Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(5):455-462. doi:10.7150/ijbs.4630 This issue Cite
Research Paper
Overexpression of microRNA-1 Causes Atrioventricular Block in Rodents
Yong Zhang1,2*, Lihua Sun1*, Yan Zhang1, Haihai Liang1, Xuelian Li1, Ruijun Cai1, Lu Wang1, Weijie Du1, Ruixue Zhang1, Jing Li1, Zhiguo Wang2, Ning Ma3, Xidi Wang3, Zhimin Du2, Baofeng Yang1,2, Xu Gao3, ![]() , Hongli Shan1,2,
, Hongli Shan1,2, ![]()
1. Department of Pharmacology (State-Province Key Laboratories of Biomedicine- Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China;
2. Institute of Cardiovascular Research, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China;
3. Department of Biochemistry, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China.
* The first two authors made equal contribution to this work.
Received 2012-5-22; Accepted 2013-4-29; Published 2013-5-9
Abstract
The present study was designed to investigate whether microRNAs (miRNAs) are involved in atrioventricular block (AVB) in the setting of myocardial ischemia (MI). A cardiac-specific miR-1 transgenic (Tg) mouse model was successfully established for the first time in this study using microinjection. miR-1 level was measured by real-time qRT-PCR. Whole-cell patch clamp was employed to record L-type calcium current (ICa,L) and inward rectifier K+ current (IK1). Expression of connexin 43 (Cx43) protein was determined by western blot analysis. Alternations of [Ca2+]i was detected by laser scanning confocal microscopy in ventricular myocytes. The incidence of AVB was higher in miR-1 Tg mice than that in wild-type (WT) mice. The normalized peak current amplitude of ICa,L was lower in ventricular myocytes from miR-1 Tg mice as compared with WT mice. Similarly, the current density of IK1 was decreased in miR-1 Tg mice than that in WT mice. Compared with WT mice, miR-1 Tg mice exhibited a significant decrease of the systolic [Ca2+]i in ventricular myocytes but a prominent increase of the resting [Ca2+]i. Moreover, Cx43 protein was downregulated in miR-1 Tg mice compared to that in WT mice. Administration of LNA-modified antimiR-1 reversed all the above changes. miR-1 overexpression may contribute to the increased susceptibility of the heart to AVB, which provides us novel insights into the molecular mechanisms underlying ischemic cardiac arrhythmias.
Keywords: atrioventricular block (AVB), miR-1, L-type calcium current ICa,L, inward rectifier K+ current IK1, connexin 43.
Introduction
microRNAs (miRNAs) are a class of ~22 nucleotide non-coding RNAs that regulate gene expression at the posttranscriptional level. Recent studies have revealed that miRNAs play important roles in cardiovascular development and diseases [1-5]. In particular, the muscle-specific, cardiac-enriched miRNA miR-1 has been shown to control cardiac excitability by regulating expression of ion channel genes [5-7]. This property of miR-1 confers it the ability to modulate cardiac electrical activities and the associated arrhythmogenic potential. For example, our previous study demonstrated that miR-1 expression was significantly up-regulated in post-myocardial ischemia (MI) hearts from both patients and rats and this upregulation promotes ischemic arrhythmias by slowing cardiac conduction via repressing KCNJ2 and GJA1 genes encoding inward rectifier K+ channel subunit and gap junctional channel connexin 43 (Cx43), respectively [6]. A subsequent study showed that forced expression of miR-1 by adenovirus promotes arrhythmogenic disturbances in Ca2+ cycling of cardiomyocytes by enhancing the functional activity of RyR2 channels [7]. Noteworthy is that in these studies, knockdown of miR-1 is able to prevent or reverse the arrhythmogenic actions, indicating miR-1 as a target for anti-arrhythmic therapy. This notion has indeed been evidenced by our recent studies showing the antiarrhythmic efficacy of beta-adrenergic receptor blockers and tanshinone IIA with downregulation of miR-1 as a mechanism [8, 9].
However, in all these above studies upregulation of miR-1 was transient. Whether long-term overexpression of miR-1 affects cardiac excitability and arrhythmogenesis remained unknown. To resolve this issue, we generated a Tg mouse line with cardiac-specific overexpression of miR-1 and investigated the impact of miR-1 overexpression on cardiac electrical activities and the targeting mechanisms.
