Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(4):361-369. doi:10.7150/ijbs.6081 This issue Cite
Research Paper
Complete Mitochondrial Genome Sequence Data Provides Genetic Evidence That the Brown Dog Tick Rhipicephalus sanguineus (Acari: Ixodidae) Represents a Species Complex
Guo-Hua Liu1,2, Fen Chen1,3, Yi-Zhou Chen1,4, Hui-Qun Song1, Rui-Qing Lin1,4, Dong-Hui Zhou1, Xing-Quan Zhu1,2, ![]()
1. State Key Laboratory of Veterinary Etiological Biology, Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, Gansu Province, China,
2. College of Veterinary Medicine, Hunan Agricultural University, Changsha, Hunan Province, China,
3. Department of Preclinical Medicine, Xiangnan University, Chenzhou, Hunan Province, China,
4. College of Veterinary Medicine, South China Agricultural University, Guangzhou, Guangdong Province, China.
Received 2013-2-12; Accepted 2013-4-16; Published 2013-4-22
Abstract
Ticks are blood-sucking ectoparasites of great medical and veterinary significance that can transmit bacteria, protozoa, fungi and viruses, and cause a variety of human and animal diseases worldwide. In the present study, we sequenced the complete mitochondrial (mt) genome of Rhipicephalus sanguineus from China (RSC) and compared with that of R. sanguineus from USA (RSU). Nucleotide sequence difference in the full mt genome was 11.23% between RSC and RSU. For the 13 protein-coding genes, comparison revealed sequence divergences at both the nucleotide (9.34-15.65%) and amino acid (2.54-19.23%) levels between RSC and RSU. In addition, sequence comparison of the conserved mt cox1 and cytb genes among multiple individual R. sanguineus revealed substantial nucleotide differences between RSC and RSU but limited sequence variation within RSC. Phylogenetic analysis of ticks based on the amino acid sequence data of 13 protein-coding genes revealed that R. sanguineus from China and R. sanguineus from USA represent sister taxa (likely separate species). Taken together, the findings support the recently proposal that R. sanguineus tick may represents a species complex of at least two closely related species.
Keywords: Rhipicephalus sanguineus, species complex, mitochondrial genome, mitochondrial DNA, phylogenetic analysis.
Introduction
Ticks are obligate blood-sucking ectoparasites of a wide range of animals and humans, which currently are considered as the second most important transmitters of a range of pathogens [1]. There are three tick families: the Ixodidae (hard ticks), the Argasidae (soft ticks), and the Nuttalliellidae (restricted to southern Africa) [2,3]. The Ixodidae is the most important tick family of medical and veterinary importance, which consists of the prostriate and metastriate ticks [4]. They are able to maintain and transmit many pathogens, including bacteria, helminths, protozoa, and viruses [2,5].
Rhipicephalus sanguineus (Ixodidae: Rhipicephalus), commonly known as the brown dog tick, is the most widespread tick of the world, mainly within latitudes 35°S and 50°N [6]. This tick usually parasitizes dogs, but it can be often found on cattle, horses, goats, cats, wild animals, and humans [5]. In China, R. sanguineus is widely distributed and is the dominant tick species in working dogs [7]. More importantly, R. sanguineus is a vector of many pathogens affecting humans and animals, such as Babesia canis, Ehrlichia canis and Rickettsia conorii, and causes considerable public health problems in humans and considerable economic losses to animals globally [6].
Identification and differentiation of R. sanguineus from other closely-related ticks have traditionally been based on morphological features (e.g., mean size, genital aperture, sexual dimorphism and capitulum) [8]. However, it is not always possible to accurately identify and differentiate R. sanguineus from other closely-related ticks based on morphological descriptions at some developmental stages, such as larva and nymph. Furthermore, the size of R. sanguineus may vary widely among different populations or geographical origins [9]. Over the last years, there has been considerable debate as to the specific taxonomic status of R. sanguineus [10-17]. For example, R. sanguineus was considered as a species complex of about 10 closely related species based on traditional morphological approach [5,6]. However, the systematic status of the members of the R. sanguineus group is still unclear, resulting in their misidentification [18,19]. To date, phenotypic techniques have considerable limitations for the precise identification and differentiation of all the members belonging to this group [6]. Molecular analytical tools, employing appropriate genetic markers, in particular mitochondrial (mt) DNA markers, have proven valuable complementary tools for overcoming this limitation and have been used to identify and differentiate tick species [15].
The metazoan mt genome, ranging in length from 14 to 18 kb approximately, is typically circular and usually contains 36-37 genes, including 12-13 protein-coding genes, 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes and 2 non-coding control region (NCR) and some intergenic spacers [20,21]. Mitochondrial DNA (mtDNA) sequences are useful molecular markers for the identification and differentiation of organisms, particularly for population genetic and systematic investigations among animal species [22-29]. Therefore, the objectives of the present study were: (i) to characterize the mt genome of R. sanguineus from China (RSC), (ii) to compare this mt genome with that of R. sanguineus from USA (RSU), (iii) to test the hypothesis that R. sanguineus is a complex of some closely related species by phylogenetic analysis based on the amino acid sequence data of mt protein-coding genes.
