Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(6):541-549. doi:10.7150/ijbs.5763 This issue Cite
Research Paper
Overexpression of DOC-1R Inhibits Cell Cycle G1/S Transition by Repressing CDK2 Expression and Activation
Qi Liu, Xing Liu, Jinlan Gao, Xiuyan Shi, Xihua Hu, Shusen Wang, Yang Luo ![]()
The Research Center for Medical Genomics, MOH Key Laboratory of Cell Biology and Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang 110001, China.
Received 2012-12-24; Accepted 2013-5-31; Published 2013-6-9
Abstract
DOC-1R (deleted in oral cancer-1 related) is a novel putative tumor suppressor. This study investigated DOC-1R antitumor activity and the underlying molecular mechanisms. Cell phenotypes were assessed using flow cytometry, BrdU incorporation and CDK2 kinase assays in DOC-1R overexpressing HeLa cells. In addition, RT-PCR and Western blot assays were used to detect underlying molecular changes in these cells. The interaction between DOC-1R and CDK2 proteins was assayed by GST pull-down and immunoprecipitation-Western blot assays. The data showed that DOC-1R overexpression inhibited G1/S phase transition, DNA replication and suppressed CDK2 activity. Molecularly, DOC-1R inhibited CDK2 expression at the mRNA and protein levels, and there were decreased levels of G1-phase cyclins (cyclin D1 and E) and elevated levels of p21, p27, and p53 proteins. Meanwhile, DOC-1R associated with CDK2 and inhibited CDK2 activation by obstructing its association with cyclin E and A. In conclusion, the antitumor effects of DOC-1R may be mediated by negatively regulating G1 phase progression and G1/S transition through inhibiting CDK2 expression and activation.
Keywords: DOC-1R, CDK2, G1/S transition, cyclin E, cyclin A, CKI.
Introduction
DOC-1R (deleted in oral cancer-1 related), a homolog of DOC-1 (deleted in oral cancer-1), was identified and cloned in 1999 from chromosome 11q13 where the tumor suppressor gene MEN1 and several oncogenes, including CCND1/cyclin D1/PRAD1, are located [1]. Human DOC-1R encodes a protein with 126 amino acids and a molecular mass of 14 kDa. Comparative studies have shown that DOC-1R is highly conserved from Xenopus Laevis to human and that DOC-1R mRNA is ubiquitously expressed in all examined mouse and human tissues. Thus, DOC-1R is suggested to play an important role in vertebrate species [1, 2]. However, to date, our understanding of the function of DOC-1R is limited. DOC-1R is a MAP kinase substrate that controls the microtubule organization of metaphase II mouse oocytes. A previous study reinforced the importance of this gene pathway in controlling spindle stability during metaphase II arrest [3]. Moreover, in our previous study, we demonstrated that ectopic expression of DOC-1R significantly inhibited the growth and colony formation of NIH3T3 cells [4]. However, the antiproliferative activity and the underlying mechanisms of DOC-1R gene remain largely unknown. Thus, in this study, we first investigated the effects of DOC-1R expression in HeLa cells and then explored the altered expression of cell cycle-related proteins. We found that DOC-1R inhibited G1/S transition and repressed cyclin-dependent kinase 2 (CDK2) expression. Moreover, DOC-1R associated with CDK2 and interfered with CDK2 activation via its association with cyclin E and A.
Indeed, CDK2 is one of the essential regulators in cell cycle progression, and the activity of CDK2 is regulated by two cyclins, cyclin E and A [5-7]. Specifically, cyclin E binds to CDK2 to promote the transition of cells from G1 to S phase [8, 9], whereas cyclin A binds to CDK2 to progress the cell cycle through the S phase and G2/M transition [10, 11]. Thus, our findings of DOC-1R regulation of CDK2 expression and activation may significantly impact research in the field, especially G1/S cell cycle transition and CDK regulation, and could be important in the development of novel strategies in future treatment of different human cancers.
Materials and Methods
Cell lines and culture
A human cervical cancer cell line (HeLa) and a human embryonic kidney cell line (HEK-293) obtained from ATCC and the retroviral packaging GP2-293 cell line from Clontech (San Diego, CA, USA) were maintained in DMEM with 10% FBS. In addition, the Chinese hamster ovary CHO cell line was obtained from ATCC and maintained in RPMI 1640 with 10% FBS. All of these cells were cultured at 37°C in a humidified incubator with 5% CO2 and 95% air.
