: Supplementary Tables 1 and 2). Selected transcripts from the 201 gene list, organized by putative function, are shown in Table 2, and discussed below.
Clustered heatmap of transcripts associated with chemotherapy-induced heart failure. Transcript profiles of blood from women with breast cancer who did (Group A) or did not (Group B) develop low EFs were compared to identify differentially-expressed genes (DEGs) by a combined fold-change (>1.5 fold) and t-test (p<0.05) filtering. To exclude transcripts that were associated with heart failure in women not receiving doxorubicin, women with breast cancer and normal cardiac performance were sampled prior to chemotherapy (Group D) and compared to women with low EFs but who never received chemotherapy (Group C). The resulting 201 transcripts were clustered by their expression pattern across the 4 groups with each row indicating one transcript. Red indicates increased expression, blue indicates decreased expression, with yellow indicating little or no change.
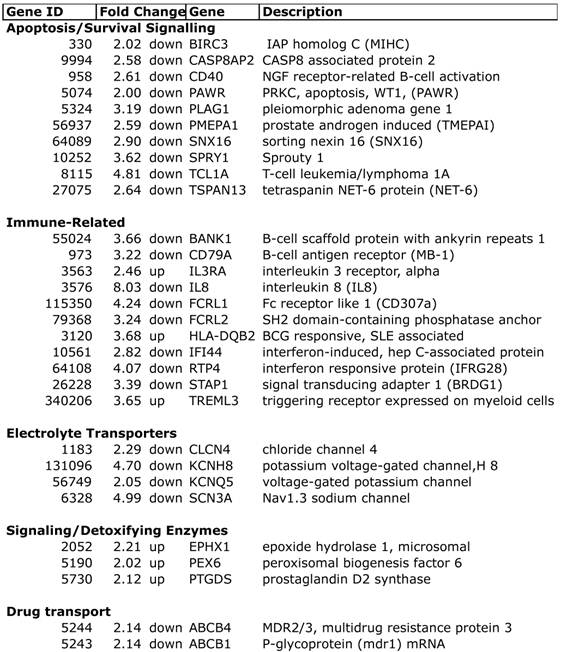
Selected differentially expressed transcripts in chemo-induced low ejection fractions.
Functional annotation: Analysis of the DEG list in DAVID and IPA helped to identify relationships between the transcripts. In the 'biological processes' category, 18 terms were statistically overrepresented, with transcripts mainly categorized into cellular activation, cellular proliferation, immune response, differentiation, signal transduction, and drug response/transport. With additional manual annotation, several general pathways with potential relevance to heart failure emerged.
Apoptosis: Among the DEGs unique to chemo-induced low EFs, there were notable changes in apoptosis-related transcripts. There was decreased expression of BIRC3, which is a member of the inhibitor of apoptosis protein family (cIAP2, IAP-C, MIHC), which are ubiquitin ligases that target the degradation of the TNF receptor interacting protein (RIP)[17]. Conversely, there was decreased expression of the proapoptotic caspase 8 associated protein 2 (CASP8AP2, FLASH, Flice-associated huge protein), which facilitates caspase 8 activation by TNF receptor or fas [18]. Interestingly, CASP8AP2 has been identified as mediating the cardiac ischemic preconditioning effect of mir210 [19]. PLAG1, decreased more than 3-fold, is a proto-oncogene transcription factor that regulates numerous apoptotic signals [20].
Notably, women with chemo-induced low EFs had a 4.8-fold decrease in the oncogene T-cell leukemia/lymphoma 1A (TCL1A). TCL1A is expressed in both cardiac and skeletal muscle, and is a known co-activator for AKT, one of the major pro-survival factors for cardiomyocytes. Additionally, genome-wide SNP analysis has previously identified TCL1A SNPs strongly associated with musculoskeletal adverse events in women receiving aromatase inhibitors (AI) [21]. The variant allele at rs11849538 is quite common (22-34% heterozygous, 6% homozygous), and creates a new estrogen receptor-binding site [21].