Materials and Methods
Parasites and DNA extraction
Adult ticks representing R. sanguineus from China (RSC) were obtained from the skin of infected pet dogs at an animal hospital in Guangzhou, Guangdong Province, China. The ticks were washed in physiological saline, identified preliminarily to species based on morphological characters and predilection sites [8], fixed in 70% (v/v) ethanol and stored at -20 °C until use. Total genomic DNA was isolated from individual tick using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (Wizard® SV Genomic DNA Purification System, Promega). The identity of these ticks were further ascertained as R. sanguineus by PCR amplification and subsequent sequencing of the region spanning the first internal transcribed spacer (ITS-1), the 5.8S and the second internal transcribed spacer (ITS-2) as reported previously [30]. The ITS-2 sequence of the representative female R. sanguineus China sample (sample code FSF1) had 99.2% similarity with that of R. sanguineus from Australia (GenBank accession no. AF271283) [31].
Long-PCR amplification and sequencing
Four primers (Table 1) were designed based on mtDNA sequences of R. sanguineus (USA isolate, ie, RSU, GenBank accession No. NC_002074) [32] by using Program Primer Premier 5.0 (PREMIER Biosoft International). Long-PCR reactions (25 μl) were performed in 2 mM MgCl2, 0.2 mM each of dNTPs, 2.5 μl 10× rTaq buffer, 2.5 μM of each primer, 1.25 U rTaq polymerase (Takara), and 1 μl of DNA sample in a thermocycler (Biometra) under the following conditions: 92 °C for 2 min (initial denaturation), then 92 °C for 10 s (denaturation), 45-50 °C for 30 s (annealing), and 60 °C for 4 min (extension) for 6 cycles, followed by 92 °C for 10 s, 45-50 °C for 30 s, and 62 °C for 4 min for 25 cycles and a final extension at 60 °C for 10 min. Samples containing no DNA (no-DNA controls) were included in each amplification run, and in neither case were amplicons detected in the no-DNA controls (data not shown). Each amplicon (5 µL) was examined by agarose (1%) gel electrophoresis, stained with ethidium bromide and photographed using a gel documentation system (UVItec). PCR products were sequenced by Sangon Company (Shanghai, China) from both directions using a primer walking strategy.
Sequences of primers used to amplify Long-PCR fragments from Rhipicephalus sanguineus.
| Name of primer | Sequence (5' to 3') |
|---|---|
| RS1F | GGTCAATGTTCAGAAATTTGTGG |
| RS1R | ACATGATCTGAGTTCAAACC |
| RS2F | TCCGGTTTGAACTCAGATC |
| RS2R | TCAGCATTAGTTCATTCCTC |
| RS3F | CTAAAGTAGAGGAATGAACTAATGCTG |
| RS3R | AAAAGGTAGTAGAAAATGTAAAGTGAA |
| RS4F | ACTTATTGAATTTGGGGAGG |
| RS4R | CACAAATTTCTGAACATTGACC |
Gene annotation and sequence analysis
Sequences were assembled manually and aligned against the complete mt genome sequence of RSU using the computer program Clustal X 1.83 [33] to identify gene boundaries by comparison to those of RSU [32]. The open-reading frames and codon usage profiles of protein-coding genes were analysed by the Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the invertebrate mitochondrial code. Translation initiation and translation termination codons were identified based on comparison with the mt genome of RSU. The amino acid sequences inferred for the mt genes of RSC were aligned with those of RSU by using Clustal X 1.83. Based on pairwise comparison, amino acid identity (%) was calculated for homologous genes. Codon usage was examined based on the relationships between the nucleotide composition of codon families and amino acid occurrence, where the genetic codons are partitioned into AT rich codons, GC-rich codons and unbiased codons. The tRNA genes were identified using tRNAscan-SE [34], or by eye inspection, and rRNA genes were identified by comparison with the mt genome of RSU [32].
Phylogenetic analyses
Phylogenetic relationship among 15 tick species (Table 2), plus the mtDNA sequence of RSC obtained in the present study, was reconstructed based on amino acid sequences of 13 protein-coding genes using one Acariformes species (Panonychus citri, GenBank accession no. NC_014347) as the outgroup. Each gene was translated into amino acid sequence using the invertebrate mitochondrial genetic code in MEGA 5 [35], and aligned based on its amino acid sequence using default settings, and ambiguously aligned regions were excluded using Gblocks online server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) with the default parameters [36] using less stringent selection criteria. The amino acid sequences of all the 13 protein-coding genes were then concatenated into single alignment for phylogenetic analyses. Three different inference methods, namely maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI) were used for phylogenetic analyses. MP analysis was performed using PAUP* 4.0b10 [37], with indels treated as missing character states. A total of 1,000 random addition searches using tree bisection-reconnection (TBR) branch swapping were performed for each MP analysis. Bootstrap probability (BP) was calculated from 1000 bootstrap replicates with 10 random additions per replicate in PAUP. PhyML 3.0 [38] was used for ML analyses of mt amino acid and nucleotide sequences, and ProtTest 10.2 for the amino acid sequences and jModelTest for nucleotide sequences [39,40] were used to choose the model of protein evolution (MtArt+G+F model for the amino acid sequences and GTR+I+G model for cox1 and cytb nucleotide sequences) based on the Akaike information criterion (AIC) with its parameter for the ML analysis. 100 bootstrap replicates were run and bootstrap replicates were calculated. BI was conducted with four independent Markov chains run for 1,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generations in MrBayes 3.1.1 [41]. The first 2,500 trees were omitted as burn-in and the remaining trees were used to calculate Bayesian posterior probabilities (PP). Phylograms were drawn using the TreeView program version 1.65 [42].
