Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(4):425-434. doi:10.7150/ijbs.6068 This issue Cite
Research Paper
PIG3 Functions in DNA Damage Response through Regulating DNA-PKcs Homeostasis
Bing Li, Zeng-Fu Shang, Jiao-Jiao Yin, Qin-Zhi Xu, Xiao-Dan Liu, Yu Wang, Shi-Meng Zhang, Hua Guan, Ping-Kun Zhou ![]()
Department of Radiation Toxicology and Oncology, Beijing Institute of Radiation Medicine, 27 Taiping Road, Haidian District, Beijing 100850, China.
Received 2013-2-12; Accepted 2013-4-22; Published 2013-5-3
Abstract
The p53-inducible gene 3 (PIG3) recently has been reported to be a new player in DNA damage signaling and response, but the crucial mechanism remains unclear. In the present study, the potential mechanism of PIG3 participation in the DNA damage response induced by ionizing radiation (IR) was investigated in multiple cell lines with depleted expression of PIG3 transiently or stably by the small interference RNA and lentivirus-mediated shRNA expression strategies. PIG3 knockdown led to an abnormal DNA damage response, including decreased IR-induced phosphorylation of H2AX, Chk1, Chk2 and Kap-1 as well as a prolonged G2-M arrest and aberrant mitotic progression. Notably, PIG3 knockdown resulted in a striking depression of cellular DNA-PKcs protein level, and was accompanied by a downregulation of ATM. Re-expression of PIG3 effectively rescued the depression of DNA-PKcs in PIG3-depleted cells. This negative regulation of DNA-PKcs by depleting PIG3 seemed to take place at the translational level but not at the levels of transcription or protein degradation. However, a compensatory feedback of increased mRNA expression of DNA-PKcs was formed in PIG3-depleted cells after a few passages or cell cycles of subculture, which led the recovery of the DNA-PKcs protein level and the consequent recovered efficiency of the DNA damage response. These results provide a new insight into the mechanism of PIG3's functioning in DNA damage signaling and the regulation network of cellular DNA-PKcs expression homeostasis.
Keywords: DNA damage response, cell cycle progression, checkpoint, PIG3, DNA-PKcs, ATM.
Introduction
The p53-inducible gene 3 (PIG3) was originally indentified by Polyak et al in the analysis of p53-induced genes related to the onset of apoptosis (1). PIG3 is highly homologous to NADPH oxidoreductase TED2 in plants and zeta-Crystalline in mammalian cells, and is considered as a proapoptosis marker. In some p53 mutants, induction of cell cycle arrest, but not apoptosis was found to be associated with a lack of induction of PIG3 expression (2). Silencing of the transcription factor hCAS/CSE1L, which regulated PIG3 expression, led to decreased PIG3 transcription and concurrently decreased apoptosis (3). Also, PIG3 has been reported to have oxidoreductase enzymatic activity (4), this provides direct evidence of PIG3's participation in the generation of ROS in cells. Additionally, PIG3 mediates cancer cell death through the GPx3 pathway, and knocking down PIG3 or blocking the interaction between PIG3 and GPx3 would abolish the increase in ROS and apoptosis (5).
Recently, PIG3 has been shown to participate in the DNA damage response as well (6 - 8). Our previous study demonstrated that PIG3 was induced in a dose-dependent manner by ionizing radiation (IR) (6). The IR-increased expression of PIG3 is p53 dependent (7). This was the first evidence suggesting the involvement of PIG3 in the IR-induced DNA damage response. We also observed that PIG3 formed specific ionizing radiation-induced foci (IRIF) colocalized with γ-H2AX foci and partially with pKAP-1(7). J-H Lee et al extensively investigated the participation of PIG3 in DNA damage checkpoints after UV irradiation or after treatment using the radiomimetic drug neocarzinostatin (NCS) (8). PIG3 knockdown can suppress intra-S phase and G2/M checkpoints (8). J-H Lee et al also have provided the first evidence showing that PIG3 is closely associated with the induction and maintenance of the phosphorylation of H2AX and some other DDR proteins after DNA damage induced by NCS (8).
The DNA double-strand break (DSB) is considered to be the most severe type of DNA damage induced by ionizing radiation, and this form of DNA damage must be repaired immediately to prevent cell death. There are two mechanistic pathways in the cells to repair DSB: homologous recombination (HR) and non-homologous end joining (NHEJ) (9, 10). HR is the principle mechanism for DSB repair in bacteria and lower eukaryotes such as yeast, while NHEJ is the main mechanism used in higher eukaryotes. Some of the NHEJ components also participate in other biological processes with DNA breaking signal, such as V(D)J recombination and exogenous gene concatemer formation (10). Phosphorylation of H2AX at Ser 139 is a critical molecular event, even at the very early stage of DNA damage signaling. It has been widely reported that H2AX can be phosphorylated at Ser139 by the phosphatidylinositol 3-kinase-like kinase (PIKK) family members, including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), the ataxia telangiectasia mutated gene (ATM), and ATM and Rad3 related genes (ATR) (11 - 14). ATM and DNA-PK mainly respond to DNA double strand breaks (DSB) induced by ionizing radiation, whereas ATR is activated after UV radiation or replication fork collapse (9, 15). Once DSB occur, ATM and DNA-PKcs are activated by autophosphorylating or phosphorylating each other. The activated ATM and DNA-PKcs can directly or indirectly phosphorylate a series of downstream substrates including H2AX, and trigger the cellular DNA damage responses (12, 16). The functions of the PIKK family members, to some extent, overlap with each other. For instance, DNA-PKcs shares a number of substrates with ATM in DNA damage (16). DNA-PKcs also has been shown contributing to the regulation of ATM expression level, and interfering with DNA-PKcs expression will lead subsequently to ATM transcription inhibition (17). We have previously reported that DNA-PKcs plays a dominant role in DNA damage-induced or cell cycle progression-associated H2AX phosphorylation (13).
