International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
The structure of c-kit receptor
The distribution of c-kit...
The signaling pathway of c-kit...
The activation mechanism of...
SCF/c-kit downstream signal...
The c-kit receptor mutation in...
The c-kit receptor mutations in...
The c-kit receptor mutations in...
The c-kit receptor mutations in...
The c-kit receptor-targeted...
Prospects
Abbreviations
Acknowledgements
References
Author biography
Introduction
The structure of c-kit receptor
The distribution of c-kit...
The signaling pathway of c-kit...
The activation mechanism of...
SCF/c-kit downstream signal...
The c-kit receptor mutation in...
The c-kit receptor mutations in...
The c-kit receptor mutations in...
The c-kit receptor mutations in...
The c-kit receptor-targeted...
Prospects
Abbreviations
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(5):435-443. doi:10.7150/ijbs.6087 This issue Cite
Review
The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases
Jing Liang1, Yan-Ling Wu2 ![]() , Bing-Jia Chen1, Wen Zhang1
, Bing-Jia Chen1, Wen Zhang1 ![]() , Yoshimasa Tanaka3, Hiroshi Sugiyama4
, Yoshimasa Tanaka3, Hiroshi Sugiyama4
1. Department of Biopharmaceutical Science, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, China.
2. Virus Inspection Department, Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, China.
3. Center for Innovation in Immunoregulative Technology and Therapeutics, Graduate School of Medicine, Kyoto University, Kyoto, 606-8501, Japan.
4. Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan.
Received 2013-2-17; Accepted 2013-4-15; Published 2013-5-8
Citation:
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int J Biol Sci 2013; 9(5):435-443. doi:10.7150/ijbs.6087. https://www.ijbs.com/v09p0435.htm
Other stylesAbstract
As an important member of tyrosine kinase family, c-kit receptor causes specific expression of certain genes, regulates cell differentiation and proliferation, resists cell apoptosis, and plays a key role in tumor occurrence, development, migration and recurrence through activating the downstream signaling molecules following interaction with stem cell factor (SCF). The abnormality of SCF/c-kit signaling pathway is closely related to some certain tumors. The discovery of c-kit receptor-targeted drugs has promoted clinical-related cancer's diagnosis and treatment. In this paper, we review recent research progress on c-kit receptor-mediated signal transduction and its potential therapeutic application as a target in tumor-related diseases.
Keywords: c-kit receptor, signal transduction, tumor, targeted therapy
Introduction
The c-kit receptor (CD117) is a transmembrane protein with tyrosine kinase activity encoded by the oncogene c-kit. It is an important member of type III receptor tyrosine kinase family; other tyrosine kinase receptor molecules include macrophage colony-stimulating factor receptor (M-CSFR), platelet-derived growth factor (PDGF) and flk2/flk3 receptor and so on. The ligand for c-kit is stem cell factor (SCF), a hematopoietic cytokine, which plays an important role in maintaining the survival of hematopoietic cells, promoting hematopoietic cell proliferation and differentiation, and regulating growth and development of hematopoietic cells [1]. Generally, SCF dimer forms complexes with two molecules of the extracellular domain of c-kit to activate downstream signal transduction and then regulate a variety of cells biological behavior, such as normal cells proliferation and differentiation, tumor occurrence, development, migration and recurrence [2-4]. Recent studies show that the abnormal expression genes and abnormal expression products caused by c-kit mutations are the key reason for gastrointestinal stromal tumour [5]. The c-kit as a target for tumor treatment is a hot topic at the current moment. Here we describe the major structural, functional features of c-kit.
The structure of c-kit receptor
The human receptor tyrosine kinase (RTK) c-kit (also referred to as stem cell factor receptor or CD117) was identified and characterized in 1987 as the cellular homologue [6] of the viral oncogene v-kit which was isolated in 1986 from a feline retrovirus [7]. The c-kit gene is the allele of the white spot dominant gene. Human c-kit cDNA was first isolated from the fetal brain cDNA library (Fig. 1). Human c-kit proto-oncogene locates on chromosome 4q11∼12 and has a total length of 90 kb. The coded product c-kit receptor (CD117) is a type I transmembrane glycoprotein of relative molecular mass of 145 kDa and belongs to the type III receptor tyrosine kinase family. C-kit is highly conserved in the evolution. And the c-kit receptor is composed of 976 amino acids (aa) divided into an extra-cellular domain with 519 aa, a trans-membrane domain with 23 aa, an intracellular tail of 433 aa consisting of a juxta-membrane domain and a tyrosine kinase domain inserted by about 80 amino acid residues [5] (Fig. 2). The extra-cellular domain contains five immunoglobulin-like domains (D1∼D5), D1∼D3 of which as the key component of c-kit binding to SCF, and D4∼D5 of which are the essential region of dimerization [8]. The membrane region near the dimerization domain (exon 8 and exon 9), the intra-cellular domain close to the membrane and the kinase domain (exon 17) are often prone to mutations. In addition, the tyrosine kinase domain which contains autophosphorylation tyrosine kinase functions as homodimers with phosphatidylinositol kinase. Subsequently downstream signal transduction molecules can be activated through the formation of the c-kit/SCF complex, and then regulate gene expression and cell growth, proliferation and differentiation.