Mitochondrial genome sequences of ticks sequenced completely.
| Family | Species | The size of mtDNA (bp) | GenBank accession numbers |
|---|---|---|---|
| Ixodidae | Amblyomma triguttatum | 14740 | NC_005963 |
| Amblyomma elaphense | 14627 | NC_017758 | |
| Aponomma fimbriatum | 14705 | NC_017759 | |
| Amblyomma sphenodonti | 14772 | NC_017745 | |
| Haemaphysails flava | 14686 | NC_005292 | |
| Ixodes hexagonus | 14539 | NC_002010 | |
| Ixodes holocyclus | 15007 | NC_005293 | |
| Ixodes persulcatus | 14539 | NC_004370 | |
| Ixodes uriae | 13717 | NC_006078 | |
| Rhipicephalus sanguineus | 14710 | NC_002074 | |
| Bothriocroton undatum | 14769 | NC_017757 | |
| Bothriocroton concolor | 14809 | NC_017756 | |
| Argasidea | Carios capensis | 14418 | NC_005291 |
| Ornithodoros moubata | 14398 | NC_004357 | |
| Ornithodoros porcinu | 14378 | NC_005820 |
Sequencing and analysis of the cox1 and cytb genes from multiple individuals of RSC
A fragment of cox1 (850 bp) was amplified by PCR with primers cox1F (5'-GGAACAATATATTTAATTTTTGG-3') and cox1R (5'-ATCTATCCCTACTGTAAATATATG-3') [43], and a fragment of cytb (550 bp) was amplified by PCR with primers cytbF (5'-ATATTCATGTAGCTCGAGGA-3') and cytbR (5'-ATTGATCGTAAAATTGCGTA-3') from multiple individuals of RSC (Table 3). P. citri (accession no. NC_014347) was used as the outgroup for phylogenetic analyses. All cox1 and cytb sequences were aligned using Clustal X 1.83, and the alignment was modified manually, and then subjected to phylogenetic analysis using the same three methods as described above.
Rhipicephalus sanguineus samples collected from Guangdong Province, China, and accession numbers of their partial mitochondrial cox1 and cytb sequences.
| Sample codes | Geographical origin | Accession numbers | |
|---|---|---|---|
| cox1 | cytb | ||
| GTF2 | Tianhe, Guangdong | JX416303 | JX416319 |
| GTF3 | Tianhe , Guangdong | JX416304 | JX416320 |
| GBF1 | Baiyun, Guangdong | JX416299 | JX416315 |
| GBM1 | Baiyun, Guangdong | JX416300 | JX416316 |
| GPF1 | Panyu, Guangdong | JX416305 | JX416321 |
| GPF2 | Panyu, Guangdong | JX416306 | JX416322 |
| GHF1 | Haizhu, Guangdong | JX416301 | JX416317 |
| GHM1 | Haizhu, Guangdong | JX416302 | JX416318 |
| CHF1 | Conghua, Guangdong | JX416297 | JX416313 |
| CHM1 | Conghua, Guangdong | JX416298 | JX416314 |
| FSF1 | Foshan, Guangdong | JX416293 | JX416309 |
| FSM1 | Foshan, Guangdong | JX416294 | JX416310 |
| DGF1 | Dongguan, Guangdong | JX416295 | JX416311 |
| DGM1 | Dongguan, Guangdong | JX416296 | JX416312 |
| SZM1 | Shenzhen, Guangdong | JX416307 | JX416323 |
| SZM2 | Shenzhen, Guangdong | JX416308 | JX416324 |
Results
Genome content and organization
The complete mt genome of RSC was 14,714 bp in size (Figure 1), and the mtDNA sequence was deposited in GenBank (accession no. JX416325). The RSC mt genome contains 13 protein-coding genes (cox1-3, nad1-6, nad4L, atp6, atp8 and cytb), a small subunit ribosomal RNA gene (rrnS), a large subunit ribosomal RNA gene (rrnL), 22 transfer RNA genes, and two non-coding regions (Table 4). The details of gene locations are given in Table 4. The nucleotide compositions of the complete mtDNA sequence of RSC are biased toward A and T, with T being the most favored nucleotide and G the least favored, in accordance with the mt genome of RSU [32]. The content of A+T is 77.37% for RSC (37.69% A, 39.68% T, 10.04% G and 12.59% C), and 77.97% for RSU (37.65% A, 40.32% T, 9.93% G and 12.10% C). The mt genes of RSC overlap a total of 44 bp in 12 locations which range from 1 to 8 bp, and the overlapping has also been found in RSU mt genome [32]. The RSC mt genes are separated by intergenic spacer sequences of a total of 94 bp in length, which are located in 16 regions and range from 1 to 26 bp in size. The longest intergenic region (26 bp) is located between tRNA-Gln and tRNA-Phe genes.