Although it is clear that PIG3 participates in the regulation of H2AX phosphorylation and DNA damage response (7, 8), it remains unknown how this oxidoreductase PIG3 functions in DNA damage signaling. In the present study, small interference RNA (siRNA) and lentivirus-mediated shRNA expression strategies were used to knock down PIG3 transiently or stably, and the participation of PIG3 in the DNA damage response induced by ionizing radiation was further confirmed. Notably, the DNA-PKcs was found to be a critical down-stream target of PIG3. The DNA-PKcs protein level was significantly attenuated in the early several passages of PIG3-depleted cells. However, a feedback of compensatory augmentation of the DNA-PKcs transcriptional expression took place in PIG3-depleted cells after a certain number of cell cycles or passages of continuous culture. Besides the defect in DNA damage signaling, PIG3-depleted cells also showed a prolonged cell cycle, similar with the phenotype we observed in DNA-PKcs knock down or inactivated cells. When the attenuated level of DNA-PKcs was compensated by the feedback-increased transcriptional expression, the defect in DNA damage signaling and abnormal cell cycle progression were well corrected in PIG3-depleted cells. These results provide more insights into the mechanism of PIG3 contributing to the DNA damage response, and help to further understand the regulation network of cellular DNA-PKcs homeostasis.
Materials and Methods
Cell culture
HeLa, MCF-7 cells, and ATM defective ATS4 and AT5BIVA cells were maintained in DMEM (HyClone) supplemented with 10% fetal bovine serum (HyClone), 100 units per ml penicillin and 100 μg/ml streptomycin in a humidified incubator at 37°C with 5% CO2. PIG3 knocked-down MCF-7 and HeLa cell lines were established by infection with PIG3 shRNA expressing viral particles and selection using puromycin. In detail, 48 hours after viral particles infection, puromycin was added to the medium at 2 μg/ml to kill the uninfected cells. About 6 days later, cells resistant to puromycin were collected and their PIG3 level was assessed by immunoblotting.
Antibodies
The antibodies against PIG3 (H300, sc-30068), DNA-PKcs (H-163, sc-9051), β-actin (C4, sc-47778) and ATM (1A1, sc-73615) were purchased from Santa Cruz Biotechnology. Antibodies against p-Chk2-T68 (C13C1, #2197), Chk2 (#2662), Chk1 (2G1D5, #2360), KAP-1 (#4123s) and pKAP-1/S824 (#4127) were purchased from Cell signaling Technology. Polyclonal rabbit antibody against phosphor-Chk1-s317 (AF2054) was from R&D System. γ-H2AX (ser 139)(JBW301, #05-636) antibody was purchased from Millipore.
siRNA and transfection
To knock down PIG3 expression, a PIG3 siRNA molecule of 21-nucleotides oligomers from the published data (8) was synthesized. The sequences of this siRNA are as follows: 5'-GGACUUAAUGCAGAGACAAdTdT-3' (sense) and 5'-UUGUCUCUGCAUUAAGUCCdTdT-3' (antisense). The siRNA was transfected into cells using lipofectamine2000 according to the manufacturer's instruction. At 48 hours post transfection cells were harvested and underwent following test. A viral shRNA vector based on the same target sequence of PIG3 was constructed by Shanghai GenePharma Co. Ltd. and viral particles were packaged.
Plasmid constructs and transfection
To generate the full-length construct of PIG3, a pair of primers containing EcoR I and Xho I restriction sites were used in polymerase chain reaction. The sequences of these primers are as follow: 5' tgcgaattcatgttagccgtgcactttg 3' (forward) and 5' tatctcgagtcactggggcagttcc 3' (reverse). The PCR product was digested by EcoR I and Xho I and then inserted into a pCMV-TAG-2B vector. The PIG3 sequence was confirmed by sequencing. For RNAi resist PIG3 expression vector, the codons at siRNA target site were synonymous mutant and constructed into pCMV-TAG-2B. To construct DNA-PKcs' promoter reporter plasmid, sequence between -939 and +1 on the DNA-PKcs gene was inserted into pGL3-basic. Cells were transfected with plasmid using lipofectamine2000 (Invitrogen) according to the manufacturer's instruction.
Flow cytometric analysis of the cell cycle
After washing twice with PBS solution, cells were collected and fixed using 75% ethanol at -20°C for at least 24 h. Then the cells were washed twice with PBS solution, incubated with 20 μl RNase A (Sigma, St. Louis, MO, USA) (1 mg/ml) for 30 min at 37°C, and stained with 25 μg/ml PI (Sigma) for 30 min at room temperature. The cell cycle distributions were then analyzed using flow cytometry and more than 10,000 cells per sample were counted. All experiments were repeated three times.