Immune-related: Considering that whole blood was profiled, it is reasonable that the majority of differentially-expressed transcripts fall into immune-related categories, particularly T-cell and B-cell activation and proliferation. However, recent data has suggested that hyper-activated innate immune responses could contribute to the progression of CHF (reviewed in [22]). Notably, there was a 2.5-fold increase in the interleukin-3 receptor (IL3R), and an 8-fold decrease in IL-8, a change twice as large as any other transcript. Expression levels in general, however, did not support a general activation of innate immunity in circulating cells, with consistent down-regulation of B-cell activation markers (BANK1, CD79A), Fc-like receptors (FCRL1, FCRL2), and two interferon-responsive transcripts (IFI44, RTP4/IFRG28). However, elevated expression of HLA-DQB2 could suggest some type of autoimmune reaction triggered by chemotherapy, because SNPs in DQB2 have been associated with rheumatoid arthritis [23] and lupus [24].
Electrolyte transporters: Another potential explanation for the increased cardiotoxicity in some women would be if there were inherited or acquired defects in membrane transporters for electrolytes, which could create performance deficits that would then be exacerbated by chemotherapy, leading to cell death. There were noticeable changes in four electrolyte channels: chloride channel 4 (CLCN4), Nav1.3 sodium channel (SCN3A), and two voltage-gated potassium channels (KCNH8, KCNQ5).
Signaling/Detoxifying Enzymes: Interestingly, the microsomal epoxide hydrolase (EPHX1), which is a known xenobiotic metabolizing enzyme for by-products of doxorubicin, was elevated 2.2-fold in the chemo-CHF group. Prior genotyping studies observed that EPHX1 polymorphisms were associated with both breast cancer risk and overall survival after therapy [25]. PEX6 was elevated 2-fold and plays a key role in the assembly of the peroxisome. Defects in PEX6 lead to elevated free radicals and cellular necrosis [26], suggesting that elevated PEX6 may be a compensatory response to increased free radical levels. Prostaglandin D2 synthase (PTGDS) mRNA was elevated 2.1-fold in the chemo-CHF cases, and the PTGDS protein has recently been reported as a circulating biomarker in coronary artery disease [27; 28] and PTGDS mRNA is elevated in epicardial adipose tissue of patients with coronary disease [29].
Drug Transport: Among the most straightforward explanations for increased cardiotoxicity would be elevated cardiac levels of doxorubicin, possibly secondary to decreased multidrug resistance proteins in the cardiac tissue. Thus, it is interesting that both multidrug resistance protein 1 (MDR1, p-glycoprotein, ABCB1) and MDR2/3 (ABCB4) mRNAs in blood were decreased more than 2-fold in the chemo patients that developed low EFs. MDR1 is a well-established efflux pump for doxorubicin [30]. Elevated levels of MDR1 in tumor cells [31], and promoter SNPs that increase MDR1 expression, are commonly part of the chemotherapy-resistant phenotype [32]. Acute cardiotoxicity of doxorubicin has previously been linked to G671V variants of MDR1 in case-control studies [33]. While it is well known that doxorubicin is a substrate for MDR1, recent in vitro analysis also suggests that this coding variant decreases the efflux of free radical adducts from cells [34]. Thus, if cardiac levels of MDR1 and MDR2/4 were reduced in parallel to the blood levels, the cells may experience both elevated doxorubicin levels and elevated retention of the induced free radicals.
Free Radical Levels
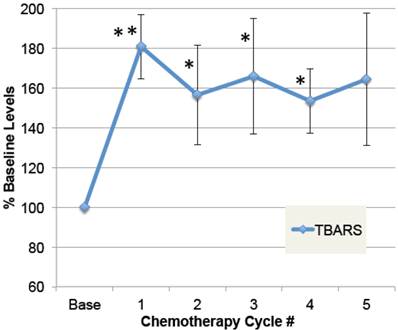
To determine whether doxorubicin caused elevations in free radical formation, a substudy was conducted on patients in which plasma samples were obtained pre-chemo, and 24 hours after at least five chemo cycles. As shown in Figure 3, plasma TBARS increased up to 2.5-fold after chemotherapy in some patients, with average levels elevated 64% in the four patients evaluated (ANOVA p≤0.01). Despite elevated free radicals, none of these patients developed cardiomyopathy or low EF within the study period. This confirms prior data demonstrating elevated plasma TBARS post-chemo [35], and extends it to demonstrate that the elevations are sustained over five chemo cycles.