The mitochondrial genome of R. sanguineus China isolate. Gene scaling is only approximate. All genes have standard nomenclature including the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the two leucine- and serine-specifying tRNA (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGN and UCN, respectively). ''NCR1'' refers to a long non-coding region and ''NCR2'' refers to a short non-coding region.
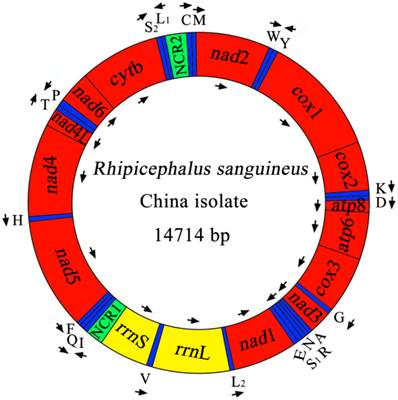
Mitochondrial genome organization of Rhipicephalus sanguineus China isolate (RSC) and USA isolate (RSU) [32].
| Genes/regions | Positions and nt sequence lengths (bp) | Strand | Initiation and stop codons | ||
|---|---|---|---|---|---|
| RSC | RSU | RSC | RSU | ||
| tRNA-Met (M) | 1-64 (64) | 1-64 (64) | H | ||
| nad2 | 81-1022 (942) | 81-1022 (942) | H | ATA/TAA | ATT/TAA |
| tRNA-Trp (W) | 1025-1083 (59) | 1029-1089 (61) | H | ||
| tRNA-Tyr (Y) | 1087-1147 (61) | 1093-1153 (61) | L | ||
| cox1 | 1141-2679 (1539) | 1147-2685 (1539) | H | ATT/TAA | ATT/TAA |
| cox2 | 2684-3359 (676) | 2690-3365 (676) | H | ATG/T | ATG/T |
| tRNA-Lys (K) | 3357-3422 (66) | 3363-3428 (66) | H | ||
| tRNA-Asp (D) | 3423-3483 (61) | 3429-3490 (62) | H | ||
| atp8 | 3485-3643 (159) | 3492-3650 (159) | H | ATT/TAA | ATT/TAA |
| atp6 | 3637-4302 (666) | 3644-4309 (666) | H | ATG/TAA | ATG/TAA |
| cox3 | 4313-5087 (775) | 4321-5095 (775) | H | ATG/T | ATG/T |
| tRNA-Gly (G) | 5092-5151 (60) | 5100-5159 (60) | H | ||
| nad3 | 5150-5494 (345) | 5158-5502 (345) | H | ATA/TAA | ATA/TAA |
| tRNA-Ala (A) | 5502-5564 (63) | 5511-5573 (63) | H | ||
| tRNA-Arg (R) | 5567-5625 (59) | 5576-5634 (59) | H | ||
| tRNA-Asn (N) | 5624-5685 (62) | 5634-5694 (61) | H | ||
| tRNA-Ser AGN (S1) | 5684-5738 (55) | 5693-5747 (55) | H | ||
| tRNA-Glu (E) | 5741-5803 (63) | 5751-5810 (60) | H | ||
| nad1 | 5802-6743 (942) | 5809-6750 (942) | L | ATA/TAA | ATA/TAA |
| tRNA-LeuUUR (L2) | 6742-6801 (60) | 6749-6809 (61) | L | ||
| rrnL | 6802-7995 (1194) | 6810-7999 (1190) | L | ||
| tRNA-Val (V) | 7996-8052 (57) | 8000-8057 (58) | L | ||
| rrnS | 8058-8743 (686) | 8063-8749 (687) | L | ||
| Non-coding region (NCR1) | 8744-9050 (307) | 8750-9054 (305) | |||
| tRNA-Ile (I) | 9051-9111 (61) | 9055-9115 (61) | H | ||
| tRNA-Gln (Q) | 9114-9180 (67) | 9118-9184 (67) | L | ||
| tRNA-Phe (F) | 9207-9264 (58) | 9209-9266 (58) | L | ||
| nad5 | 9265-10923 (1659) | 9267-10923 (1657) | L | ATT/TAA | ATT/T |
| tRNA-His (H) | 10924-10984 (61) | 10924-10984 (61) | L | ||
| nad4 | 10991-12298 (1308) | 10991-12292 (1302) | L | ATT/TAA | ATA/TAA |
| nad4L | 12301-12576 (276) | 12298-12573 (276) | L | ATG/TAA | ATG/TAA |
| tRNA-Thr (T) | 12579-12639 (61) | 12576-12636 (61) | H | ||
| tRNA-Pro (P) | 12640-12703 (64) | 12637-12700 (64) | L | ||
| nad6 | 12691-13140 (450) | 12688-13137 (450) | H | ATA/TAA | ATA/TAA |
| cytb | 13145-14221 (1077) | 13142-14218 (1077) | H | ATG/TAA | ATG/TAA |
| tRNA-SerUCN (S2) | 14222-14285 (64) | 14219-14283 (65) | H | ||
| tRNA-LeuCUN (L1) | 14288-14348 (61) | 14286-14347 (62) | L | ||
| Non-coding region (NCR2) | 14349-14651 (303) | 14348-14650 (303) | |||
| tRNA-Cys (C) | 14652-14711 (60) | 14651-14707 (57) | H | ||
Annotation