Dual-Luciferase reporter assay
To measure DNA-PKcs' promoter activity, the Promega dual-luciferase reporter assay system was applied according to the manufacturer's instruction. Briefly, 10 days post viral particles infections, cells were planted into 24 wells plates. 24 hours later, pGL3-basic-DNAPKcs and RL-CMV were transfected into cells using lipofectmin2000 according to the manufacturer's instruction. 36 hours post-transfection, cells were harvested and the firefly luciferase and renilla luciferase activity were measured respectively.
Immunoblotting analysis
Cells lysates or immunoprecipitation products were denatured, resolved by SDS-PAGE, and subjected to immunoblotting analyses. When preparing the cell lysates, the cell pellets were treated with lysis buffer, and total protein was isolated. The protein (50 μg) was resolved using SDS-PAGE (8%), and then transferred onto a polyvinylidene fluoride (PVDF) membrane for immune-hybridization analysis.
Results
Involvement of PIG3 in DNA damage signaling to ionizing radiation
PIG3 has been found to participate in the DNA damage response (DDR) induced by UV radiation and the radiomimetic drug neocarzinostatin recently (8). We acquired similar results by treating PIG3 knockdown cells and control cells with ionizing radiation. After 4 Gy of γ-irradiation, the phosphorylation of the DDR proteins H2AX, Chk1 and Chk2 was much weaker or of shorter duration in PIG3 knockdown HeLa cells than in control cells (Fig. 1A). The phosphorylated H2AX is considered as the damage sensor of DNA double strand breaks, and phosphorylation of chk1 and chk2 is essential to G1/S and G2/M checkpoints activation after DNA damage. Knockdown of PIG3 also changed the cell cycle arrest pattern induced by IR. As shown in Fig. 1B & C, PIG3 knockdown cells (HeLa-shPIG3) arrested at G2-M phases and reached the peak 4 h earlier as compared to the control HeLa-shNC cells. However, it was much later for the PIG3 knockdown cells to be released from the IR-induced G2-M arrest.
Knockdown of PIG3 changes the DNA damage response and cell cycle progression of HeLa cells. (A) The phosphorylation of Chk1, Chk2 and H2AX was detected by immunoblotting analysis at various time points after 4 Gy of γ-ray irradiation in PIG3-depleted (HeLa-shPIG3) and control (HeLa-shNC) cells. (B, C) Cell cycle distribution of PIG3-depleted cells and control cells was detected by Flow cytometry assay after 4 Gy of γ- irradiation. (D) HeLa PIG3-depleted cells and control cells were synchronized in G1 phase by TdR double blocking method and then released, and cell cycle distributions at the indicated time points were examined. (E) 5×103 cells were plated at day 0 and cell numbers were counted every day to generate a growth curve. The data are the mean ± S.D. from three tests. (F) HeLa PIG3-depleted cells and control cells were synchronized at G1 phase by TdR double blocking method and then released, the p53 and cyclin B1 levels at the indicated times were examined by immunoblotting.
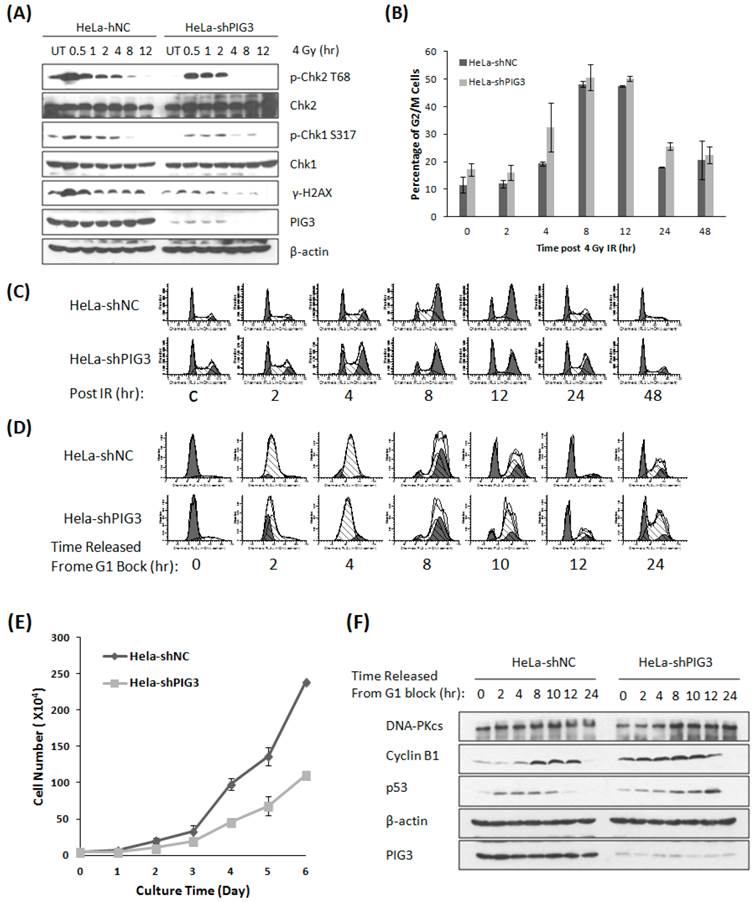
Knockdown of PIG3 elongates the G2-M phase