Materials and Methods
Generation of miR-1 Tg mice
A fragment (264 bp) containing the precursor miR-1-2 (pre-miR-1-2) sequence was amplified by polymerase chain reaction (PCR) (accession No. NT_039674). The fragment was then subcloned into the SalI and HindIII sites of the Bluescript vector (Promega, Madison, WI) carrying the cardiac-specific α-myosin heavy chain (α-MHC) promoter and human growth hormone poly(A)+ signal. The plasmid was digested at the SpeI site to release the premiR-1 sequence flanked by 5'end α-MHC promoter and 3'end poly (A). The fragment was separated on an agarose gel and purified by QIAEX II gel extraction kit (Qiagen No. C04539; Valencia, CA). The DNA sample was prepared at a concentration of 2 ng/μL ready for injection. A Tg mouse line carrying a mismatched premiR-1 sequence was also generated for negative control experiments. Sexually immature female mice (4 to 5 weeks of age) were superovulated by consecutive PMS and HCG hormone injections to obtain sufficient quantity of eggs (>250) for injection. These female mice were mated with vasectomized males immediately after the HCG injection. Eggs were harvested the next day from the ampulla of the oviduct of the mated females and treated with hyaluronidase to remove nurse cells. Fertilized eggs were then stored in M16 media (37°C, 5% CO2) until injection. Each egg was individually microinjected with the DNA fragment, and the eggs that did not survive injection were removed. Pseudopregnant female mice were prepared by mating with the vasectomized males. On the day of microinjection, the pseudopregnant females were anesthetized with 0.5% pentobarbital (80 mg/kg intraperitoneal injection). Anesthesia was monitored by reaction to foot pinching at 15 min intervals and respiration monitoring and maintained by supplementary infusions of pentobarbital. The injected eggs were then implanted in a group of 10 to 15 bilaterally into the oviduct of these animals. The animals were allowed to recover from anesthesia on a warming plate and then brought back to the animal room. They were kept under sterile conditions throughout their pregnancy. The genomic DNA was prepared from tail tissue of the Tg mice and subjected to PCR verification for the presence of miR-1 transgene. The forward primer was designed to recognize α-MHC (position 5250 to 5268): 5'-CCTTACCCCACATAGACCT-3'and the reverse primer was for miR-1 (position 58 to 39): 5'-CTGTAGATACTTTCTCCCT-3'. The PCR profiling was composed of an initial denaturing step at 94 °C for 2 min and 35 cycles of 94 °C for 20 sec, 60 °C for 7 sec, and 72 °C for 20 sec, followed by a final extension step at 72 °C for 5 min. All animal procedures were approved by the Ethical Committee for Animal Experiments, Harbin Medical University, and confirmed with the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, Ad Hoc Animal Research Committee.
Electrocardiograms and assessment of arrhythmias
Mice were anesthetized with pentobarbital (80 mg/kg, intraperitoneal injection). Cardiac arrhythmia in miR-1 Tg mice was monitored using a standard lead II ECG for a continuous period of 2 h under anesthesia. Incidence of arrhythmias was evaluated.
Isolation of mouse ventricular myocytes
The heart was quickly excised from mice under deep anesthesia with pentobarbital (120 mg/kg, intraperitoneal injection) and was retrogradely perfused by using a Langendorff perfusion system for 5 min at 37 °C, at a perfusion rate of 7 mL/min with Ca2+-containing Tyrode's solution [6, 9]. The perfusate was then switched to a Ca2+-free Tyrode's solution for 5 min, followed by perfusion with the same solution to which collagenase (type II, 100-150 kU/L, Worthington, Lakewood, NJ) and 1% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) had been added. The left ventricular tissue was then excised from the softened hearts, minced, and placed in a KB medium at 4 °C for about 1 h before electrophysiological experiments. This procedure typically yielded 50% rod-shaped Ca2+-tolerant cardiomyocytes. Only single rod-shaped cells with clear cross-striation and without spontaneous contraction were used for experiments.
Whole-cell patch-clamp recording
The whole-cell patch-clamp techniques were used to record ionic currents in the voltage-clamp mode using an Axopatch 200B amplifier controlled by a personal computer using a Digidata1200 acquisition board driven by pCLAMP software (Version 9.02, Axon Instruments) [6, 9]. Borosilicate glass electrodes had tip resistances of 2~4 MΩ when filled with the internal pipette solution. Data were high-pass filtered 10 kHz. Calcium and potassium currents were recorded at room temperature (22~23 °C). The measured currents were normalized by whole-cell capacitance. Series resistance compensation was typically 50~80%. Data were analyzed with Graphpad Prism 5.0 software.