The subtypes of c-kit receptor have been characterized. These involve the presence or absence of the Gly-Asn-Asn-Lys (510-GNNK-513) sequence in the extra-cellular domain adjacent to the trans-membrane domain and the presence or absence of a Ser residue at position 715 in the inter-kinase domain [9], but the correlation between the GNNK+/- subtypes and the Ser+/- subtypes is unclear. The finding that the GNNK+/- subtypes of c-kit possess different signal transduction properties may be due to the differential recruitment and activation of Src family kinases [9, 10]. Furthermore, the Ser+/- subtypes likely have different roles has to be studied. In addition, there is s-kit formation which may be the antagonist of c-kit, which can inhibit the combination of c-kit and SCF or regulate the biological activity of the local SCF.
Figure 1
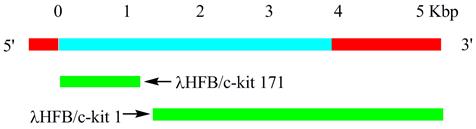
Human c-kit cDNA clones. Human c-kit cDNA clones (green bars) from human fetal brain (HFB) λgt10 cDNA libraries are shown with a schematic diagram of the predicted c-kit mRNA. Cloning sequences (blue bar) and untranslated regions (red bar) are indicated.
Figure 2
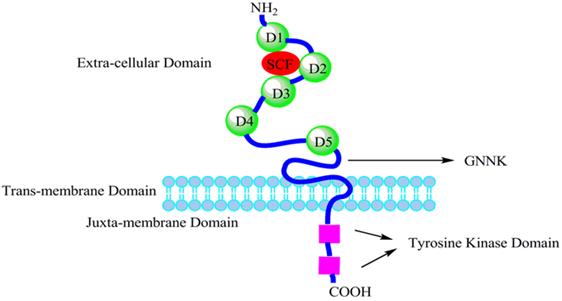
Schematic representation of the structure of c-kit. The extra-cellular domain consists of five Ig-like domains (D1-D5). The sequence GNNK (a tetrapeptide sequence) is either present or absent in the extra-cellular domain near the plasma membrane of c-kit (GNNK+ or GNNK-). The intracellular domain contains the tyrosine kinase domain, which is split into two parts by the amino acid residues insert sequence.
The distribution of c-kit receptor
C-kit receptor is widely distributed in hematopoietic cells and other tissue cells. In healthy people, approximately 1% to 4% of bone marrow stem cells and 60% to 75% of CD34+ positive hematopoietic cells express c-kit receptor. C-kit-dependent cell types include hematopoietic cells, germ cells, mast cells, melanoma cells and the gastrointestinal tract cajal cells. Adult mice oocytes express c-kit receptor while the surrounding granulosa cells express stem cell factor SCF. Studies reported that 66.3% of the germ cells and the sperm express c-kit receptor, which is in the fact that each type of germ cells can be isolated from human semen by using anti-CD117 monoclonal antibody [11]. It has also been found that c-kit function defects can cause abnormal peripheral nerve regeneration and spatial memory impairment [12]. Interestingly, the approximately 85% of the B cell progenitors also express c-kit receptor, but it is gradually disappearing with cell differentiation and maturation. The approximately 30% of CD4-, CD8-, CD3- B cells present the expression of c-kit receptor. In addition, c-kit receptor was also found in skin appendages, the breast epithelial cells and the small neurons in the brain. C-kit+ cells from human fetal liver have been studied, the data suggest that c-kit receptor plays a role in the liver stem cell growth and development [13].
The signaling pathway of c-kit receptor
C-kit signaling plays a very important role in regulation of the red blood cell production, lymphocyte proliferation, mast cell development and function, melanin formation, and gamete formation. Specific binding of SCF can induce homologous dimerization, and drives downstream signal transduction pathway. It subsequently regulates gene expression and cell growth, proliferation and differentiation.
The activation mechanism of c-kit receptor
The activation mechanism of c-kit receptor was studied by Satoru Yuzawa, et al [14]. The c-kit extra-cellular domain composed of five Ig-like domains (D1∼D5) can be divided into three separate functional units depending on the biochemical functions (Fig. 3): D1-D2-D3 is a particular unit binding to SCF; the second functional unit is formed by this SCF binding unit and a D4; on the basis of second functional unit, the third functional unit is constituted through a flexible joint to connect D5. Further, the strong electrostatic environment of D4 can induce repulsion between D4-D4 and maintain the inactive monomer. As both D4-D4 and D5-D5 binding affinity is respectively lower in the absence of SCF, c-kit extra-cellular domains are not easy to form dimers. When the local concentration of the SCF-mediated c-kit receptor reaches a certain value, the adjacent D4s will overcome the electrostatic repulsion and combine into the D4-D4 form. The transverse D4-D4 and D5-D5 interaction may accumulate and stabilize the two adjacent membrane proximal external regions of the c-kit extra-cellular domain. In case of SCF ligand and c-kit receptor combination there may be two steps: first, SCF integrates with the corresponding domain of c-kit by electrostatic attraction between SCF and D1-D2-D3 functional unit [15]; second, SCF conformation changes can mediate other functions to make the SCF/c-kit complex structure more stable. Also, SCF-induced c-kit receptor dimerization can cause a series of changes on D4 and D5 domains, but D1-D2-D3 domains which bind SCF are almost unchanged, leading to that the specific conformation that the C-terminal of D5 will move to the cell membrane can occur during the dimerization. It is worth noting that Arg381 or Glu386 point mutation of D4 domain can make a huge impact on c-kit receptor tyrosine auto-phosphorylation and downstream signal transduction pathways.