The phosphorylation of chk1 and chk2 is not only associated with cell cycle checkpoint activation post DNA damage, but also plays a role in normal cell cycle progression. To investigate whether PIG3 is involved in normal cell cycle regulation, we used the thymidine (TdR) double-blocking method to synchronize cells in G1 phase for the test of cell cycle progression. After TdR double-blocking, almost 90% cells stopped in G1 phase, and the synchronized cells entered S phase 2 - 4 hours after released for both PIG3 knockdown and control cells. However, we observed a striking difference between PIG3-deplepted cells and control cells at 10 hours after release from G1 block, where PIG3-deplepted HeLa cells still had the highest ratio of G2-M phases populations and a relative lower peak of G1 population, indicating a longer G2-M progression (Fig. 1D). A similar elongated G2-M progression was also detected in PIG3-depleted MCF7 cells (data not shown). Higher percentage of cells in S/G2/M has been reported indicating cells are more actively proliferating (18), however, when growth curve was generated, we found that knocking down PIG3 causes decreased cell proliferation capability (Fig. 1E). This result might suggest the disorder of G2-M progression. We further detected the p53 and cyclin B1 expression status after G1 block, since p53 and its transcriptional target p21 play important roles in G2 arrest induced by DNA damage (19, 20), and cyclin B1 is essential for the entry of mitosis (21). Interestingly, the PIG3-depleted cells have a constitutive higher level of cyclin B1 as compared to the control cells, which is much notable at 0 - 4 hours after released (Fig. 1F). The increased cyclin B1 level was comparable between two cells at 8 - 12 hours after released (cells progressed into G2-M phase), but with a higher level of p53 in the PIG3-depleted cells. p53 has been reported to down regulate cyclin B1 expression and inhibit cyclin B1/Cdk1 complex activity to prevent the initiate of mitosis after ionizing radiation (20). Higher level p53 in this case may reflect a compensatory mechanism to restore the cyclin B1 to the comparable level.
Knockdown of PIG3 leads to a decreased DNA-PKcs protein level
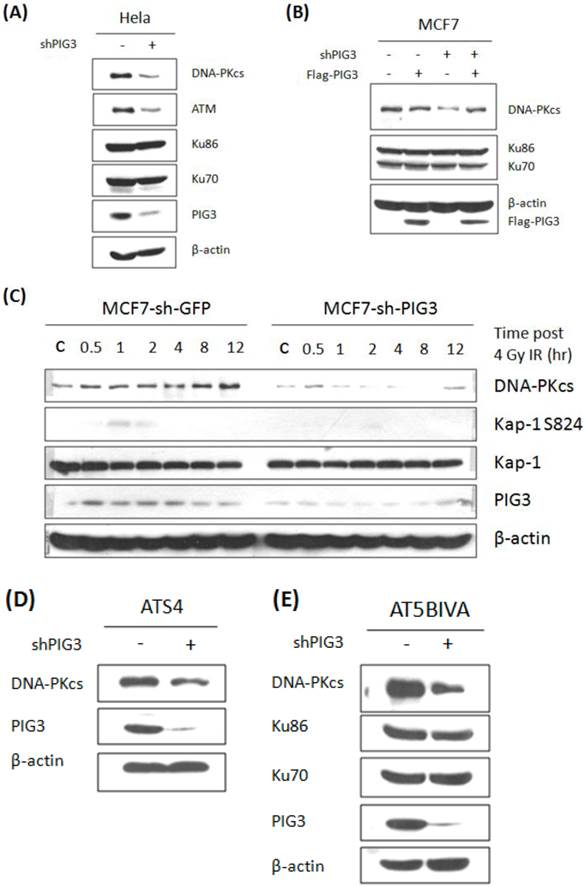
We next tested the protein levels of ATM and DNA-PKcs, both of which can phosphorylate H2AX at S139, in PIG3-depleted and control cell lines. As shown in Fig. 2A, ATM and DNA-PKcs were significant lower in PIG3 knockdown HeLa cells than in control cells. The same result can also be seen in PIG3-depleted MCF7 cells (Fig. 2B, compare lane 1 with lane 3). To examine the off-target effect of shRNA, a siRNA resistant Flag-tag PIG3 expression vector was generated and transfected into cells. Immunoblotting analysis showed that re-expression of PIG3 can effectively rescue the decreased DNA-PKcs level in PIG3-depleted MCF7 cells (Fig. 2B, lane 4), indicating that the defeat of DNA-PKcs is indeed caused by PIG3 depletion. Likewise, a significantly induced DNA-PKcs after 4 Gy IR was seen in the control MCF7 cells, but barely seen in the PIG3-depleted MCF7 cells (Fig. 2C). Moreover, the phosphorylated Kap-1 S824, a substrate of DNA-PKcs, was also much weaker in the PIG3-depleted MCF7 cells as compared to control cells.