Recombinant vector construction, transient transfection and cell infection
In this study, pLXSN-FLAG-DOC-1R carrying DOC-1R cDNA was constructed. Briefly, FLAG-tagged DOC-1R cDNA was amplified by PCR and subcloned into a pLXSN retroviral vector (Clontech). For generation of pGST-DOC-1R and its truncated mutants, DOC-1R cDNA was amplified and subcloned into a pGEX-5X-1 vector (GE Healthcare, Piscataway, NJ). In addition, the CDK2 coding sequence was subcloned into a pDsRED-N1 vector (Clontech) and the DOC-1R coding sequence was subcloned into pEGFP-C3 vector (Clontech) using a standard gene subcloning protocol. All vectors were verified by sequencing.
For transient gene transfection, the cells were cultured and transfected with recombinant DNAs or shRNA plasmids using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instruction. For the generation of recombinant retrovirus, pLXSN-FLAG-DOC-1R or a control vector was transfected into packaging GP2-293 cells with pVSVG using Lipofectamine 2000 reagent according to the manufacturer's instruction. Two days after transfection, supernatant of the cultured cells was collected, filtered with 0.45 μM membrane filters and harvested to infect HeLa cells for 48 h in the presence of 8 μg/ml polybrene.
Cell cycle analysis
HeLa cells were trypsinized, collected by centrifugation, and fixed in 70% ethanol at -20°C overnight. The fixed cells were then washed twice with PBS and incubated with propidium iodide (5 mg/ml with 0.1% RNase A) solution for 30 min at 4°C. After that, cell cycle distributions were analyzed and recorded using FACSCalibur and CellQuest software (BD, San Jose, CA).
BrdU incorporation assay and indirect immunofluorescence
HeLa cells were seeded onto a glass coverslip and grown overnight for transfection. Twenty four hours after transfection, cells were labeled with 20 μg/ml BrdU for 1 h and fixed with 3.7% paraformaldehyde for 15 min. After denaturing with 2M HCl for 30 min followed by neutralization with 0.1 M sodium biborate for 2 min, cells were digested with pre-warmed 0.1% trypsin for 10 min, blocked with 10% serum for 30 min and incubated with primary antibodies (anti-BrdU antibody and anti-FLAG antibody, Sigma-Aldrich, St. Louis, MO, USA) for 1 h. After washing three times with PBS, the coverslips were incubated for 45 min with secondary antibodies (goat anti-rabbit IgG-FITC and goat anti-mouse IgG-R, Santa Cruz Biotechnology, Santa Cruz, CA, USA) [3]. Next, the slides were thoroughly washes with PBS, and images were visualized and recorded using a Leica DMIRE2 microscope.
CDK2 kinase assay
To measure CDK2 activity, cell lysates (150 μg of each sample) were immunoprecipitated with an anti-CDK2 antibody as described above. After three washes with PBS and two washes with kinase buffer (50 mM HEPES-pH7.5, 10 mM MgCl2, 2.5 mM EGTA, 1 mM DTT), pellets were resuspended in 15 μl kinase buffer followed by incubation with 10 μg histone H1 and 0.002 μM ATP at 30°C for 90 min. After the addition of Kinase-Glo reagent (Promega, Madison, WI), samples were incubated for 10 min at room temperature. At last, luminescence was recorded with a luminometer as described previously [12].
Protein extraction and Western blot
Total cellular protein was extracted using a lysis buffer containing 50 mM Tris-HCl, 300 mM NaCl, 1% NP-40 and 5 mM EDTA. The protein concentration was estimated using the Bradford method. Next, equal amounts (50 μg) of protein samples were subjected to SDS-PAGE and transferred onto PVDF membrane (Millipore, Billerica, MA, USA). For Western blot, the membranes were incubated in 5% nonfat skimmed milk in TBS and then with the primary antibodies. After washing with TBS/tween 20 solution, the membranes were further incubated with the secondary antibody, and the positive signal of protein bands was developed with a super signal chemiluminescent substrate (Thermo Fisher Scientific, Scoresby, VIC, Australia).
Reverse transcription PCR (RT-PCR) and real-time quantitative RT-PCR (qRT-PCR) analysis
Total cellular RNA was extracted from DOC-1R overexpressing or control cells using a TRIzol reagent (Invitrogen) according to the manufacturer's protocols. After quantification, 1 μg of RNA sample was reversely transcribed into cDNA using an AMV reverse transcriptase kit from TaKaRa Company (Dalian, China) according to the manufacturer's instruction. PCR was then performed with GAPDH as an internal control. For qRT-PCR, reactions were performed with SYBR Green PCR Mater Mix (TaKaRa) in triplicate. The 2-ΔΔCT method was used to calculate relative changes in CDK2 expression. Melting curve analysis confirmed that only the expected product was amplified.