As shown in Table 4, the start codons of 13 protein-coding genes are ATN codon, which is typical of most metazoan mt genomes. The start codons inferred in the mt genome of RSC are ATA, ATT and ATG, and all reading-frames of RSC mt genome ended with T and TAA as termination codons. Note that nad5 gene uses TAA as termination codons in RSC, whereas it uses T as termination codons in RSU. The 22 tRNA genes in RSC mt genome vary in length from 55 to 66 nucleotides with differences in stem and loop sizes of dihydrouridine (D) and TΨC loops. The 22 tRNA genes are located on both strands. Of these 22 tRNA genes, 14 tRNA genes are encoded on the H-strand and 8 on the L-strand. All of the 22 tRNA genes can be folded into normal cloverleaf structure, except for tRNA-SerAGN and tRNA-Cys which lacks DHU arm. The rrnL is located between tRNA-LeuUUR and tRNA-Val (V), and the rrnS is located between tRNA-Val and the non-coding region (NCR1). The lengths of the rrnL and rrnS genes of RSC are 1194 bp and 686 bp. The A+T contents of the rrnL and rrnS of RSC are 81.41% and 79.01%. Two AT-rich or non-coding regions (NCR1, NCR2) were inferred in the mt genome of RSC. For this genome, the long NCR (designated as NCR1; 307 bp) is located between the rrnS and tRNA-Ile, and has an A+T content of 64.82%. The short NCR (designated as NCR2; 303 bp) is located between the tRNA-LeuCUN and tRNA-Cys, and has an A+T content of 65.02%.
Comparative analyses between R. sanguineus from China and USA
The complete mt genome sequence of RSC was 14714 bp in length, 4 bp longer than that of RSU. The arrangement of mt genes (i.e., 13 protein genes, 2 rRNA genes and 22 tRNA genes) and NCR is identical in the two mt sequences. A comparison of the nucleotide sequences of each mt gene and NCR, as well as the amino acid sequences, conceptually translated from all protein genes of the two R. sanguineus isolates (ie, RSC and RSU), is given in Table 5. Sequence difference across the complete mt genome between RSC and RSU was 11.23%. The magnitude of nucleotide sequence variation in each gene and NCR between RSC and RSU ranged from 9.34% (cox3)-15.65% (nad3). Difference in amino acid sequences of RSC and RSU mt genomes was 9.3%. The amino acid sequence differences ranged from 2.54-19.23%, with COX1 being the most conserved protein, and ATP8 the least conserved. There were 334 amino acid substitutions (for an alignment length of 3593 and 3591 positions, respectively) in the 13 proteins of RSC and RSU mt genomes, and more than half of which were in the proteins NAD5 (n = 77), NAD2 (n = 60) and NAD4 (n = 54).
Differences in mitochondrial nucleotides and predicted amino acids sequences between Rhipicephalus sanguineus China isolate (RSC) and USA isolate (RSU).
| Gene/region | Nucleotide sequence length | Nucleotide difference (%) | Number of aa | aa difference (%) | ||
|---|---|---|---|---|---|---|
| RSC | RSU | RSC/RSU | RSC | RSU | RSC/RSU | |
| atp6 | 666 | 666 | 12.31 | 221 | 221 | 5.59 |
| nad1 | 942 | 942 | 11.25 | 313 | 313 | 7.99 |
| nad2 | 942 | 942 | 14.65 | 313 | 313 | 19.17 |
| nad3 | 345 | 345 | 15.65 | 114 | 114 | 14.04 |
| nad4 | 1308 | 1302 | 12.92 | 435 | 433 | 12.41 |
| nad4L | 276 | 276 | 14.70 | 91 | 91 | 14.13 |
| nad5 | 1659 | 1657 | 13.86 | 552 | 552 | 13.95 |
| nad6 | 450 | 450 | 12.67 | 149 | 149 | 12.08 |
| cox1 | 1539 | 1539 | 10.79 | 512 | 512 | 2.54 |
| cox2 | 676 | 676 | 10.07 | 225 | 225 | 3.57 |
| cox3 | 775 | 775 | 9.34 | 258 | 258 | 4.26 |
| cytb | 1077 | 1077 | 10.21 | 358 | 358 | 4.47 |
| atp8 | 159 | 159 | 13.84 | 52 | 52 | 19.23 |
| rnnL | 1194 | 1190 | 12.98 | - | - | - |
| rrnS | 686 | 687 | 9.17 | - | - | - |
| Non-coding | 610 | 608 | 7.38 | - | - | - |
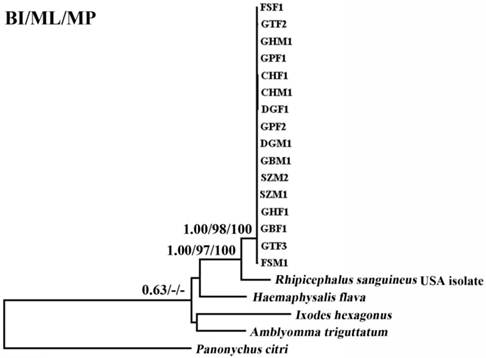
Two regions (~ 850 bp and ~ 550 bp) in the cox1 and cytb genes were used to examine the magnitude of genetic variation within 16 individuals of RSC. A comparison of the partial cox1 (pcox1) and partial cytb (pcytb) sequences among 16 RSC individuals revealed that sequence variations were 0-2.87% for pcox1, and 0-1.89% for pcytb. However, comparison of the pcox1 and pcytb sequences between RSC and RSU shows significant differences (10.8-11.5% and 10.4-11.4%, for pcox1, and pcytb, respectively), supporting that RSC and RSU may represent distinct tick species.