Inactivation of DNA-PKcs would lead to down regulation of ATM (17). To further elucidate the upstream or downstream relationship for the association of PIG3 with DNA-PKcs and ATM, PIG3-depleted cell lines were generated based on two ATM defective cell lines ATS4 and AT5BIVA cells. Down-regulation of DNA-PKcs could also be seen in both ATM defective cell lines through shRNA-mediated depletion of PIG3 (Fig. 2D & E), suggesting that PIG3-depletion causing DNA-PKcs depression is independent of ATM. However, the decreased ATM could be the consequence of DNA-PKcs depression by depleting PIG3.
Intrinsic nature of recovering DNA damage response capability in PIG3 knockdown cells
The effect of PIG3 on DNA damage response can be well explained by its role of regulating DNA-PKcs. However, this hypothesis was challenged by our further observation: the defect of DNA damage response in PIG3-depleted cell line could be recovered quickly after a certain number of cell cycles or passages in continuous subculture. The irradiation experiment in PIG3-depleted MCF7cell line was repeated after about 5 passages of subculture. Surprisingly, there was no longer a decrease in the phosphorylation level of Chk1 and Chk2 post 4 Gy or 8 Gy of γ-irradiation in PIG3-depleted cells, it was even stronger than in control cells, although PIG3 still remained depressed status at this time (Fig. 3A). Meanwhile, the DNA-PKcs and ATM levels had recovered to normal levels or even higher. The UV irradiation experiment was also performed, but there was no attenuation of the phosphorylation of chk1 or chk2 at this time (Fig. 3B). The DNA-PKcs protein level was also re-checked in the PIG3-depleted HeLa cell line after a longer period of subculture. As shown in Fig. 3C, DNA-PKcs recovered while PIG3 levels still remained depressed status.
To further confirm this special regulation pattern of PIG3 on DNA-PKcs expression, a lentivirus-mediated shRNA expression system based on the same siRNA sequence was constructed and used. As shown in Fig. 3D, DNA-PKcs as well as ATM levels decreased at about eight to ten days after lentivirus infection and then recovered in a few days later. A similar result was also observed in the PIG3 knockdown human normal fibroblast HFS cells (Fig. 3E). It is obvious that the role of PIG3 in regulating the DNA damage response is dependent on its regulation of DNA-PKcs.
Knockdown of PIG3 leads to depression of DNA-PKcs protein. (A) DNA-PKcs and ATM protein levels in PIG3-depleted and control HeLa cells were tested by immunoblotting analysis. (B) A wild type PIG3 expressing plasmid or empty vector were transfected into control and PIG3-suppressed MCF7 cells and 48 hours later the DNA-PKcs level was examined by immunoblotting analysis. (C) DNA-PKcs, pKAP-1 and PIG3 were detected in PIG3-depleted and control MCF7 cells at different time after 4 Gy irradiation. (D, E) PIG3 was knocked down in ATM defective cell lines ATS4 and AT5BIVA, then DNA-PKcs level was detected by immunoblotting analyses.
DNA damage response and DNA-PKcs expression in PIG3-depleted cells after a number of cell cycles or passages of subculture. (A) The phosphorylation and protein level of a number of DDR proteins were detected by immunoblotting analysis at various time points post 8 Gy of γ-irradiation in PIG3-depleted (MCF7-shPIG3) and control (MCF7-shNC) cells. (B) The level of p-Chk2 T68 and p-Chk1 S317 was detected by immunoblotting analysis at various time points after 10 J / m2 of UV irradiation in PIG3-depleted (MCF7-shPIG3) and control (MCF7-shNC) cells. (C) The DNA-PK complex was detected in HeLa cells after more than 5 passages of subculture after PIG3 knockdown. (D) HeLa cells were infected with the viral particles expressing PIG3 shRNA, and 48 hours later puromycin was added into the medium to kill uninfected cells. At 7 days post-infection, cells stopped dying and were collected at the indicated days. PIG3 and DNA-PKcs levels were detected by SDS-PAGE. (E) Human fibroblast cells HFS were infected by the viral particles of expressing PIG3 shRNA. PIG3 and DNA-PKcs levels were tested at the indicated days post infection.