Plasma levels of free radical modifications to protein. A small group of patients were followed prior to doxorubicin-based chemotherapy and then after multiple cycles of chemotherapy. Plasma samples, pre-chemo, and 24 hours after each chemo cycle, were specially preserved, stored at -80˚ C, and then analyzed for products of oxidative modification. Thiobarbituric-acid reactive substances (TBARS) in plasma are shown for pre-chemo (Base) and 5 subsequent chemo cycles (error bars = s.e.m.). The main effect of chemotherapy on TBARS, expressed as a percent of baseline values, was evaluated by ANOVA (p=0.01), and asterisks indicate where post-hoc t-tests were significant compared to pre-chemo baseline (** p≤0.01, * p≤0.05, n=4).
Genotyping Analysis
The almost 5-fold reduction in TCL1A mRNA levels in the chemo-CHF group suggested that it might contribute to increased cardiac apoptosis. Genome-wide SNP analysis has previously identified TCL1A SNPs strongly associated with musculoskeletal adverse events in women receiving aromatase inhibitors (AI) [21]. The variant allele (G) at rs11849538 is quite common (HapMap variant allele frequency of 8-43%), and creates an estrogen receptor-binding site. However, genotyping for this SNP in 18 patients, including all of the women with chemo-induced low EFs (Group A), did not detect differential variant allele frequencies, with essentially all groups near 40% (not shown).
Effect of MDR1 inhibition on response to doxorubicin in vitro
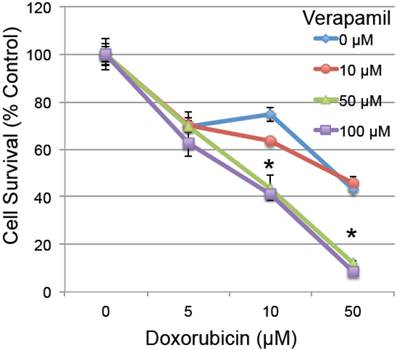
To determine whether the reduced MDR1 could have a functional effect on cardiomyocyte survival, H9C2 rat embryonic cardiomyocytes were pretreated with verapamil (0-100 µM), a known MDR1 inhibitor with an effective IC50 of 120 µM [36], 2 hours prior to low doses of doxorubicin (0-50 µM). Pretreatment with verapamil significantly enhanced the toxicity of doxorubicin (ANOVA p<0.001 for both main effects and interaction), which was significant in pairwise testing at the combined doses of 50-100 µm verapamil and 10-50 µM doxorubicin in H9C2 cardiomyocytes (Figure 4). This confirms prior studies indicating that inhibition of MDR1 activity by verapamil increases the toxic effect of doxorubicin in cardiomyocytes [37-40].
The effect of MDR1 inhibition on doxorubicin-induced cell death. Rat H9C2 cardiomyocytes were pretreated with the MDR1 inhibitor verapamil for 2 hours at the specified doses (0-100 µM). Cells were then treated with increasing doses of doxorubicin (0-50 µM) and cell survival was determined 20 hours later by measuring their metabolic reduction of MTT, with the product measured by O.D. at 570 nm (n=3/group, error bars = s.e.m.). Asterisk indicates a significant change between verapamil treated and untreated (p≤0.05).
Discussion
The general mechanisms of chemo-induced heart failure have been extensively studied and two major etiologies have emerged: 1) direct damage to the cardiomyocyte's DNA, triggering apoptosis, and 2) induction and accumulation of cellular free radicals causing toxicity and cardiomyocyte death. However, the particular inter-individual variations that affect the susceptibility to chemo-induced CHF are poorly understood. Pretreatment variables, such as age and left ventricular EF have been identified as predictors of the adverse cardiac response to epirubicin, suggesting that a pre-existing, but compensated cardiac performance deficit may increase the susceptibility to chemotherapy-induced damage [16].