Figure 3
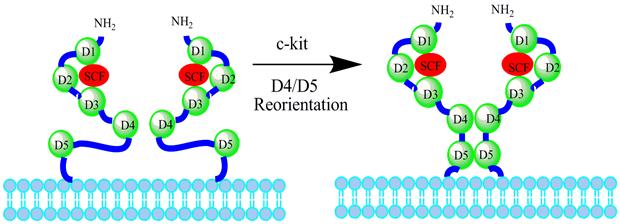
Interactions of the c-kit dimer. When the dimerization of the c-kit is induced by SCF, D1-D3 remains unchanged, whereas D4 and D5 become reoriented to reach each other by overcoming the electrostatic repulsion. Left structure shows the model of c-kit dimer with two free c-kit molecules. Right structure shows that the D4-D4 and D5-D5 domains are in close proximity in actual SCF-induced c-kit dimer.
SCF/c-kit downstream signal transduction pathway
C-kit receptor downstream signal transduction pathways have been described in different cell lines. Mast cells are the most commonly used cell type for signal study. Transient transfection systems can also be used, such as HEK293 cells or Cos cells. Furthermore, some scientists use chimeric objects, for example the fusion chimera combines epidermal growth factor (EGF) receptor and c-kit receptor extracellular part. SCF/c-kit downstream signal transduction pathways are very complex and currently known signal transduction pathways are as follows (Fig. 4).
Ras/Erk signal transduction pathway [16]: Ras/Erk pathway plays a very important role in cell differentiation and survival. That RTK collaborates with GDP/GTP exchange factor Sos can activate Ras. Sos and adapter protein Grb2 form a complex through SH2, and then raise to membrane to act on Ras. Activated Ras can activate the Ser/Thr kinase Raf-1 which can then activate the dual specificity kinases Mek1 and Mek2. Phosphorylation of Mek1 and Mek2 can re-activate Erk1 and Erk2. Subsequently, dimerization Erks move to the nucleus and regulate gene activities.
PI3K signal transduction pathway [17]: After c-kit receptor activation, PI3K dimerizes through SH2 and then recruits to membrane. SH2 which contains an adapter protein of 85 kDa connects 110 kDa esterase subunits to c-kit receptor. As PI3K produces a series of biological signals on the membrane, signal transduction molecules downstream of PI3K are activated and cell survival and anglogenesis are regulated.
PLC-γ signaling transduction pathway [18]: Phospholipase (PLC) PLC-γ contains two SH2 domains, a SH3 domain, a PH domain and a catalytic domain. PLC-γ can catalyze phosphoinositide PIP2 to generate second messenger diacylglycerol (DAG) and soluble inositol 1,4,5-trisphosphate (IP3). DAG can activate PKC; IP3 then binds to the endoplasmic reticulum to stimulate the release of Ca+.
Figure 4
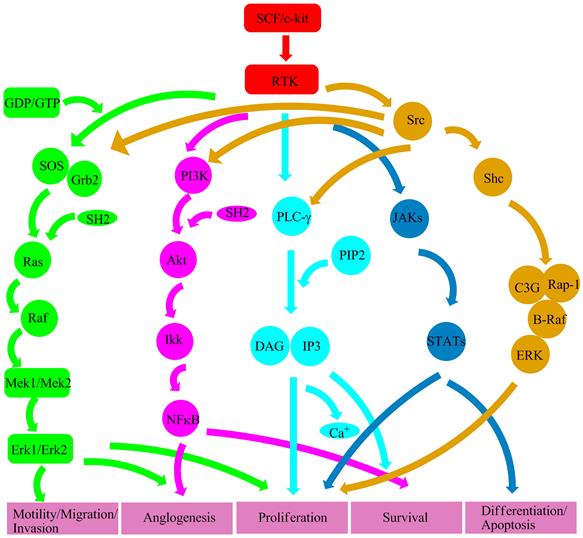
A proposed structure of RTK downstream signal transduction pathways. Ras/Erk signal (green color) leads to activation of the RAS-RAF-MEK-ERK pathway. PI3K signal (purple color) is associated with anglogenesis and cell survival. PLC-γ signal (light blue color) regulates cell proliferation and survival. Src signal (yellow color) plays a key role in several signal transduction pathways involved in cell survival, proliferation, motility, migration, invasion and anglogenesis. JAK/STAT signal (blue color) implicates in cell proliferation, differentiation and apoptosis.
Src kinase signal transduction pathway [19]: Src kinase is related to a series of cell functions including cell survival, angiogenesis, proliferation, motility, migration and invasion. Src kinase which contains SH2, SH3, the N-terminal kinase domain and a fourteen alkylation site plays the role of anchoring to membrane. And Src kinase activation has to do with GNNK sequence. More importantly, GNNK- form can be faster and more intense to activate this kinase.
JAK/STAT signaling pathway [20]: JAKs are cytoplasmic tyrosine kinases. The c-kit receptor can be fast and transient to activate JAK2 [21]. Then, activated JAKs can make the transcription factor STATs phosphorylation and dimerization. Finally, dimerized STATs transfer to the nucleus and regulate cell proliferation, differentiation and apoptosis.