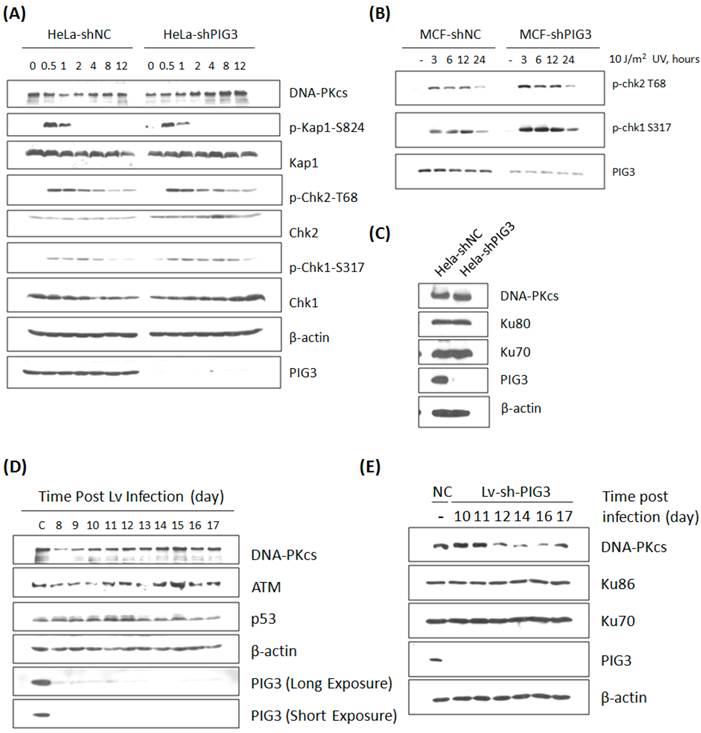
A compensatory feedback of increased transcription of DNA-PKcs
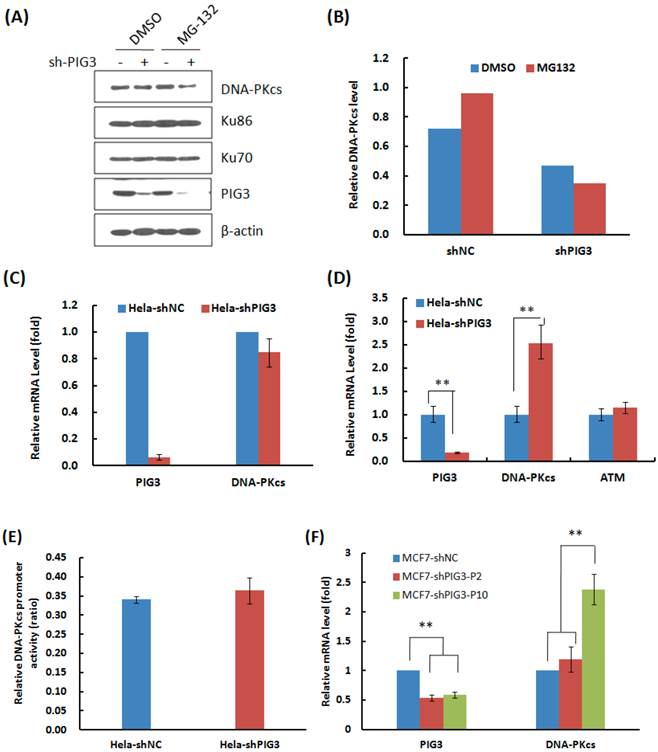
Why DNA-PKcs was attenuated at the early several cell cycles or passages after PIG3 knockdown, but recovered after a certain passages of subcultures? We firstly tested whether knockdown of PIG3 accelerated the degradation of DNA-PKcs. As shown in Fig. 4A & B, the proteasome inhibitor MG132 effectively increased the accumulation of DNA-PKcs in the control HeLa cells (compare lane 1 and lane 3 in Fig. 4A), but did not increase the level of DNA-PKcs in PIG3-depleted cells (compare lane 2 and lane 4 in Fig. 4A). It seems that PIG3 may affect the production of DNA-PKcs, but not affect the degradation of DNA-PKcs.
The mRNA expression of DNA-PKcs then was detected by the quantitative PCR. As shown in Fig. 4C, there was no obvious change in the mRNA level in the early several cell cycles of subculture, when the DNA-PKcs protein level was attenuated, after PIG3 depletion. The dual-luciferase reporter assay also showed no obvious changes in DNA-PKcs promoter activity at the earlier passages of subculture (Fig. 4E). However, a 2.5 fold increase in DNA-PKcs mRNA was detected in PIG3-depleted HeLa cells after a longer subculture of more than 15 days (Fig. 4D), which was accompanied by the recovery of DNA-PKcs protein levels. The similar pattern of DNA-PKcs mRNA level changes was also displayed in PIG3-depleted MCF7 cells, i.e. no obvious change in the early passages but significant increase after a longer subculture for more cell cycles (Fig. 4F). It seems that the attenuation of DNA-PKcs had been rescued by a compensatory feedback of increased DNA-PKcs transcription activity.
The effects of PIG3 depletion on DNA-PKcs degradation and mRNA expression. (A) The effect of proteasome inhibitor MG132 on the accumulation of DNA-PKcs protein. HeLa cells were infected with the viral particles expressing PIG3 shRNA, 48 hours later puromycin was added into the medium to kill uninfected cells. At 10 days post-infection, the cells were subjected to protein degradation inhibition by MG132. (B) Quantification of the relative DNA-PKcs protein level. The intensity of immunoblotting hybridization signals from Figure 4A was quantified. The data are the intensity ratio of DNA-PKcs and β-actin for each lane (sample). (C) The mRNA amount of DNA-PKcs in HeLa cells was detected by quantitative real-time PCR at 10 days after PIG3 shRNA expressing viral particle infection, as indicated in (A). (D) The mRNA amount of PIG3, DNA-PKcs and ATM in HeLa-shPIG3 and HeLa-shNC cells was detected by quantitative real-time PCR after more than 5 passages of longer time subculture. ** P < 0.01. (E) The relative activity of DNA-PKcs' promoter was detected by dual-luciferase reporter assay at 10 days after PIG3 shRNA expressing viral particles infection. (F) The mRNA levels of PIG3 and DNA-PKcs were detected in PIG3-depleted MCF7 cells at the early passages of culture (MCF7-shPIG-P2) and more passages of subculture (MCF7-shPIG-P10). ** P < 0.01.