Several lines of evidence suggest that the survival/apoptosis pathways are a major determinant of the cardiac outcome after doxorubicin. In rat neonatal cardiomyocytes, overexpression of the apoptosis regulator ARC inhibits doxorubicin-induced death, while siRNA to ARC increases apoptosis induced by doxorubicin [41], suggesting that doxorubicin induces death via caspase-dependent mechanisms. In a rat model, intracoronary AKT1 gene delivery mitigates doxorubicin-induced reductions in fractional shortening, and improves LV function at five weeks post-treatment [42]. The apparent increase in cardiotoxicity associated with obesity led to pre-clinical studies in which adiponectin was shown to reduce cardiac dysfunction after doxorubicin by its ability to stimulate AKT activity [43]. Likewise, microarray analysis of cardiac tissue in mice developing LV dysfunction after doxorubicin treatment showed changes in transcripts associated with hypertrophy (STARS), ATP levels (SNF1-kinase), and apoptosis, (AXUD1, a downstream target of AXIN1) [44]. Thus, the cardiac levels of TCL1A may be a critical determinant of AKT activity in the cardiomyocytes, and thus a key factor in susceptibility to doxorubicin cardiotoxicity. It is interesting to speculate that the reduced blood levels of TCL1A might be related to the known effect of Epstein-Barr Virus (EBV) latent membrane protein1 (LMP1), which suppresses TCL1A levels via miRNA 29-b [45].
The second major pathway of cardiac damage, free radical injury, is influenced by changes in antioxidant systems, and variations in the efflux pumps for doxorubicin and the free radicals that it creates. Polymorphisms in ABCB1/MDR1, such as C3435T, are known to affect mRNA levels in breast cancer specimens [46] and may be related to therapeutic outcome from anthracyclines [32; 47], possibly by affecting mRNA stability [48]. However, the relationship between MDR1 polymorphisms and mRNA expression or drug transport remains complicated [49]. For instance, cardiac levels of MDR1 are affected by an exon 21 polymorphism, but not by the C3435T SNP [50]. Transgenic mice overexpressing MDR1 selectively in the myocardium are protected from damage inflicted by doxorubicin [51]. Further, knockout of heat shock factor 1 (HSF1) increased the expression of MDR1 in the heart, increased efflux of doxorubicin, and reduced mortality [40]. MDR1 levels could be modulated by AKT activity because signaling through phosphoinositide 3-kinase (PI3K) and AKT can increase MDR1 expression [52], which is consistent with the reduction in both TCL1A and MDR1 in the present studies. Recent data in experimental models has suggested that one important factor might be the impact of the chemotherapy on the cardiac stem/progenitor cells. When syngeneic cardiac progenitors were reintroduced post-chemo in mice, cardiac performance was restored [53]. It is worth noting that MDR1 protein is considered one of the key markers of cardiac stem cells [54].
The present studies have several limitations that deserve recognition. First, this is a relatively small sample size, with only five women who have chemo-induced low EFs: a consequence of the fact that oncologists have been aware of this complication and reduce the chemo dosages to render this a relatively rare side effect. Second, of necessity, the studies employed a surrogate marker for expression profiling, peripheral blood, which may or may not reflect the expression profile in the heart. Based on the present studies, it would be important to investigate whether cardiac MDR1 levels, or MDR-like efflux activity, are predictive of cardiomyopathy after chemotherapy. Third, because of the low incidence of chemo-induced HF, it was impractical to sample patients prior to the initiation of chemo, and then follow them to determine differences between women who did, of did not develop low EFs. Rather, it was more practical to sample patients with low EFs after chemotherapy, and compare them with patients with normal EF. The general effect of low EF on expression profiles was then determined, and subtracted, by analyzing non-chemo patients with low EF.