The c-kit receptor mutation in tumors
Human c-kit receptor loss of function mutations can cause autosomal mottle disease [22]. This syndrome is characterized by hair and skin pigmentation abnormalities, deafness and megacolon. Gain of function mutations can be found in GIST (>90%), mast cell tumor (>70%), nasal T-cell lymphomas (>17%), seminoma / dysgerminoma (> 9%) [23] and acute myeloid leukemia (>68%) [24]. Mutations often occur in membrane proximal immunoglobulin-like domain (D5, exon 8 and exon 9), near membrane domain (exon 11) and the tyrosine kinase domain (exon 17) and include missing, point mutation, duplication and insertion that can lead to c-kit receptor activation. More evidence demonstrated that exon 11 and exon 17 mutations can reduce the body's self-inhibition which results in sustained c-kit receptor activation [25]. But the mechanism of exon 8 and exon 9 mutations has yet to be studied. Exon 8 deletion or insertion mutations (either missing or replaced Asp419) are found in acute myeloid leukemia (AML). And the repeat sequences Ala503∼Tyr502 and Ala502∼Phe506 are found in gastrointestinal stromal tumors (GIST). Almost all of the activation mutations of proto-oncogene occur in the D5-D5 surface and these mutations can enhance the affinity of adjacent D5-D5 domain [14]. In addition, the paracrine or autocrine activation of c-kit receptor may play an important role in many other human malignancies, such as ovarian cancer, small cell lung cancer and other tumors [23, 26].
The c-kit receptor mutations in GIST
The gastrointestinal stromal tumors (GIST) originates in gastrointestinal tract Cajal cells and approximately 70% of these tumors occur in the stomach, 20-30% in the small intestine, and less than 10% in the esophagus, colon, appendix, and rectum combined. A majority of GIST are benign; 10-30% are malignant. The c-kit receptor is positively immunostained in 95% GIST samples and it has become an important indicator for the diagnosis of GIST [27]. In case of mutations, c-kit and PDGFR mutations are the main reasons in GIST. In the approximately 20% to 80% of GIST patients, moreover, occurred c-kit gene gain of function mutations [28]. Also WeiLien Wang et al. reported that in 419 cases of GIST, mutations in c-kit exons 9,11, 13 and 17 occurred in 73%, 10%, 3% and 1%, respectively [29]. The c-kit mutations were classified as several forms, common deletions, point mutations, and occasionally insert mutation, counting up to 150 kinds of mutations which occur mainly in membrane proximal domain (exon 11), and these mutations show the extreme diversity of this region. However, mutations followed mainly occur in extracellular domain (exon 9), rarely occur in the kinase domain (exon 13 and exon 17) and ATP binding domain (exon 14). Significantly, different mutations may suggest different biological effects and different treatment options. The clinical manifestations of GIST are associated with the tumor site. Small intestine GIST was easily deteriorated and had a death rate of 39 percent which was twice more than stomach GIST [30]. Chizuru Sogawa et al. found that 111In labeled anti-c-kit monoclonal antibody probe had a very good sensitivity and specificity to detect GIST in nude mice [31]. While Chisato Yoshida et al. reported that 64Cu labeled anti-c-kit monoclonal antibody probe for PET image was clearer than 111In labeled one [32]. Because of the genetic differences between human and mouse, the animal also deserve a deeper investigation.
The c-kit receptor mutations in leukemia
Leukemia is a primary malignant tumor of the hematopoietic system with treatment difficulties and high recurrence rate. Effective forecasting and monitoring means to improve leukemia efficacy and prolong the lives of patients is of great significance. Studies have shown that c-kit receptor was expressed in 68% of acute myeloid leukemia (AML) patients and in 80% of chronic myelogenous leukemia in blast phase patients but in only 2% of acute lymphoid leukemia patients [24]. Further research also found that c-kit receptor positive AML patients survived significantly shorter than the c-kit receptor negative AML patients and c-kit receptor high expressed patients at low complete remission rate [33]. And approximately 30% of AML patients acquired c-kit receptor gene mutations mainly occurred in exon 8 and exon 17, which, especially the latter, are closely related to poor prognosis of AML. Furthermore, these mutations were revealed such as c-kit mutations in exon 8 and Asp816 mutations in both inv (16) AML and t (8; 21) (q22; q22) AML [34], and c-kit Asp816Val and Asp816His mutations [35]. Therefore, c-kit receptor plays an important role in AML complete remission and recurrence. The c-kit receptor positive expressed patients have low complete remission rate and tend to relapse despite complete remission. Hence, the expression of c-kit can be used as an indicator of
The c-kit receptor mutations in other tumors
The c-kit receptor is also expressed in bone marrow tumor, germ cell tumors, mast cell cancer, prostate cancer, glial cancer, small cell lung cancer, some non-small cell lung cancer and other tumors. In some sarcomas, such as angiosarcoma and Ewing's sarcoma, different levels of c-kit expression are shown, but whether c-kit is expressed in synovial sarcoma, leiomyosarcoma and malignant fibrous histiocytoma is still controversial. Several study results have indicated that the lymph nodes in Hodgkin's lymphoma express c-kit receptor but the non-Hodgkin's lymphoma have no c-kit receptor expression except for degenerative large cell lymphoma [36]. 24.1% of c-kit positive expression was observed in adenocarcinoma which was less than the level of 45.8% in normal colorectal tissues [37]. In addition, approximately 36% of primary malignant melanomas are c-kit immunohistochemical positive. In 20 cases of primary anal mucosal melanoma samples tested by Antonescu et al. it was observed that 15% of the samples had point mutations and 1/3 of the mutated samples increased gene copy numbers [38]. Moreover, the mutation types of melanoma usually are substitution and mutations rarely occur in exon 9 which is the difference between GISTS. Recent data have ascertained that different parts of the melanoma have different c-kit gene copy numbers; non-skin melanoma c-kit gene copy numbers are higher than skin melanoma. In addition, Aishan Li et al. reported the fact that 69.2% of small cell lung cancer (SCLC) organizations also expressed c-kit receptor, and exon 9 and exon 11 mutations were detected by immunohistochemistry method [39].