The KB solution contained (in mM) glutamic acid 70, taurine 15, KCl 30, KH2PO4 10, MgCl2 0.5, EGTA 0.5, HEPES 10, and glucose 10 at pH 7.4 adjusted by KOH. The standard Tyrode's solution contained (in mM) NaCl 126, KCl 5.4, MgCl2 1, CaCl2 1.8, NaH2PO4 0.33, glucose 10, and HEPES 10 at pH 7.4 adjusted by NaOH. This solution was used as an extracellular solution for potassium currents studies. The pipette solution for potassium currents recordings contained (in mM) KCl 20, K-aspartate 110, MgCl2 1.0, HEPES 5, EGTA 10, Na2ATP 5 at pH 7.2 adjusted by KOH. For K+ currents recordings, INa was inactivated by using a holding potential (HP) of -40 mV. Calcium currents were blocked by the use of CdCl2 (0.2 mM, Sigma) in the extracellular solution. The extracellular solution for L-type Ca2+ current (ICa,L) recording contained (in mM) Tris-Cl 136, CsCl 5.4, CaCl2 2, MgCl2.6H2O 1, HEPES 10, and glucose 5, pH 7.4 with Tris-OH. The internal solution for ICa,L recording contained (in mM) CsCl 20, MgCl2.6H2O 1, MgATP 5, EGTA 10, and CsCl.H2O 110, aspartate 110, pH adjusted to 7.2 with CsOH.
Determination of miR-1 levels in Tg mice
Total RNA samples were extracted using Trizol from mice left ventricular tissues. miR-1 level was quantified by the TaqMan® MicroRNA Reverse Transcription Kit (Cat.#4366596, Applied Biosystems, Carlsbad, CA) and the TaqMan® MicroRNA Assay (target sequences: UGGAAUGUAAAGAAGUAUGUAU, Cat.#002222, Applied Biosystems, Carlsbad, CA) [6, 8, 9]. We used U6 (Cat.#001973, Applied Biosystems, Carlsbad, CA) as an internal control. qRT-PCR was performed on 7500 FAST Real-Time PCR System (Applied Biosystems, Carlsbad, CA) for 40 cycles.
Western blot
The membrane protein samples were extracted from mouse ventricles for immunoblotting analysis of Cx43. Mice left ventricles were quickly rinsed in a standard Tyrode's solution, snap frozen in liquid nitrogen and stored at -80 °C for western blot analysis. The protein content was determined with BCA Protein Assay Kit (Bio-Rad, Mississauga, ON, Canada). Equal amounts of protein (60 μg) were loaded on a 10% SDS-PAGE gel. The lysates were resolved by electrophoresis (70 V for 30 min and 100 V for 1.5 h) and transferred onto nitrocellulose membranes. After blocking in 5% nonfat milk for 2 h at room temperature the membranes were treated with anti-Cx43 or anti-Kir2.1 antibody (1:200). Next day, the membrane was washed in PBS for three times (15 min/each) and incubated for 1.5 h with the fluorescence-conjugated anti-rabbit IgG (1:4000) in the blocking buffer. GAPDH was used as an internal control for equal input of protein samples. All primary and secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Western blot bands were quantified using Odyssey v1.2 software by measuring the band intensity (Area × OD) for each group and normalized by GAPDH. The final results are expressed as fold changes by normalizing the data to the control values.
Measurement of intracellular Ca2+ concentration ([Ca2+]i)
Intracellular Ca2+ concentration ([Ca2+]i) was assessed using fluo-3/AM (5 µM/L, Life Technologies, Grand Island, NY) [10]. Pluronic F-127 (Invitrogen, Eugene, OR), a mild detergent, is used as a dispersing agent to facilitate the loading and the final concentration in the loading medium is 0.04%. Fluorescent changes of fluo-3/AM-loaded cells were detected using laser scanning confocal microscope (Olympus FV-300) with 488 nm for excitation from an argon ion laser and 530 nm for emission. Resting [Ca2+]i was investigated in fluo-3/AM-loaded cells in standard Tyrode's solution at 23-24 °C. To measure peak [Ca2+]i, 60 mmol/L KCl was added between 1st and 2nd scan, and changes of [Ca2+]i in response to KCl stimulation was recorded. The fluorescent intensities before (FI0) and after (FI) KCl administration were measured. Qualitative changes in [Ca2+]i were inferred from the ratio of FI/FI0.
In vivo transfer of locked nucleic acid (LNA)-modified antimiR-1 oligomers
The LNA-modified oligonucleotides were synthesized as unconjugated LNA/DNA mixers with a complete phosphorothioate backbone. Mice were caudally administered 5 mg/kg LNA-antimiR-1 intravenous injection daily on day 1, 3 and 5. A vehicle control was administered with the same volume of phosphate buffered saline (PBS). The experiments were performed three days after LNA-antimiR-1 injection.
Statistics
Data are expressed as means ± SEM. Statistical analysis was performed using the Student's t-test. Differences were considered to be significant when p<0.05.