Conclusions
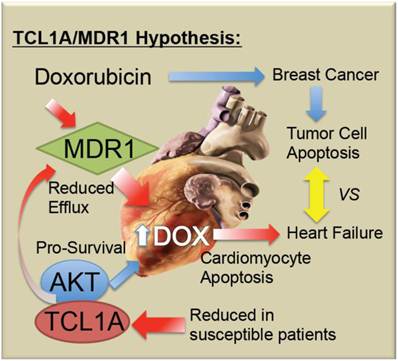
Doxorubicin-induced cardiotoxicity remains a devastating complication of a potentially life-saving drug. As summarized in Figure 5, it is proposed that chemo-induced cardiomyopathy may be due to a genetic predisposition or progressively acquired tendency to reduced TCL1A levels, reducing AKT activity and thereby causing increased apoptotic sensitivity to doxorubicin. Furthermore, it is possible that coincidental or AKT-related changes in MDR1 levels in the myocardium decrease the efflux of doxorubicin, thereby causing its accumulation, along with toxic free radicals in the cardiomyocytes. Future studies will be needed to determine whether screening for blood levels of TCL1A and MDR1 mRNA are helpful to identify women at greatest risk of chemo-induced heart failure.
Schematic model of TCL1A and MDR1 effects on the cardiac response to chemotherapy. Doxorubicin and related anthracyclines intercalate on DNA and induce DNA damage, as well as inducing free radical production, combining to preferentially induce the death of rapidly dividing tumor cells. However, cardiomyocytes may also undergo apoptosis, leading to decreased cardiac performance, and additional stress on the remaining cardiomyocytes. The present studies suggest that reduced cardiac expression of TCL1A leads to a reduced survival and protective effect of AKT1, making the cells more sensitive to apoptosis. Further, reduced cardiac expression of TCL1A/AKT could lead to reduced ABCB1/MDR1, and ABCB4, drug efflux pumps for doxorubicin, which would be expected to increase the intracellular levels of doxorubicin, as well as reducing the ability of the cardiomyocytes to export the free radicals that doxorubicin induces.
Abbreviations
CHF, congestive heart failure; DEG, differential expressed genes; EF, ejection fraction; EGF, epidermal growth factor; ESP, end-stage pressure; ESV, end-systolic volume; FBS, fetal bovine serum; HSF1, heat shock factor 1; LV, left ventricular; MDR1, multidrug resistance protein 1; mRNA, messenger RNA; PI3K, phosphoinositide 3-kinase; siRNA, small interfering RNA; SNP, single nucleotide polymorphisms; SW, stroke work; TBARS, thiobarbituric acid reactive substances; TCL1A, T-cell leukemia/lymphoma 1A.
Supplementary Material
Additional File 1Supplementary Figure 1 presents an echocardiography from typical patients with, and without, chemo-induced cardiomyopathy.
Supplementary Table 1 lists all 260 transcripts differentially expressed between Group A and Group B, with a sub-listing of the 59 transcripts differentially expressed in both chemo-dependent (A vs. B) and chemo-independent low EF (C vs. D). Supplementary Table 2 lists all 1531 transcripts differentially expressed between Group C and Group D. All original Affymetrix .cel files, as well as the summarized RMA raw data, and per gene normalized data have been deposited at NCBI Gene Expression Omnibus (GEO) under the Accession # GSE40447.
Acknowledgements
This work was supported by The GWU Heart and Vascular Institute. The authors are grateful to all of the research volunteers who selflessly participated as subjects in this study despite their serious medical situations. The heart graphic incorporated into Figure 5 is courtesy of Patrick J. Lynch, medical illustrator; C. Carl Jaffe, MD, cardiologist, Yale University, via WikiCommons.
Competing interests
TM holds an equity interest in Cellgenex, Inc. a privately-held genomics company developing RNA-based diagnostics tests in the cardiovascular area. However, the present studies are not supported by Cellgenex, thus there are no competing interests that would affect the results of these studies. The other authors declare no competing interests.