The c-kit receptor-targeted therapies
Cancer is a major disease threatening human health and the whole world has been paying close attention to its treatment. Traditional chemotherapy is not completely effective for much tumor treatment because of their nonspecific blocking tumor cells division and damage to normal cells. Also the GIST patients with complete resection still have a very high risk of recurrent. The appearance and development of c-kit antibodies, especially the monoclonal antibodies, promote the research of c-kit receptor for continuous activation mechanism. It is generally believed that, the dimerization of c-kit receptor is closely related to its continuous activation and thereby it is considered to be c-kit receptor-related tumor pathogenesis. With in-depth broadband research to c-kit receptor, therefore, molecular targeted therapy due to its specificity and low toxicity is increasingly becoming a hot spot.
Imatinib mesylate is one of the first examples of the successful solid tumor treatment with a tyrosine kinase-targeted drug in humans, which is first designed for chronic myelogenous leukemia. It can inhibit enzymatic activity of several tyrosine kinases including c-kit and the platelet-derived growth factor receptor. Later imatinib was gradually applied to treat GIST with good results [40]. GIST patients treated with imatinib had a control rate of 80% to 90%, most showed partial remission, 12% showed complete remission [41]. The sensitivity of imatinib is closely related to c-kit receptor gene. Xiaofeng Jiang et al. demonstrated that imatinib could inhibit c-kit mutated mucosal melanoma cells proliferation with induced apoptosis and reduced downstream signaling regulatory molecules (p42/44, AKT, MTOR, STAT1, STAT3, P70S6K, S6K, etc.) but the wild-type had no changes [42]. Yongsheng Ma et al. also reported that different types of the c-kit mutation showed different sensitivity to c-kit kinase inhibitors and classifications of mutation exhibited prediction in selecting drugs [43]. Unfortunately, most of patients will gradually develop resistance to imatinib. 9% to 14% of GIST patients treated with imatinib in the first three months will have an early resistance; while the average progression-free survival would develop with disease progression after 24 months and then have acquired resistance to imatinib [44]. The resistance mechanism may be ascribed to: the occurrence of c-kit gene secondary mutations [45], c-kit gene amplification in a large number, the absence of wild-type allele, or lack of blood concentration of imatinib. Because of resistance to imatinib, a novel and effective inhibitor with no/little resistance has to be explored.
Sunitinib is the second generation of multi-target tyrosine kinase receptor inhibitor approved by the FDA in 2006 and the second-line drugs for imatinib-resistant GIST patients that may extend survival. The sensitivity of sunitinib is also related to the state of c-kit gene. More recently, Ashida et al. investigated six acral melanoma cell lines, indicating that sunitinib was better sensitive to c-kit D820Y mutant cells with the secondary resistance to imatinib while the wild type cells did not change [46]. However, although Sunitinib can delay tumor process, the vast majority of patients will eventually develop resistance [47]. Hence, we need to discover more effective second-line and third-line drugs against resistance, such as, sorafenib, dovitinib, PKC412, axitinib, INNO-406, KRN951, dasatinib, pazopanib, ABT-869 and other multi-target tyrosine kinase inhibitors have been being tested in recent years. A phase II clinical trial has shown that dasatinib has good activity to imatinib-resistant or non-resistant chronic myelogenous leukemia patients [48]. While ABT-869 can inhibit Ewing's sarcoma cell proliferation by blocking c-kit receptor signaling pathway [49].
Cancer as a very complex disease can not be effectively eradicated by tyrosine kinase inhibitor treatment strategies and there are some side effects with treatment. Thus in recent years, in order to search for the new generation of more effective anticancer agents, novel treatment strategies with small molecules directed to new targets have gradually emerged. Researchers always attempted to discover that such small molecule targeted drugs can maximize efficacy with minimal dose and maximally reduce the adverse reactions of kinase inhibitors. With the in-depth understanding of the relationship between telomeric G-quadruplex and tumors it was found that G-rich promoter region of gene DNA sequence can be induced to form G-quadruplex in vitro/vivo which can inhibit telomerase activity in approximately 85% of cancers. In case of oncogene c-kit, the parallel crystal structure of a c-kit promoter G-quadruplex was first reported, indicating that as well as the observation of potassium and magnesium ions, a highly significant difference in the dimensions of the large cleft in the structure may be a potential target for small molecules [50]. To date, the research on the small molecule ligands targeting the c-kit gene promoter region attracts more attention. For example it was reported that 6-substituted indenoisoquinoline [51] (Fig. 5A), benzo[α]phenoxazine derivatives [52] (Fig. 5B), peptide nucleic acid (PNA) [53] and other small molecule ligands, which functions as potential anti-cancer drugs, can stabilize G-quadruplex and consequently effectively block the transcription of c-kit gene.