Results
miR-1 causes atrioventricular block in mice
We established a miR-1 Tg mouse line that produces stable overexpression of miR-1 specifically in the myocardium as indicated by the significantly increased miR-1 level in Tg relative to WT mice (Fig. 1A & 1B; p<0.05). We also confirmed using western blot analysis that Cx43 and Kir2.1 protein levels were significantly downregulated in myocardium from Tg mice compared with that in WT mice (Fig. 1C; p<0.05, n=6), consistent with our previous finding that miR-1 represses Cx43 and Kir2.1 [6, 8, 9].
miR-1 overexpression contributes to the increased susceptibility of the heart to AVB. (A) Verification of the successful transgene. Genomic DNA was prepared from tail tissues of the Tg mice and subjected to PCR verification for the presence of miR-1 transgene. (B) Verification of overexpression of endogenous miR-1 in Tg mice. *p<0.01 vs. WT; n=6 mice for each bar. (C) Expression of connexin 43 (Cx43) protein level was downregulated in miR-1 Tg mice. Top, examples of western blot bands; bottom, quantitation as mean ± s.e.m. (n=6). *P <0.05; unpaired Student's t-test. (D) The representative raw traces of ECG from a miR-1 Tg mouse compared with the WT mouse. (E) AVB incidence expressed as percentage of animals in Tg mice. **p<0.01 vs. WT; n=14 mice for each bar. (F) Representative raw traces of ECG from a miR-1 Tg before and after LNA-miR-1 was injected. (G) qPCR analysis showing the miR-1 level after LNA-modified anti-miR-1 injection.
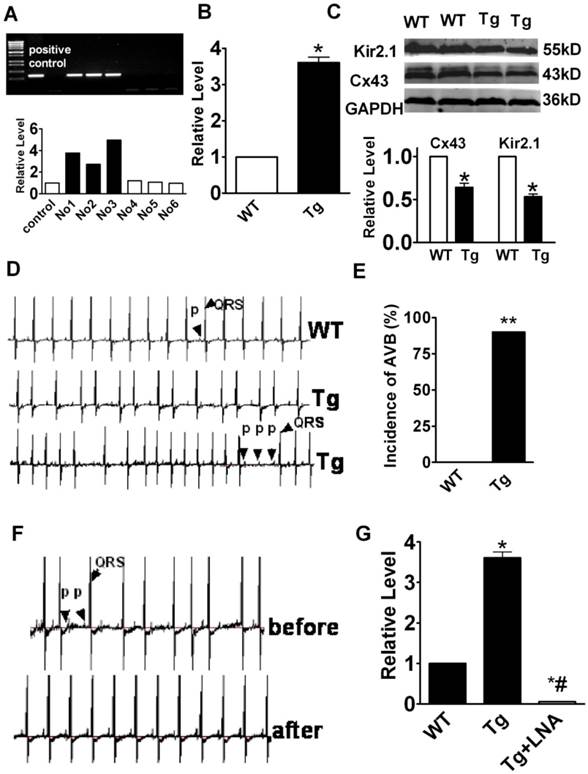
Most notably, the miR-1 Tg mice developed frequent AVB of varying degrees with prolongation of PR interval (first degree AVB), dropped ventricular beats (second degree of AVB), and even dissociation between P waves and QRS complexes (third degree AVB). Representative ECG recordings showing typical characteristics of AVB from miR-1 Tg are presented in Fig. 1D. While the incidence of AVB was as high as 90%, it was absent in WT mice (Fig. 1D & 1E).
Knockdown of miR-1 attenuates atrioventricular block in mice
To see if the AVB was indeed caused by miR-1 overexpression, we used locked nucleic acid-modified anti-miR-1 antisense (LNA-anti-miR-1) to normalize miR-1 level by knocking down its expression [11] and investigated the effects on AVB in Tg mice. LNA-anti-miR-1 was injected via caudal vein to miR-1 Tg mice. ECG recordings showed that the LNA-anti-miR-1 significantly decreased the propensity of AVB in miR-1 Tg mice (Fig. 1F). The efficacy of miR-1 knockdown by the LNA-anti-miR-1 was verified by the drastic decrease in miR-1 level in the myocardium of miR-1 Tg mice (Fig. 1G).