References
1. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. New Engl J Med. 1998;339(13):900-905
2. Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Int Med. 1996;125(1):47-58
3. Chien KR. Herceptin and the heart--a molecular modifier of cardiac failure. New Engl J of Med. 2006;354(8):789-790
4. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. New Engl J Med. 2001;344(11):783-792
5. Kobayashi S, Ito A, Okuzaki D, Onda H, Yabuta N, Nagamori I, Suzuki K, Hashimoto H, Nojima H. Expression profiling of PBMC-based diagnostic gene markers isolated from vasculitis patients. DNA Res. 2008;15(4):253-265
6. Arasappan D, Tong W, Mummaneni P, Fang H, Amur S. Meta-analysis of microarray data using a pathway-based approach identifies a 37-gene expression signature for systemic lupus erythematosus in human peripheral blood mononuclear cells. BMC Med. 2011;9:65
7. Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, Sakuma M, Shi C, Zago AC, Garasic J. et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113(19):2278-2284
8. Rosenberg S, Elashoff MR, Beineke P, Daniels SE, Wingrove JA, Tingley WG, Sager PT, Sehnert AJ, Yau M, Kraus WE. et al. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Ann Intern Med. 2010;153(7):425-434
9. Liew CC. Expressed genome molecular signatures of heart failure. Clin Chem Lab Med. 2005;43(5):462-469
10. Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quinones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30(6):1527-1533
11. Yu CM, Gorcsan J 3rd, Bleeker GB, Zhang Q, Schalij MJ, Suffoletto MS, Fung JW, Schwartzman D, Chan YS, Tanabe M. et al. Usefulness of tissue Doppler velocity and strain dyssynchrony for predicting left ventricular reverse remodeling response after cardiac resynchronization therapy. Am J Cardiol. 2007;100(8):1263-1270
12. Mak IT, Weglicki WB. Antioxidant activity of calcium channel blocking drugs. Methods Enzymol. 1994;234:620-630
13. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44-57
14. Andolfatto S, Namour F, Garnier AL, Chabot F, Gueant JL, Aimone-Gastin I. Genomic DNA extraction from small amounts of serum to be used for alpha1-antitrypsin genotype analysis. Eur Respir J. 2003;21(2):215-219
15. Heo DS, Park JG, Hata K, Day R, Herberman RB, Whiteside TL. Evaluation of tetrazolium-based semiautomatic colorimetric assay for measurement of human antitumor cytotoxicity. Cancer Res. 1990;50:3681-3690
16. Schaadt B, Kelbaek H. Age and left ventricular ejection fraction identify patients with advanced breast cancer at high risk for development of epirubicin-induced heart failure. J Nucl Cardiol. 1997;4(6):494-501
17. Park SM, Yoon JB, Lee TH. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004;566(1-3):151-156
18. Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, Yonehara S. The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature. 1999;398(6730):777-785
19. Kim HW, Haider HK, Jiang S, Ashraf M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J Biol Chem. 2009;284(48):33161-33168
20. Voz ML, Mathys J, Hensen K, Pendeville H, Van Valckenborgh I, Van Huffel C, Chavez M, Van Damme B, De Moor B, Moreau Y. et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene. 2004;23(1):179-191
21. Ingle JN, Schaid DJ, Goss PE, Liu M, Mushiroda T, Chapman JA, Kubo M, Jenkins GD, Batzler A, Shepherd L. et al. Genome-wide associations and functional genomic studies of musculoskeletal adverse events in women receiving aromatase inhibitors. J Clin Oncol. 2010;28(31):4674-4682