Prospects
C-kit is a suitable target for future drug development for the treatment of a multitude of human malignancies related to c-kit. However, some mechanism on cancer-related signal transduction downstream of c-kit is not very clear such as c-kit overexpression mechanisms in these tumors, and the correlation between c-kit mutations. And because of its diverse causes, irregular development, unstable phenotype, complex proliferation and differentiation mechanisms, multiple cellular signal transduction pathways and other features, the treatment of cancer is very difficult. The treatment with single target drugs achieves rather difficultly expected results. Although the multi-target drugs have made great progress in cancer therapy, many kinase inhibitors also inhibit the normal signal transduction pathways. Collectively, the low toxicity and multi-target anti-cancer drugs are the trend to study. Moreover G-rich region of c-kit receptor promoter has become an important potential target. As there exists the differential resistance for different cancer patients, besides efficiency and low toxicity target drugs we should further need to consider about other characteristics of tumors to implement joint and individualized treatment programs.
Figure 5
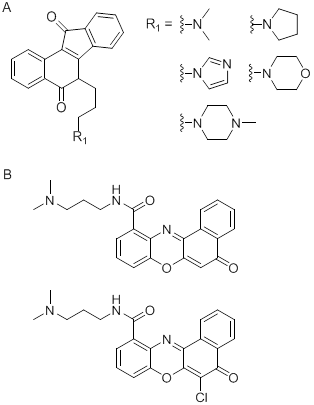
Structure of indenoisoquinoline (A) and benzo[α]phenoxazine derivatives (B).
Abbreviations
SCF: stem cell factor; GIST: gastrointestinal stromal tumors; GNNK: presence or absence of the four amino acids 510-513,Gly-Asn-Asn-Lys; M-CSFR: macrophage colony-stimulating factor receptor; HFB: human fetal brain; EGF: epidermal growth factor; Ras: a guanosine-nucleotide-binding protein (G protein) / single-subunit small GTPase; ERK: extracellular regulated protein kinases; RTK: receptor tyrosine kinase; Sos: son of sevenless; Grb2: growth factor receptor-bound protein 2; Raf: the Ser/Thr protein kinase; Mek: mitogen-activated protein kinase; Erk: extracellular signal-regulated kinase; PTK: protein tyrosine kinase; PI3K: phosphatidylinositol 3-kinase; SH2: Scr homology2; IKK: InB kinase; NF-κB: Nuclear factor kappa B; Akt/PKB: protein kinase B; PLC: Phospholipase; PH: pleckstrin homology; PIP2: phosphatidylinositol-4,5 bisphosphate; IP3: inositol 1,4,5-trisphosphate; DAG: diacylglycerol; PKC: protein kinase C; Src: proto-oncogene tyrosine-protein kinase; JAK: janus kinase; STAT: signal transducers and activators of transcription; PDGFR: platelet-derived growth factor receptor; AML: acute myeloid leukemia; SCLC: small cell lung cancer.
Acknowledgements
We gratefully acknowledge the financial support from Zhejiang Provincial Natural Science Foundation of China (LY12B02019), the Public Technology Research and Social Development Project of Zhejiang Province (2011C23004), the Qianjiang Talents Program of Zhejiang Province (2009R10002) and the Program for Zhejiang Leading Team of Science and Technology Innovation (2011R50021).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen SQ, Xiong AQ. The progress and implication of stem cell factor. Basic Medical Sciences and Clinics. 2002;22(5):385-390
2. Philo JS, Wen J, Wypych J. et al. Human Stem Cell Factor Dimer Forms a Complex with Two Molecules of the Extracellular Domain of Its Receptor, Kit. J Biol Chem. 1996;271(12):6895-902
3. Nishikawa S, Kusakabe M, Yoshinaga K. et al. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991;10(8):2111-2118
4. Huizinga JD, Thuneberg L, Kluppel M. et al. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373(6512):347-349
5. Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J Pathol. 2011;223(2):251-261
6. Yarden Y, Kuang WJ, Yang-Feng T. et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6(11):3341-3351
7. Besmer P, Murphy JE, George PC. et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320(6061):415-421
8. Lemmon MA, Ferguson KM. A new twist in the transmembrane signaling tool-kit. Cell. 2007;130(2):213-215
9. Crosier PS, Ricciardi ST, Hall LR. et al. Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood. 1993;82(4):1151-1158
10. Voytyuk O, Lennartsson J, Mogi A. et al. Src Family Kinases Are Involved in the Differential Signaling from Two Splice Forms of c-Kit. J Biol Chem. 2003;278(11):9159-9166
11. Jiang XL, Fan LQ, Zhu WB. et al. Research on expression of CD117 on germ cells in human semen. China Journal of Modern Medicine. 2005;15(2):208-211
12. Kimura Y, Jones N, Kluppel M. et al. Targeted mutations of the juxtamembrane tyrosines in the Kit receptor tyrosine kinase selectively affect multiple cell lineages. Proc Natl Acad Sci USA. 2004;101(16):6015-6020
13. Ling LZ, Ni YH, Hu YL. et al. Characteristics of C-kit+ cells derived from the human fetal liver. Journal of Clinical Rehabilitative Tissue Engineering Research. 2009;13(1):111-116
14. Yuzawa S, Opatowsky Y, Zhang Z. et al. Structural Basis for Activation of the Receptor Tyrosine Kinase KIT by Stem Cell Factor. Cell. 2007;130:323-334
15. Mueller TD, Zhang JL, Sebalda W. et al. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim Biophys Acta. 2002;1592(3):237-250
16. Yasuda T, Kurosaki T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle. 2008;7(23):3634-3640
17. Sansal I, Sellers WR. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J Clin Oncol. 2004;22(14):2954-2963
18. Hendriks RW. Drug discovery: New Btk inhibitor holds promise. Nat Chem Biol. 2011;7(1):4-5
19. Summy JM, Gallick GE. Treatment for Advanced Tumors: Src Reclaims Center Stage. Clin Cancer Res. 2006;12(5):1398-1401
20. Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23(48):8017-8023