ICa,L and IK1 current densities were decreased in miR-1 Tg mice
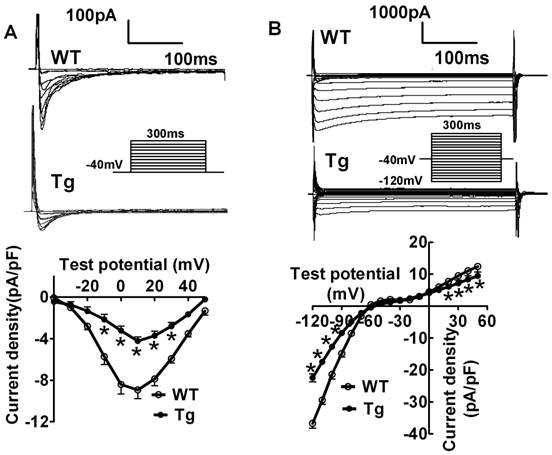
To shed light on the possible mechanisms underlying miR-1-induced AVB, we evaluated the changes of L-type Ca2+ current (ICa,L) that is known to be the major governor of AV conduction [12-16]. We found that the current density of ICa,L was remarkably decreased in miR-1 Tg mice. The decrease was voltage-dependent at potentials ranging from -30 mV to +30 mV (Fig. 2A; p<0.05, n=12 cells from 8 mice). Additionally, the current density of inward rectifier K+ current (IK1), the main determinant of cardiac membrane potential and thereby conduction, was also found smaller in miR-1 Tg mice compared with that in WT (Fig. 2B; p<0.05, n=11 cells from 8 mice).
Verification of ventricular electrical remodeling in miR-1 Tg mice. (A) Whole-cell patch-clamp results for L-type Ca2+ current ICa in isolated ventricular cardiomyocytes from mice. Typical ICa recordings (top) and current density (pA/pF)-voltage (mV) relationships of ICa (bottom) elicited by 300-ms pulses stepped from -40 to +60 mV in 10 mV increments from the holding potential -40 mV in cells, *p<0.05 vs Ctl; n=12 cells for each group. (B) Whole-cell patch-clamp results for inward rectifier K+ current IK1 in isolated ventricular cardiomyocytes from mice (top), average data for current density (pA/pF)-voltage (mV) relationships of IK1 (bottom) elicited by 100-ms pulses stepped from -120 to 0 mV in 10 mV increments in cells. *p<0.05 vs Ctl; n=11 for each group.
miR-1 induces aberrant intracellular Ca2+ handling
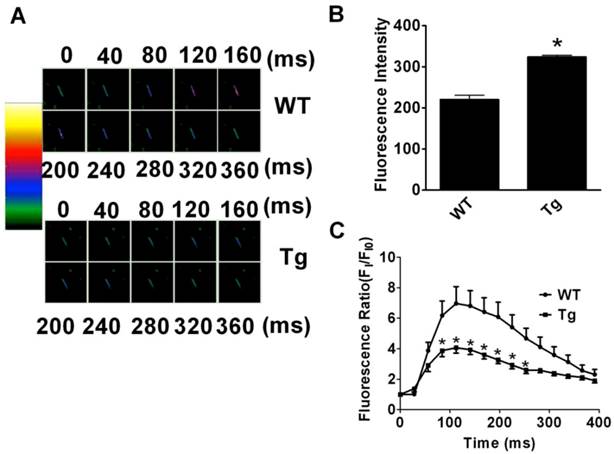
Changes of resting [Ca2+]i and peak [Ca2+]i upon stimulation with KCl (60 mM/L) to evoke Ca2+ entry were investigated in fluo-3/AM loaded cardiac myocytes (Fig. 3A). The fluorescence measurements revealed that the resting [Ca2+]i was remarkably higher in cardiomyocytes from miR-1 Tg mice than that in WT mice (Fig. 3B; p<0.01). However, the peak [Ca2+]i induced by KCl exposure was smaller in cardiac myocytes from miR-1 Tg mice (Fig. 3C; n=11 from 8 mice) compared with that in myocytes from WT (p<0.05; n=30 from 6 mice). These results indicated an abnormal handling of intracellular Ca2+ in cardiomyocytes from miR-1 Tg mice.
Changes of Intracellular Calcium ([Ca2+]i) in Cardiac Myocytes in miR-1 Tg mice. (A) The images of change of intracellular of Ca2+. Different color indicates the different fluorescence intensity. The colorful zoom of yellow indicates the maximum change of Ca2+, red and blue indicates the decreased Ca2+ in the state of recovery, and green indicates the minimum of change of Ca2+ compared with the other three colors. (B) The resting level of intracellular fluorescent intensity (FI) value (indicator of resting [Ca2+]i ) was significantly elevated in isolated ventricular myocytes from miR-1 Tg mice compared with WT control. *p<0.05 vs Ctl; n=13 cells from 4 mice for control and n=20 cells from 4 mice for Tg group. (C) Changes of [Ca2 +]i elevation induced by 60 mmol·L -1 KCl in isolated mouse ventricular myocytes. Qualitative changes in [Ca2+]i were inferred from the ratio of FI/FI0. miR-1 overexpression reduced the [Ca2 +]i responded to KCl.