Discussion
The DNA damage response is a comprehensive signaling process including damage sensing, mediators' activation, cell cycle arrest and DNA repair. PIG3 has been recently reported to participate in DNA damage response induced by UV radiation and the radiomimetic drug neocarzinostatin (8), and by ionizing radiation (7). One of the mechanistic pathways for PIG3 to function in DDR is to prompt a large scale chromatin relaxation in response to DNA damage signaling (7). It is well known that PIKK family members, including DNA-PKcs, ATM and ATR, play important roles in this DNA damage signaling process. Once IR-induced DSB occurs, ATM or DNA-PKcs binds to DNA end and then is activated (9, 22). Thereafter, the activated DNA-PKcs or ATM can phosphorylate a series of downstream targets involved in DDR and DNA repair. Here we present the first evidence to demonstrate that PIG3 functions as a new regulator of DNA-PKcs in the response to DNA damage induced by IR. In the early several cell cycles or passages of PIG3-depleted cells, the DNA-PKcs protein level was remarkably depressed, and accompanied by a decreased ATM level as well as a decreased IR-induced phosphorylation of a number of DDR proteins, including H2AX, Chk1, Chk2, Kap-1, which are substrates of the DNA-PKcs and ATM. A defect of DNA DSB rejoining capacity was also detected in the PIG3-depleted HeLa cells by using pulse-field gel electrophoresis (data not shown). Furthermore, the decreased DNA-PKcs was effectively rescued by re-expressing PIG3 in the PIG3-depleted cells.
H2AX, Chk1, Chk2 and Kap-1 are common substrates for DNA-PKcs and ATM, sharing a consensus sequence Ser-Thr-Gln-Gln (23, 24). DNA-PKcs has previously been reported to be related with ATM transcription activity, and inactivation of DNA-PKcs led to decreased levels of ATM mRNA and protein (17). In this report, DNA-PKcs was deregulated by knocking down PIG3 in both ATM efficient and deficient cell lines. Therefore, it seems that the decreased ATM in PIG3 knockdown cells is attributed to the depression of DNA-PKcs. The depression of DNA-PKcs can well explain the phenotype changes, in terms of abnormal DNA damage responses, in PIG3-depleted cells. It is easy to image that, after PIG3 knockdown, DNA-PKcs was depressed, and subsequently resulted in the attenuation of ATM. Loss of these two critical kinases seriously impacts the integrity of DNA damage response network, and would lead to decreased accumulation of damage sensors such as 53BP1, Mre11, Rad50 and Nbs1 at the site of DNA damage, as reported by Lee et al (8).
The depression of DNA-PKcs influences the phosphorylation of Chk1 and Chk2 upon IR-induced DNA damage as well, which in turn produces inefficient cell cycle arrest. The other clue indicating the role of PIG3 on DNA-PKcs regulation is that PIG3-depleted cells show a prolonged progression of G2-M phases. We have previously reported that inactivation of DNA-PKcs led to a prolonged G2/M checkpoint and abnormal mitotic progression in the cells after exposure to ionizing radiation (24). We also found that depression or inactivation of DNA-PKcs by specific siRNA or inhibitor Nu7026 would delay the mitotic exit even without DNA damage stress (data not shown), which is similar to the cell cycle change phenotype caused by PIG3 knockdown. Interestingly, depletion of PIG3 resulted in the decrease of DNA-PKcs, concomitant with an increase of cyclin B1 protein level. This result is in consistent with the previous report that shRNA-mediated depletion of DNA-PKcs resulted in an increase of cyclin B1 protein (24).
In addition, PIG3 has recently been reported to be down-regulated in cumulus cells (CCs) from aneuploid oocytes (25). CCs envelop the oocyte and continuously communicate with each other. Fragouli et al reported that TP53I3/PIG3 is one of the two genes being significantly down-regulated in CCs from chromosomally abnormal oocytes. Considering that knockdown of DNA-PKcs would lead to the increase of multipolar spindles and an aneuploid population (24), this down-regulation of PIG3 may provide an indirect clue to elucidate the connection between PIG3 and DNA-PKcs.
Although the PIG3-DNA-PKcs pathway fits the phenotype of PIG3 depletion so well, we observed that the DNA-PKcs level recovered after a certain number of cell cycles or passages of subculture, even at that time while PIG3 still remained depressed. Interestingly, the cells recovered their activity of DNA damage response along with the recovery of DNA-PKcs. This phenomenon further supports the association between PIG3 and DNA-PKcs. The MG-132 treatment experiment indicated that PIG3 doesn't affect the degradation of DNA-PKcs or its stability. There is also no obvious change in the transcription (mRNA) level during the period of DNA-PKcs depression. Therefore, it seems that PIG3 may regulate the translation activity of DNA-PKcs through an as yet unknown mechanism. Notably, a compensatory feedback of increased transcriptional activity of DNA-PKcs occurred after a certain number of cell cycles or passages of subculture after PIG3 depletion, which largely is attributed to the recovery of the cellular DNA-PKcs level. Importantly, the DNA damage response was also recovered, at least partially, along with the recovered DNA-PKcs.