22. Shi Y, Moon M, Dawood S, McManus B, Liu PP. Mechanisms and management of doxorubicin cardiotoxicity. Herz. 2011;36(4):296-305
23. Kochi Y, Yamada R, Kobayashi K, Takahashi A, Suzuki A, Sekine A, Mabuchi A, Akiyama F, Tsunoda T, Nakamura Y. et al. Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum. 2004;50(1):63-71
24. Barcellos LF, May SL, Ramsay PP, Quach HL, Lane JA, Nititham J, Noble JA, Taylor KE, Quach DL, Chung SA. et al. High-density SNP screening of the major histocompatibility complex in systemic lupus erythematosus demonstrates strong evidence for independent susceptibility regions. PLoS Genet. 2009;5(10):e1000696
25. Khedhaier A, Hassen E, Bouaouina N, Gabbouj S, Ahmed SB, Chouchane L. Implication of Xenobiotic Metabolizing Enzyme gene (CYP2E1, CYP2C19, CYP2D6, mEH and NAT2) polymorphisms in breast carcinoma. BMC Cancer. 2008;8:109
26. Jungwirth H, Ring J, Mayer T, Schauer A, Buttner S, Eisenberg T, Carmona-Gutierrez D, Kuchler K, Madeo F. Loss of peroxisome function triggers necrosis. FEBS Lett. 2008;582(19):2882-2886
27. Inoue T, Eguchi Y, Matsumoto T, Kijima Y, Kato Y, Ozaki Y, Waseda K, Oda H, Seiki K, Node K. et al. Lipocalin-type prostaglandin D synthase is a powerful biomarker for severity of stable coronary artery disease. Atherosclerosis. 2008;201(2):385-391
28. Miwa Y, Oda H, Shiina Y, Shikata K, Tsushima M, Nakano S, Maruyama T, Kyotani S, Eguchi N, Urade Y. et al. Association of serum lipocalin-type prostaglandin D synthase levels with subclinical atherosclerosis in untreated asymptomatic subjects. Hypertens Res. 2008;31(10):1931-1939
29. Guauque-Olarte S, Gaudreault N, Piche ME, Fournier D, Mauriege P, Mathieu P, Bosse Y. The transcriptome of human epicardial, mediastinal and subcutaneous adipose tissues in men with coronary artery disease. PLoS One. 2011;6(5):e19908
30. Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human "MDR1" gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA. 1987;84(9):3004-3008
31. Krech T, Scheuerer E, Geffers R, Kreipe H, Lehmann U, Christgen M. ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett. 2012;315(2):153-160
32. George J, Dharanipragada K, Krishnamachari S, Chandrasekaran A, Sam SS, Sunder E. A single-nucleotide polymorphism in the MDR1 gene as a predictor of response to neoadjuvant chemotherapy in breast cancer. Clinical Breast Cancer. 2009;9(3):161-165
33. Wojnowski L, Kulle B, Schirmer M, Schluter G, Schmidt A, Rosenberger A, Vonhof S, Bickeboller H, Toliat MR, Suk EK. et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112(24):3754-3762
34. Jungsuwadee P, Zhao T, Stolarczyk EI, Paumi CM, Butterfield DA, St Clair DK, Vore M. The G671V variant of MRP1/ABCC1 links doxorubicin-induced acute cardiac toxicity to disposition of the glutathione conjugate of 4-hydroxy-2-trans-nonenal. Pharmacogenet Genomics. 2012;22(4):273-284
35. Panis C, Herrera AC, Victorino VJ, Campos FC, Freitas LF, De Rossi T, Colado Simao AN, Cecchini AL, Cecchini R. Oxidative stress and hematological profiles of advanced breast cancer patients subjected to paclitaxel or doxorubicin chemotherapy. Breast Cancer Res Treat. 2012;133(1):89-97
36. Kars MD, Iseri OD, Gunduz U, Molnar J. Reversal of multidrug resistance by synthetic and natural compounds in drug-resistant MCF-7 cell lines. Chemotherapy. 2008;54(3):194-200
37. Gruber A, Larsson R, Nygren P, Bjorkholm M, Peterson C. A non-P-glycoprotein-mediated mechanism of vincristine transport which is affected by resistance modifiers and present in chemosensitive cells. Leukemia. 1994;8(6):985-989