LNA-anti-miR-1 restored ion channel abnormalities in Tg mice
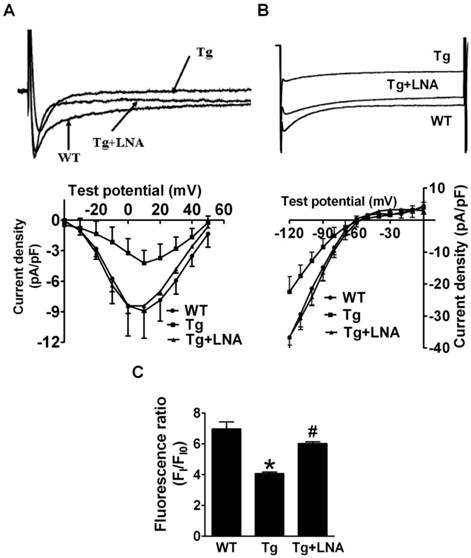
The LNA-anti-miR-1 was found to reverse the decreased ICa,L and IK1 in miR-1 Tg mice (Fig. 4A & 4B; p<0.05). Additionally, the peak [Ca2+]i induced by KCl was also decreased in cardiomyocytes from miR-1 Tg mice compared to that from WT littermates, which was reversed by LNA-anti-miR-1 (Fig. 4C).
Effects of miR-1 knockdown on AVB and the associated electrical remodeling. (A) Reduced ICa,L density in cardiomyocytes from miR-1 Tg mice was recovered by injection of LNA-modified anti-miR-1. Upper panel: ICa,L trace recorded at the test potential of 10 mV. Lower panel: I-V relationship of ICa,L. (B) Reduced IK1 density in cardiac myocytes of miR-1 Tg mice was also recovered by injection of LNA-modified anti-miR-1. Upper panel: IK1 trace recorded at the test potential of -120 mV. Lower panel: I-V relationship of IK1. (C) Effects of LNA--modified anti-miR-1on the changes of [Ca2+]i of cardiac myocytes in miR-1 Tg mice. *p<0.05 vs Ctl; n=10 cells from 4 mice for control and Tg group. #p<0.05 vs Tg group; n=10 cells from 4 mice.
Discussion
Here we present the evidence for the role of long-term overexpression of miR-1 in modulating cardiac electrical activities. The major findings are that long-term miR-1 overexpression causes AVB of varying degrees in otherwise normal miR-1 Tg mice. At the subcellular level, long-term miR-1 overexpression decreases IK1 and ICa,L densities, and impairs the intracellular calcium handling process as indicated by increased resting [Ca2+]i and decreased peak [Ca2+]i, which might underlie the AVB-promoting and AVB-inducing effects of miR-1. Additionally, our study clearly showed that Cx43 and Kir2.1 expression were downregulated in miR-1 Tg mice. All these effects were reversed upon knockdown of miR-1 by LNA-anti-miR-1. These findings help us understand the cellular functions of miRNA in particular miR-1 and the molecular mechanisms underlying the generation of AVB.
Atrioventricular block (AVB) is a common complication of MI, which is relatively frequent and contributes to increased mortality of patients with cardiovascular diseases (CVD) [17-19], and prognostic significance of complete AVB in patients with AMI has been well recognized [20]. Upregulation of miR-1 in myocardial infarction has been reported by several groups. In patients with MI, miR-1 level was found upregulated by 2.8-fold [6] in one study and 3.8-fold in another with the samples of infarcted tissue and remote myocardium from 24 patients with MI [21]. A greater degree (~22 folds) of expression upregulation of miR-1 was reported in a rat model of MI [22]. It is conceivable that miR-1 overexpression accounts at least partially for the AVB occurring in MI. Unfortunately, because of the technical difficulties and limitations with our mouse model, especially the small size of AV node and the limited quantity of LNA-antimiR for larger animal species, we were unable to investigate this notion in the present study.