Phylogenetic analyses
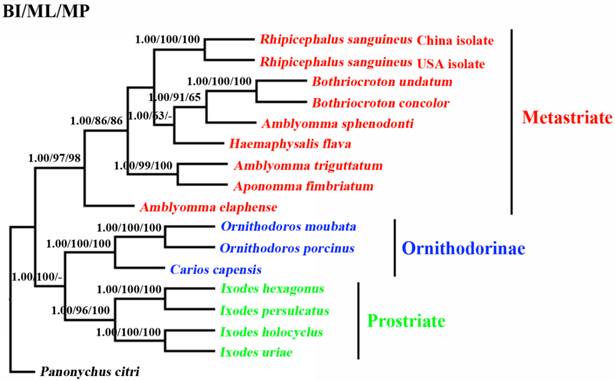
The phylogenetic relationships of 15 tick species based on concatenated amino acid sequence datasets, plus the corresponding sequences of RSC obtained in the present study, using MP, ML and BI analyses are shown in Figure 2. The topologies of the trees using MP, ML and BI were identical. In the tree, two major clades were recovered within ticks: ornithodorinae + prostriate and metastriate form monophyletic groups. Within the metastriate, RSC and RSU clustered together with high statistical support (BP=100%; PP=1), indicating that RSC is sister to RSU. Phylogenetic analyses using the combined cox1 and cytb sequences also grouped RSC and RSU into two distinct clades, and the trees produced using the three different methods were essentially the same in topology, indicating that RSC is sister to RSU (Fig. 3).
Inferred phylogenetic relationship among tick species. The concatenated amino acid sequences of 13 mitochondrial protein-coding genes were analyzed utilizing maximum parsimony (MP), maximum likelihood (ML) and Bayesian analysis (BI), using Panonychus citri as outgroup. The numbers along branches indicate bootstrap probability (BP) and posterior probability (PP) resulting from different analyses in the order: BI /MP/ML. Values lower than 50 are given as "-".
Inferred phylogenetic relationship among tick species based on combined mitochondrial cox1 and cytb gene sequences. The phylogenetic relationship was analyzed utilizing maximum parsimony (MP), maximum likelihood (ML) and Bayesian analysis (BI), using Panonychus citri as outgroup. The numbers along branches indicate bootstrap probability (BP) and posterior probability (PP) resulting from different analyses in the order: BI/ML/MP. Values lower than 50 are given as "-".
Discussion
The brown dog tick R. sanguineus is a major health threat for humans and both domestic and wild animals. However, its taxonomic status is an ongoing controversy [10-17]. In the present study, a substantial level of nucleotide differences (11.23%) was detected in the complete mt genome between a R. sanguineus isolate from China (RSC) and that from USA (RSU). These results were consistent with previous findings of sequence variations in part of the mt 12S rRNA gene and 16S rRNA gene among R. sanguineu from different regions [15,16]. A previous study has clearly indicated that sequence differences in mtDNA between closely-related species were typically 10-20% [44]. Therefore, our mtDNA data provide mt evidence that RSC and RSU represent distinct tick species.
In order to vigorously estimate the intra-specific variation within the RSC, a comparison of the pcox1 and pcytb sequences among 16 RSC individuals revealed a intra-specific variation of 0-2.87% for pcox1 and 0-1.89% for pcytb, consistent with that of a very recent study [45]. Previous studies of other ticks have also detected low intra-specific sequence variation in mt sequences. For example, the intra-specific sequence variation in Dermacentor marginatus was 0.1-1.0% for pcox1 (732 bp) and 0.2-1.2% for pnad5 (519 bp) [43], and in Haemaphysalis punctata it was 0.4-1.9% for pcox1 and 0.4-1.0% for pnad5 [43], and intra-specific sequence variation in Ixodes holocyclus and I. cornuatus was less than 0.86% for pcox1 (640 bp) [46]. However, comparison of the pcox1 and pcytb sequences between RSC and RSU revealed significantly high level of difference (10.8-11.5% for pcox1 and 10.4-11.4% for pcytb), consistent with previous studies of other Ixodes ticks, where inter-specific sequence variation was 9.1-16.0% for cox1 [46], and 13.0% for cox1 [47], and 4.2-14.1% for rrnL [48]. In spite of the molecular evidence here supporting the RSC and RSU as distinct tick species, interpretation from present study needs to be somewhat caution until detailed population genetic studies have been carried out on samples from broader geographical localities to better illuminate the taxonomy and population genetic structures of R. sanguineus ticks.