38. Estevez MD, Wolf A, Schramm U. Effect of PSC 833, verapamil and amiodarone on adriamycin toxicity in cultured rat cardiomyocytes. Toxicol In Vitro. 2000;14(1):17-23
39. Farokhpour M, Karbalaie K, Tanhaei S, Nematollahi M, Etebari M, Sadeghi HM, Nasr-Esfahani MH, Baharvand H. Embryonic stem cell-derived cardiomyocytes as a model system to study cardioprotective effects of dexamethasone in doxorubicin cardiotoxicity. Toxicol In Vitro. 2009;23(7):1422-1428
40. Krishnamurthy K, Vedam K, Kanagasabai R, Druhan LJ, Ilangovan G. Heat shock factor-1 knockout induces multidrug resistance gene, MDR1b, and enhances P-glycoprotein (ABCB1)-based drug extrusion in the heart. Proc Natl Acad Sci U S A. 2012;109(23):9023-8
41. An J, Li P, Li J, Dietz R, Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med (Berl). 2009;87(4):401-410
42. Taniyama Y, Walsh K. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol. 2002;34(10):1241-1247
43. Maruyama S, Shibata R, Ohashi K, Ohashi T, Daida H, Walsh K, Murohara T, Ouchi N. Adiponectin ameliorates doxorubicin-induced cardiotoxicity through Akt protein-dependent mechanism. J Biol Chem. 2011;286(37):32790-32800
44. Yi X, Bekeredjian R, DeFilippis NJ, Siddiquee Z, Fernandez E, Shohet RV. Transcriptional analysis of doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2006;290(3):H1098-1102
45. Anastasiadou E, Boccellato F, Vincenti S, Rosato P, Bozzoni I, Frati L, Faggioni A, Presutti C, Trivedi P. Epstein-Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene. 2010;29(9):1316-1328
46. Taheri M, Mahjoubi F, Omranipour R. Effect of MDR1 polymorphism on multidrug resistance expression in breast cancer patients. Genet Mol Res. 2010;9(1):34-40
47. Cizmarikova M, Wagnerova M, Schonova L, Habalova V, Kohut A, Linkova A, Sarissky M, Mojzis J, Mirossay L, Mirossay A. MDR1 (C3435T) polymorphism: relation to the risk of breast cancer and therapeutic outcome. Pharmacogenomics Journal. 2010;10(1):62-69
48. Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet Genomics. 2005;15(10):693-704
49. Sakaeda T. MDR1 genotype-related pharmacokinetics: fact or fiction? Drug Metab Pharmacokinet. 2005;20(6):391-414
50. Meissner K, Jedlitschky G, Meyer zu Schwabedissen H, Dazert P, Eckel L, Vogelgesang S, Warzok RW, Bohm M, Lehmann C, Wendt M. et al. Modulation of multidrug resistance P-glycoprotein 1 (ABCB1) expression in human heart by hereditary polymorphisms. Pharmacogenetics. 2004;14(6):381-385
51. Dell'Acqua G, Polishchuck R, Fallon JT, Gordon JW. Cardiac resistance to adriamycin in transgenic mice expressing a rat alpha-cardiac myosin heavy chain/human multiple drug resistance 1 fusion gene. Hum Gene Ther. 1999;10(8):1269-1279
52. Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe S, Mills GB, Unate H. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene. 2002;21(13):1945-1954
53. De Angelis A, Piegari E, Cappetta D, Marino L, Filippelli A, Berrino L, Ferreira-Martins J, Zheng H, Hosoda T, Rota M. et al. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation. 2010;121(2):276-292
54. Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. N Engl J Med. 2002;346(1):5-15
Author contact
![]() Corresponding author: Sidney W. Fu, M.D., Ph.D. Department of Medicine, Division of Genomic Medicine, The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C, Washington, DC 20037, Email: sfuedu Tel: 202-994-4767 Fax: 202-994-8924.
Corresponding author: Sidney W. Fu, M.D., Ph.D. Department of Medicine, Division of Genomic Medicine, The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C, Washington, DC 20037, Email: sfuedu Tel: 202-994-4767 Fax: 202-994-8924.