AVB can be induced by any disturbances of electrical conduction within the AV node, the bundle of His, and the bundle branches. It is known that L-type Ca2+ channels play an essential role in the maintenance of the AV conduction pathways: AV conduction through AV nodal cells is mainly mediated by ICa,L, but not by INa as in working myocardial fibers; this is because Na+ channels are largely inactivated due to depolarized membrane potential in AV nodal cells that intrinsically have small IK1 [13]. Previous study has reported that inhibition of Ca2+ channels may be a critical factor contributing to the pathogenesis of congenital heart block [14]. Impairment of Ca2+ channel function by any factors including repression by miR-1 is deemed to slowing AV conduction. Moreover, the ability of miR-1 to diminish IK1, the primary determinant of resting membrane potential, can further depolarize the membrane of AV nodal cells and the cells in the bundle of His and the bundle branches, and as such manifest the role of ICa,L in governing AV conduction. Here we found a dramatic decrease in ICa,L, which may explain the observed AVB, in miR-1 Tg mice. Though our data were obtained from ventricular myocytes because of the difficulties of isolating single AV nodal cells from mice and even rats, the molecular compositions of L-type Ca2+ channels in AV nodal cells have been found to be essentially the same as those in ventricular myocytes [23]. It is not unreasonable to speculate that the same mechanism operates in AV nodal cells. Based on our computational analysis, however, none of the cardiac-specific Ca2+ channel subunits are predicted to be the targets for miR-1. It is possible that miR-1 acts through indirect pathways to affect Ca2+ channel functions. For example, miR-1 might impair intracellular Ca2+ handling process to impose a negative influence to ICa,L [24, 25]. Indeed, our study demonstrated that overexpression of miR-1 increases resting [Ca2+]i and decreases peak [Ca2+]i, these changes may well cause reduction of ICa,L through Ca2+ induced-Ca2+ channel inactivation. Our data showing decrease in ICa,L with miR-1 overexpression is in contrast to the results reported by other group [7] who demonstrated an increase in ICa,L in cultured cardiomyocytes infected with pre-miR-1-carrying adenovirus. Though it is not exactly clear what accounts for the discrepancy, one possible explanation is that our study deals with long-term overexpression of miR-1 in the heart whereas the study by other group focused on transient expression of miR-1 in cultured myocytes. Long-term overexpression of miR-1 could well cause electrical and molecular remodeling of cardiac muscles leading to decrease in ICa,L.
In heart, the physiological role of IK1 is to accelerate phase III repolarization and to maintain the resting membrane potential (RMP). Lowered Kir2.1 expression prolongs the refractoriness of the atrial and ventricular muscle as well as the His-Purkinje bundle. The decremental conduction property was demonstrated in the His-Purkinje system after Kir2.1 expression decreased in the Tg mice. The T wave amplitude was much reduced, and the repolarization process was slowed. Gap junctions provide cell-to-cell electrical continuity for rapid transmission of cardiac action potential among all cardiac cell types [26]. A variable severity of gap junctional disruption alters electrical conduction, increasing risk of arrhythmia [27]. Recently, several connexin isoforms have been identified in heart with Cx43 being the primary isoform in ventricular cells. Dysfunction of Cx43 contributes to the perpetuation of re-entry and the degeneration of these arrhythmias [28]. Though the existence of Cx43 in AV nodal cells has been a subject of debating, several groups have indeed reported the distribution and function of Cx43 in these cells [29-31]. Under such a context, downregulation of Cx43 by miR-1 can well cause slowing or even blockade of AV conduction. Alternatively, Cx43 has been shown to exist in the His bundle and the bundle branches, the excitation conduction system critical for AV conduction and suppression of Cx43 in these cells can lead to AVB, too [32, 33].
In summary, our data demonstrate for the first time that miR-1 induces AVB via the regulation of several important ion channels. miR-1 appears to inhibit Cx43 and Kir2.1 expression, subsequently decreases IK1 and ICa,L densities, and impair the intracellular calcium handling process. In conclusion, modulation of miR-1 is emerging as a novel approach for the prevention or therapy of AVB related heart diseases.
Abbreviations
miRNAs: microRNAs; AVB: atrioventricular block; Tg: transgenic; ICa,L: L-type calcium current; IK1: inward rectifier K+ current; WT: wild-type; α-MHC: α-myosin heavy chain; PCR: polymerase chain reaction; HP: holding potential; Cx43: connexin43; FI: fluorescent intensities; LNA: locked nucleic acid; PBS: phosphate buffered saline; CVD: cardiovascular diseases.
Acknowledgements
This work was supported by the Funds for Creative Research Groups [grant number 81121003] and the Major Program [grant number 81130088] of National Natural Science Foundation of China, and the National Nature Science Foundation of China [grant numbers 31171094, 81100134].