Phylogenetic analyses of R. sanguineus using concatenated amino acid sequences of 13 protein-coding genes by three different computational algorithms (MP, ML and Bayes) also provided addition support that RSC and RSU represent closely but distinct taxa (Figures 2 and 3). Taken together, the molecular evidence presented here supports the hypothesis that R. sanguineus represents a species complex [5,6]. In the present study, the phylogenetic status of the Amblyomma spp. and prostriate ticks remain problematic. Recent study indicated that the genus Amblyomma is polyphyletic [49]. Our results support the proposal that A. sphenodonti is sister to Bothriocroton spp.. In addition, the present study also revealed that the prostriate ticks and ornithodorinae ticks are more closely related than metastriate ticks. The monophyly of the hard ticks (prostriate + metastriate) was strongly rejected by BI, MP and ML analyses with high PP and BP value, respectively. This result was not consistent with results of a recent study, which showed that prostriate ticks and the metastriate ticks were more closely-related than to ornithodorinae ticks [49]. This contradiction is probably due to the use of a limited mt genome dataset. Thus, more mt genomes of ticks need to be sequenced and employed for evolutionary relationships of ticks in further studies to re-examine the evolutionary relationships among tick species.
In conclusion, the findings of the present study provide mt evidence that RSC and RSU represent distinct tick species, supporting the hypothesis that R. sanguineus represents a species complex. The mtDNA data presented in the present study provide useful novel markers for further addressing the taxonomy and systematics of the R. sanguineus species complex.
Acknowledgements
Project support was provided by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840) and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006) to XQZ.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kiss T, Cadar D, Spînu M. Tick prevention at a crossroad: New and renewed solutions. Vet Parasitol. 2012;187:357-366
2. Mihalca AD, Gherman CM, Cozma V. Coendangered hard-ticks: Threatened or threatening? Parasit Vectors. 2011;4:71
3. Fernandes ÉK, Bittencourt VR, Roberts DW. Perspectives on the potential of entomopathogenic fungi in biological control of ticks. Exp Parasitol. 2012;130:300-305
4. Barker SC, Murrell A. Systematics and evolution of ticks with a list of valid genus and species names. Parasitology. 2004;129:15-36
5. Dantas-Torres F. Biology and ecology of the brown dog tick, Rhipicephalus sanguineus. Parasit Vectors. 2012;3:26
6. Dantas-Torres F. The brown dog tick, Rhipicephalus sanguineus (Latreille, 1806) (Acari: Ixodidae): from taxonomy to control. Vet Parasitol. 2008;152:173-185
7. Liu WC, Liu ZC, Ye JH. Prevalence of ticks in working dogs in China. J Cynol. 2012;88:19-20
8. Walker JB, Keirans JE, Horak IG. The Genus Rhipicephalus (Acari, Ixodidae). A Guide to the Brown Ticks of the World. Cambridge University Press, Cambridge. 2000:655
9. de Oliveira PR, Bechara GH, Denardi SE, Saito KC, Nunes ET, Szabó MP, Mathias MI. Comparison of the external morphology of Rhipicephalus sanguineus (Latreille, 1806) (Acari: Ixodidae) ticks from Brazil and Argentina. Vet Parasitol. 2005;129:139-147
10. Pegram RG, Clifford CM, Walker JB, Keirans JE. Clarification of the Rhipicephalus sanguineus group (Acari, Ixodoidea, Ixodidae). I. R. sulcatus Neumann, 1908 and R. turanicus Pomerantsev, 1936. Syst Parasitol. 1987;10:3-26
11. Farid HA. Morphological keys for the separation of the Rhipicephalus sanguineus group of ticks (Acarina: Ixodidae) in Egypt. J Egypt Soc Parasitol. 1996;26:453-460
12. Zahler M, Filippova NA, Morel PC, Gothe R, Rinder H. Relationships between species of the Rhipicephalus sanguineus group: a molecular approach. J Parasitol. 1997;83:302-306
13. Barker SC. Distinguishing species and populations of rhipicephaline ticks with its 2 ribosomal RNA. J Parasitol. 1998;84:887-892
14. Matsumoto K, Brouqui P, Raoult D, Parola P. Experimental infection models of ticks of the Rhipicephalus sanguineus group with Rickettsia conorii. Vect Borne Zoonotic Dis. 2005;5:363-372
15. Szabo´ MP, Mangold AJ, Joao CF, Bechara GH, Guglielmone AA. Biological and DNA evidence of two dissimilar populations of the Rhipicephalus sanguineus tick group (Acari: Ixodidae) in South America. Vet Parasitol. 2005;130:131-140
16. Moraes-Filho J, Marcili A, Nieri-Bastos FA, Richtzenhain LJ, Labruna MB. Genetic analysis of ticks belonging to the Rhipicephalus sanguineus group in Latin America. Acta Trop. 2011;117:51-55
17. Burlini L, Teixeira KR, Szabó MP, Famadas KM. Molecular dissimilarities of Rhipicephalus sanguineus (Acari: Ixodidae) in Brazil and its relation with samples throughout the world: is there a geographical pattern? Exp Appl Acarol. 2010;50:361-374