21. Brizzi MF, Zini MG, Aronica MG. et al. Convergence of signaling by interleukin-3, granulocyte-macrophage colony-stimulating factor, and mast cell growth factor on JAK2 tyrosine kinase. J Biol Chem. 1994;269(50):31680-31684
22. Spritz RA. Molecular basis of human piebaldism. Invest Dermatol. 1994;103:S137-S140
23. Heinrich M, Blanke CD, Druker BJ. et al. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20:1692-1703
24. Shen HQ, Tang YM, Yang SL. et al. Analysis of CD117 expression on leukemia cells. Chinese Journal of Hematology. 2003;24(5):516-521
25. Greenwald J, Vega ME, Allendorph GP. et al. A flexible activin explains the membran-dependent cooperative assembly of TGF-β family receptors. Mol Cell. 2004;15(3):485-489
26. Krystal GW, Hines SJ, Organ CP. Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res. 1996;56(2):370-376
27. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GIST at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13(10):1134-1142
28. Gomes AL, Gouveia A, Capelinha AF. et al. Molecular alterations of KIT and PDGFRA in GISTs evaluation of a Portuguese series. J Clin Pathol. 2008;61(12):203-208
29. Wang WL, Conley A, Reynoso D. et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67:S15-S24
30. Miettinen M, Makhlouf H, Sobin LH. et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30(4):477-489
31. Sogawa C, Tsuji AB, Sudo H. et al. c-kit-targeted imaging of gastrointestinal stromal tumor using radiolabeled anti-c-kit monoclonal antibody in a mouse tumor model. Nucl Med Biol. 2010;37(2):179-187
32. Yoshida C, Tsuji AB, Sudo H. et al. Development of positron emission tomography probe of 64Cu-labeled anti-C-kit 12A8 Fab to measure protooncogene C-kit expression. Nucl Med Biol. 2011;38(3):331-337
33. Tsao AS, Kantarjian H, Thomas D. et al. c-kit receptor expression in acute leukemias-association with patient and disease characteristics and with outcome. Leuk Res. 2004;28(4):373-378
34. Care RS, Valk PJ, Goodeve AC. et al. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol. 2003;121(5):775-777
35. Pullarkat VA, Bueso-Ramos C, Lai R. et al. Systemic mastocytosis with associated clonal hematological non-masr-cell lineage disease: analysis of clinicopathologic features and activating c-kit mutations. Am J Hematol. 2003;73(1):12-17
36. Pinto A, Gloghini A, Gattei V. et al. Expression of the c-kit receptor in human lymphoma as is restricted to Hodgkin's Disease and CD30+ anaplastic large cell lymphomas. Blood. 1994;83(3):785-792
37. Gopalakrishnan N, Jayanthi V, Devaraj H. c-Kit and Musashi-1 expression in colorectal carcinoma and its association with ethological factors. Oncol Gastroenterol Hepatol Reports. 2012;1:1-8
38. Antonescu CR, Busam KJ, Francone TD. et al. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. Int J Cancer. 2007;121(2):257-264
39. Li AS, Sun LN, Zhan ZL. Expression and mutational analysis of c-kit gene in small cell lung cancer. Chin J Cancer Prev Treat. 2007;14(12):917-919
40. Joensuu H, Roberts PJ, Sarlomo-Rikala M. et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Eng J Med. 2001;344(14):1052-1056
41. Boeden EC, Baker LH, Bell RS. et al. Soft tissue sarcomas of adults: state of the translational science. Clin Cancer Res. 2003;9(6):1941-1956
42. Jiang X, Zhou J, Yuen NK. et al. Imatinib Targeting of KIT-Mutant Oncoprotein in Melanoma. Clin Cancer Res. 2008;14(23):7726-7732
43. Ma YS, Zeng S, Metcalfe DD. et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99(5):1741-1744
44. Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemther Pharmacol. 2011;67:S25-S43
45. Debiec M, Sciot R, Lecesne A. et al. Kit mutations and dose selection for imatinib in patient with advanced gastrointestinal stromal tumors. Eur J Cancer. 2006;42(8):1093-1103
46. Ashida A, Takata M, Murata H. et al. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. Int J Cancer. 2009;124(4):862-868
47. Bauer S, Yu LK, Demetri GD. et al. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153-9161
48. Hochhaus A, Kantarjian HM, Baccarani M. et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109(6):2303-2309
49. Ikeda AK, Judelson DR, Federman N. et al. ABT-869 Inhibits Proliferation of Ewing Sarcoma cells and Suppresses PDGFRβ and c-KIT Signaling Pathways. Mol Cancer Ther. 2010;9(3):653-660
50. Wei D, Parkinson GN, Reszka AP. et al. Crystal structure of a c-kit promoter quadruplex reveals the structural role of metal ions and water molecules in maintaining loop conformation. Nucleic Acids Res. 2012;40(10):4691-4700
51. Bejugam M, Gunaratnam M, Müller S. et al. Targeting the c-Kit Promoter G-quadruplexes with 6-Substituted Indenoisoquinolines. ACS Med Chem Lett. 2010;1(7):306-310
52. McLuckie KIE, Waller ZAE, Sanders DA. et al. G-Quadruplex-Binding Benzo[α]phenoxazines Down-Regulate c-KIT Expression in Human Gastric Carcinoma Cells. J Am Chem Soc. 2011;133(8):2658-2663
53. Amato J, Pagano B, Borbone N. et al. Targeting G-Quadruplex Structure in the Human c-Kit Promoter with Short PNA Sequences. Bioconjugate Chem. 2011;22(4):654-663
Author biography
 Jing Liang is a postgraduate majoring in pharmacognosy. She obtained the Bachelor's Degree in Biotechnology in 2010 from Guilin Medical University, China. Then, she entered Prof. Zhang's group of College of Pharmaceutical Sciences, Zhejiang University of Technology, China in 2010. She is working with small molecules regulating disease-related gene to explore gene-targeted drugs under the direction of Profs W. Zhang and Y.-L. Wu. To date, she has co-authored several papers.