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A. 2005;102:18986-91
2. Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214-20
3. Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072-83
4. Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419-29
5. Yang B, Lu Y, Wang Z. Control of cardiac excitability by microRNAs. Cardiovasc Res. 2008;79:571-80
6. Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B. et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486-91
7. Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE. et al. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104:514-21
8. Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B. et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischaemic cardioprotection. Cardiovasc Res. 2009;84:434-41
9. Shan H, Li X, Pan Z, Zhang L, Cai B, Zhang Y. et al. Tanshinone IIA protects against sudden cardiac death induced by lethal arrhythmias via repression of microRNA-1. Br J Pharmacol. 2009;158:1227-35
10. Sun L, Ai J, Wang N, Zhang R, Li J, Zhang T. et al. Cerebral ischemia elicits aberration in myocardium contractile function and intracellular calcium handling. Cell Physiol Biochem. 2010;26:421-30
11. Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S. et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896-9
12. Katz AM. Calcium channel diversity in the cardiovascular system. J Am Coll Cardiol. 1996;28:522-9
13. Gilmour RF Jr, Zipes DP. Slow inward current and cardiac arrhythmias. Am J Cardiol. 1985;55:89B-101B
14. Boutjdir M, Chen L, Zhang ZH, Tseng CE, El-Sherif N, Buyon JP. Serum and immunoglobulin G from the mother of a child with congenital heart block induce conduction abnormalities and inhibit L-type calcium channels in a rat heart model. Pediatr Res. 1998;44:11-9
15. Takahara A, Wagatsuma H, Aritomi S, Konda T, Akie Y, Nakamura Y. et al. Measurements of cardiac ion channel subunits in the chronic atrioventricular block dog. J Pharmacol Sci. 2011;116:132-5
16. Zhang Q, Timofeyev V, Qiu H, Lu L, Li N, Singapuri A. et al. Expression and roles of Cav1.3 (alpha1D) L-type Ca2+ channel in atrioventricular node automaticity. J Mol Cell Cardiol. 2011;50:194-202
17. Bhalli MA, Khan MQ, Samore NA, Mehreen S. Frequency and clinical outcome in conduction defects in acute myocardial infarction. J Ayub Med Coll Abbottabad. 2009;21:32-7
18. Shirafkan A, Mehrad M, Gholamrezanezhad A. Conduction disturbances in acute myocardial infarction: a clinical study and brief review of the literature. Hellenic J Cardiol. 2009;50:179-84
19. Meine TJ, Al-Khatib SM, Alexander JH, Granger CB, White HD, Kilaru R. et al. Incidence, predictors, and outcomes of high-degree atrioventricular block complicating acute myocardial infarction treated with thrombolytic therapy. Am Heart J. 2005;149:670-4
20. Behar S, Zissman E, Zion M, Goldbourt U, Reicher-Reiss H, Shalev Y. et al. Complete atrioventricular block complicating inferior acute wall myocardial infarction: short- and long-term prognosis. Am Heart J. 1993;125:1622-7
21. Bostjancic E, Zidar N, Stajner D, Glavac D. MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol (Praha). 2010;56:27-31
22. Yin C, Wang X, Kukreja RC. Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett. 2008;582:4137-42
23. Hancox JC, Mitcheson JS. Ion channel and exchange currents in single myocytes isolated from the rabbit atrioventricular node. Can J Cardiol. 1997;13:1175-82
24. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198-205
25. Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727-41
26. Peters NS. New insights into myocardial arrhythmogenesis: distribution of gap-junctional coupling in normal, ischaemic and hypertrophied human hearts. Clin Sci (Lond). 1996;90:447-52
27. Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547-52
28. McGuire MA, de Bakker JM, Vermeulen JT, Moorman AF, Loh P, Thibault B. et al. Atrioventricular junctional tissue. Discrepancy between histological and electrophysiological characteristics. Circulation. 1996;94:571-7
29. Gourdie RG, Severs NJ, Green CR, Rothery S, Germroth P, Thompson RP. The spatial distribution and relative abundance of gap-junctional connexin40 and connexin43 correlate to functional properties of components of the cardiac atrioventricular conduction system. J Cell Sci. 1993;105( Pt 4):985-91
30. Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr Biol. 1998;8:295-8
31. Kirchhoff S, Kim JS, Hagendorff A, Thonnissen E, Kruger O, Lamers WH. et al. Abnormal cardiac conduction and morphogenesis in connexin40 and connexin43 double-deficient mice. Circ Res. 2000;87:399-405
32. Hucker WJ, McCain ML, Laughner JI, Iaizzo PA, Efimov IR. Connexin 43 expression delineates two discrete pathways in the human atrioventricular junction. Anat Rec (Hoboken). 2008;291:204-15
33. Davis LM, Rodefeld ME, Green K, Beyer EC, Saffitz JE. Gap junction protein phenotypes of the human heart and conduction system. J Cardiovasc Electrophysiol. 1995;6:813-22
Author contact
![]() Corresponding author: Dr. Hongli Shan, Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), Harbin Medical University, Heilongjiang 150081, P. R. China. E-mail: shanhonglicom.cn. Phone: 86-451-86671354; Fax: 86-451-86671354. Or Dr. Xu Gao, Department of Biochemistry, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China. E-mail: gaoxhrbmu.edu.cn. Phone: 86-451-86671354; Fax: 86-451-86667511.
Corresponding author: Dr. Hongli Shan, Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), Harbin Medical University, Heilongjiang 150081, P. R. China. E-mail: shanhonglicom.cn. Phone: 86-451-86671354; Fax: 86-451-86671354. Or Dr. Xu Gao, Department of Biochemistry, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China. E-mail: gaoxhrbmu.edu.cn. Phone: 86-451-86671354; Fax: 86-451-86667511.