18. Pegram RG, Keirans JE, Clifford CM, Walker JB. Clarification of the Rhipicephalus sanguineus group (Acari, Ixodoidea, Ixodidae). II. R. sanguineus (Latreille, 1806) and related species. Syst Parasitol. 1987;10:27-44
19. Ioffe-Uspensky I, Mumcuoglu KY, Uspensky I, Galun R. Rhipicephalus sanguineus and R. turanicus (Acari: Ixodidae): Closely related species with different biological characteristics. J Med Entomol. 1997;34:74-81
20. Wolstenholme DR. Animal mitochondrial DNA, structure and evolution. Int Rev Cytol. 1992;141:173-216
21. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
22. Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, Zhao GH, Huang SY, Zhu XQ. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012;492:110-116
23. Li H, Liu H, Cao L, Shi A, Yang H, Cai W. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int J Biol Sci. 2012;8:93-107
24. Tan HW, Liu GH, Dong X, Lin RQ, Song HQ, Huang SY, Yuan ZG, Zhao GH, Zhu XQ. The complete mitochondrial genome of the Asiatic cavity-nesting honeybee Apis cerana (Hymenoptera: Apidae). PLoS One. 2011;6:e23008
25. Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld MJ, Sun W, Ba MN, Sanon A, Binso-Dabire CL, Baoua I, Ishiyaku MF, Covas FG, Srinivasan R, Armstrong J, Murdock LL, Pittendrigh BR. Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS One. 2011;6:e16444
26. Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012;6:e1539
27. Liu GH, Wang SY, Huang WY, Zhao GH, Wei SJ, Song HQ, Xu MJ, Lin RQ, Zhou DH, Zhu XQ. The complete mitochondrial genome of Galba pervia (Gastropoda: Mollusca), an intermediate host snail of Fasciola spp. PLoS One. 2012;7:e42172
28. Liu GH, Li C, Li JY, Zhou DH, Xiong RC, Lin RQ, Zou FC, Zhu XQ. Characterization of the complete mitochondrial genome sequence of Spirometra erinaceieuropaei (Cestoda: Diphyllobothriidae) from China. Int J Biol Sci. 2012;8:640-649
29. Jex AR, Hall RS, Littlewood DT, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522-533
30. Chitimia L, Lin RQ, Cosoroaba I, Braila P, Song HQ, Zhu XQ. Molecular characterization of hard and soft ticks from Romania by sequences of the internal transcribed spacers of ribosomal DNA. Parasitol Res. 2009;105:907-911
31. Murrell A, Campbell NJ, Barker SC. Recurrent gains and losses of large (84-109 bp) repeats in the rDNA internal transcribed spacer 2 (ITS2) of rhipicephaline ticks. Insect Mol Biol. 2001;10:587-596
32. Black WC 4th, Roehrdanz RL. Mitochondrial gene order is not conserved in arthropods: prostriate and metastriate tick mitochondrial genomes. Mol Biol Evol. 1998;15:1772-1785
33. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876-4882
34. Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
35. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA 5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731-2739
36. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564-577
37. Swofford DL. Paup*: Phylogenetic Analysis Using Parsimony, version 4.0b10. Sunderland, MA: Sinauer Associates. 2002
38. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
39. Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104-2105
40. Posada D. JModelTest phylogenetic model averaging. Mol Biol Evol. 2008;25:1253-1256
41. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
42. Page RD. TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357-358
43. Chitimia L, Lin RQ, Cosoroaba I, Wu XY, Song HQ, Yuan ZG, Zhu XQ. Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial cox1 and nad5 genes. Exp Appl Acarol. 2010;152:305-311
44. Blouin MS. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527-531
45. Nava S, Mastropaolo M, Venzal JM, Mangold AJ, Guglielmone AA. Mitochondrial DNA analysis of Rhipicephalus sanguineus sensu lato (Acari: Ixodidae) in the Southern Cone of South America. Vet Parasitol. 2012;190:547-555
46. Song S, Shao R, Atwell R, Barker S, Vankan D. Phylogenetic and phylogeographic relationships in Ixodes holocyclus and Ixodes cornuatus (Acari: Ixodidae) inferred from COX1 and ITS2 sequences. Int J Parasitol. 2011;41:871-880
47. Apanaskevich DA, Horak IG, Matthee CA, Matthee S. A new species of Ixodes (Acari: Ixodidae) from South African mammals. J Parasitol. 2011;97:389-398
48. Chao LL, Wu WJ, Shih CM. Molecular analysis of Ixodes granulatus, a possible vector tick for Borrelia burgdorferi sensu lato in Taiwan. Exp Appl Acarol. 2009;48:329-344
49. Burger TD, Shao R, Beati L, Miller H, Barker SC. Phylogenetic analysis of ticks (Acari: Ixodida) using mitochondrial genomes and nuclear rRNA genes indicates that the genus Amblyomma is polyphyletic. Mol Phylogenet Evol. 2012;64:45-55
Author contact
![]() Corresponding author: Prof. Xing-Quan Zhu, State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, CAAS, Lanzhou, Gansu Province 730046, China. Phone: 86-931-8342837; Fax: 86-931-8340977; Email: xingquanzhu1com.
Corresponding author: Prof. Xing-Quan Zhu, State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, CAAS, Lanzhou, Gansu Province 730046, China. Phone: 86-931-8342837; Fax: 86-931-8340977; Email: xingquanzhu1com.