Our work reveals a novel mechanism through which PIG3 contributes to the DNA damage response. Inactivation of PIG3 leads temporarily to the attenuation of DNA-PKcs, which in turn causes a depression of ATM and a defect in DNA damage response and abnormal cell cycle procession. After a certain cell cycles or passages of subculture, there followed a feedback of compensatory increased transcription activity of DNA-PKcs, which resulted in recovery of the cellular DNA-PKcs level and ATM, and consequently the DNA damage response activities. These results provide a new insight into PIG3's role in DNA damage signaling and the regulation network of the cellular DNA-PKcs homeostatic balance.
Acknowledgements
The authors thank Professor JH Hendry (Manchester University, UK) for helpful suggestion and editorial assistance with this paper. This work was supported by the National Key Basic Research Program (973 Program) of MOST, China (Grant 2007CB914603), the Chinese National Natural Science Foundation (Grant 81071678 and 30970677), and the Distinguished Youth Scientist Foundation of NFSC, China (Grant 30825011).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300-305
2. Campomenosi P, Monti P, Aprile A, Abbondandolo A, Frebourg T, Gold B, Crook T, Inga A, Resnick MA, Iggo R, Fronza G. p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene. 2001;20:3573-3579
3. Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638-650
4. Porte S, Valencia E, Yakovtseva EA, Borras E, Shafqat N, Debreczeny JE, Pike ACW, Oppermann U, Farres J, Fita I, Pares X. Three-dimensional Structure and Enzymatic Function of Proapoptotic Human p53-inducible Quinone Oxidoreductase PIG3. J Biol Chem. 2009;284:17194-17205
5. Wang H, Luo K, Tan LZ, Ren BG, Gu LQ, Michalopoulos G, Luo JH, Yu YP. p53-induced Gene 3 Mediates Cell Death Induced by Glutathione Peroxidase 3. J Biol Chem. 2012;287:16890-16902
6. Long XH, Zhao ZQ, He XP, Wang HP, Xu QZ, An J, Bai B, Sui JL, Zhou PK. Dose-dependent expression changes of early response genes to ionizing radiation in human lymphoblastoid cells. Int J Mol Med. 2007;19:607-615
7. Qin X, Zhang SM, Li B, Liu XD, He XP, Shang ZF, Xu QZ, Zhao ZQ, Ye QN, Zhou PK. p53-dependent upregulation of PIG3 transcription by γ--ray irradiation and its interaction with KAP1 in responding to DNA damage. Chin Science Bulletin. 2011;56:3162-3171
8. Lee JH, Kang Y, Khare V, Jin ZY, Kang MY, Yoon Y, Hyun JW, Chung MH, Cho SI, Jun JY, Chang IY, You HJ. The p53-inducible gene 3 (PIG3) contributes to early cellular response to DNA damage. Oncogene. 2009;29:1431-1450
9. Zhou PK. DNA Damage, Signaling and Repair: Protecting genomic integrity and reducing the risk of human disease. Chin Science Bulletin. 2011;56:3119-3121
10. Dai J, Cui X, Zhu Z, Hu W. Non-homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryos. Int J Biol Sci. 2010;6(7):756-768
11. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605-611
12. Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, Oakley GG. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012;40(21):10780-94
13. An J, Huang YC, Xu XZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Molecular Biology. 2010;11:e18
14. Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9(17):3602-3610
15. Speroni J, Federico MB, Mansilla SF, Soria G, Gottifredi V. Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc Natl Acad Sci USA. 2012;109(19):7344-7349
16. Tomimatsu N, Mukherjee B, Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009;10:629-635
17. Peng Y. Deficiency in the Catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Research. 2005;65:1670-1677
18. Liu X, Bennett RL, Cheng X, Byrne M, Reinhard MK, May WS. PKR regulates proliferation, differentiation and survival of murine hematopoietic stem/progenitor cells. Blood. 2013 epub
19. Baus F, Gire V, Fisher D, Piette J, Dulic V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003;22:3992-4002
20. Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803-1815
21. Ohi R, Gould KL. Regulating the onset of mitosis. Curr Opin Cell Biol. 1999;11:267-273
22. Suzuki K, Kodama S, Watanabe M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J Biol Chem. 1999;274:25571-25575
23. Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538-37543
24. Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, Wang Y, Zhou PK. Inactivation of DNA-Dependent Protein Kinase Leads to Spindle Disruption and Mitotic Catastrophe with Attenuated Checkpoint Protein 2 Phosphorylation in Response to DNA Damage. Cancer Res. 2010;70:3657-3666
25. Fragouli E, Wells D, Iager AE, Kayisli UA, Patrizio P. Alteration of gene expression in human cumulus cells as a potential indicator of oocyte aneuploidy. Human Reproduction. 2012;27:2559-2568
Author contact
![]() Corresponding author: Ping-Kun Zhou, Tel: +86-1066931217; Fax: +86-1068213912; E-Mail: zhoupkac.cn.
Corresponding author: Ping-Kun Zhou, Tel: +86-1066931217; Fax: +86-1068213912; E-Mail: zhoupkac.cn.