Jing Liang is a postgraduate majoring in pharmacognosy. She obtained the Bachelor's Degree in Biotechnology in 2010 from Guilin Medical University, China. Then, she entered Prof. Zhang's group of College of Pharmaceutical Sciences, Zhejiang University of Technology, China in 2010. She is working with small molecules regulating disease-related gene to explore gene-targeted drugs under the direction of Profs W. Zhang and Y.-L. Wu. To date, she has co-authored several papers.
 Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
 Bing-Jia Chen is a postgraduate majoring in pharmacy. She obtained the Bachelor's Degree in Bio-pharmaceutical Engineering in 2012 from Zhejiang University of Technology, China. After that, she entered Prof. Zhang's group of College of Pharmaceutical Sciences, Zhejiang University of Technology, China, working with small molecules to regulate disease-related gene to explore gene-targeted drugs under the direction of Prof W. Zhang.
Bing-Jia Chen is a postgraduate majoring in pharmacy. She obtained the Bachelor's Degree in Bio-pharmaceutical Engineering in 2012 from Zhejiang University of Technology, China. After that, she entered Prof. Zhang's group of College of Pharmaceutical Sciences, Zhejiang University of Technology, China, working with small molecules to regulate disease-related gene to explore gene-targeted drugs under the direction of Prof W. Zhang.
 Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang obtained a Doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered to Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he joined Professor Sugiyama's Chemical Biology group of Kyoto University, Japan, as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers in excellent Journals including JACS, ChemBioChem etc.
Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang obtained a Doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered to Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he joined Professor Sugiyama's Chemical Biology group of Kyoto University, Japan, as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers in excellent Journals including JACS, ChemBioChem etc.
 Yoshimasa Tanaka received his Ph. D in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
Yoshimasa Tanaka received his Ph. D in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
 Hiroshi Sugiyama received his PhD in 1984, studying under Teruo Matuura at Kyoto University. After postdoctoral studies at the University of Virginia with Sidney M. Hecht, he returned to Kyoto University in 1986 as Assistant Professor and became Associate Professor in 1993. In 1996, he joined the Institute of Biomaterials and Bioengineering at Tokyo Medical and Dental University as Professor. He has been Professor of chemical biology at Kyoto University since 2003. Among the honors he has received are the Nippon IBM Award and the Chemical Society of Japan Award for Creative Work. His current research focuses on exploring gene regulation and cell biology mediated by small organic molecules and DNA nano functional structure and single molecular analysis.
Hiroshi Sugiyama received his PhD in 1984, studying under Teruo Matuura at Kyoto University. After postdoctoral studies at the University of Virginia with Sidney M. Hecht, he returned to Kyoto University in 1986 as Assistant Professor and became Associate Professor in 1993. In 1996, he joined the Institute of Biomaterials and Bioengineering at Tokyo Medical and Dental University as Professor. He has been Professor of chemical biology at Kyoto University since 2003. Among the honors he has received are the Nippon IBM Award and the Chemical Society of Japan Award for Creative Work. His current research focuses on exploring gene regulation and cell biology mediated by small organic molecules and DNA nano functional structure and single molecular analysis.
![]() Corresponding author: Yanling Wu, Virus Inspection Department of Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn; Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn. Both contributed equally to this work.
Corresponding author: Yanling Wu, Virus Inspection Department of Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn; Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn. Both contributed equally to this work.
Citation styles
APA
Liang, J., Wu, Y.L., Chen, B.J., Zhang, W., Tanaka, Y., Sugiyama, H. (2013). The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. International Journal of Biological Sciences, 9(5), 435-443. https://doi.org/10.7150/ijbs.6087.
ACS
Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int. J. Biol. Sci. 2013, 9 (5), 435-443. DOI: 10.7150/ijbs.6087.
NLM
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int J Biol Sci 2013; 9(5):435-443. doi:10.7150/ijbs.6087. https://www.ijbs.com/v09p0435.htm
CSE
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. 2013. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int J Biol Sci. 9(5):435-443.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.