GST pull-down assay
Recombinant GST, GST-DOC-1R and truncated mutated DOC-1R vectors were transferred into Escherichia coli BL-21. Expressions of these recombinant proteins were examined by SDS-PAGE. For the in vitro binding assay, GST, GST-DOC-1R and truncated proteins were gently rotated with HeLa lysates and MagneGST particles (Promega) overnight based on the manufacturer's protocol. After a thorough wash, particles were eluted in 1×SDS sample buffer for Western blot analysis of CDK2, cyclin A and E expression, respectively.
Immunoprecipitation
For immunoprecipitation analysis of CDK2 protein, cell lysates were rotated with 1 μg anti-CDK2 antibody and 20 μl protein G plus-agarose (Santa Cruz Biotechnology) as recommended by the manufacturer. At last, the pellets were washed with PBS and resuspended in 1 × SDS sample buffer. For immunoprecipitation of FLAG-DOC-1R protein, anti-FLAG-M2 affinity agarose (Sigma-Aldrich) was used as described previously [13].
Statistical analysis
Data presented were representative of at least three independent experiments for each condition. Statistical significance was determined by the Student's t test or χ2 test, respectively. A value of P<0.05 was considered to be statistically significant.
Results
Overexpression of DOC-1R inhibited G1/S transition and reduced CDK2 activity in HeLa cells
To examine whether DOC-1R participated in cell cycle control and contributed to cell growth inhibition, DOC-1R-mediated cell cycle distributions were analyzed. Flow cytometry data demonstrated the delay of G1 phase progression and decreased S phase entry following DOC-1R overexpression (P<0.01; Fig. 1A).
DOC-1R expression inhibited G1/S transition and suppressed CDK2 kinase activity. (A) Cell cycle analysis. HeLa cells were transiently transfected with pFLAG-DOC-1R and then subjected to flow cytometry assay at 48 h post-transfection. *P<0.01 and **P<0.05. (B) BrdU incorporation assay. HeLa cells with the same conditions as above were subjected to BrdU incorporation assay. *P<0.001. (C) CDK2 kinase assay. Immunoprecipitated CDK2 protein from HeLa cell lysates was resuspended and subjected to a CDK2 kinase assay. The data are expressed as mean ± S.D. RLU, relative light units, *P<0.01.
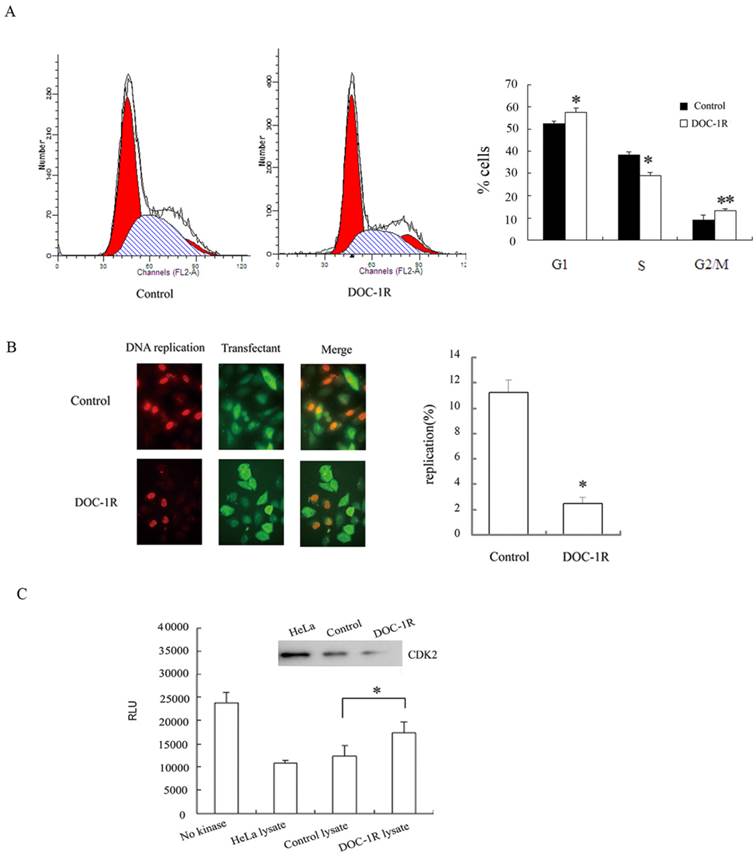
Furthermore, BrdU incorporation assays were performed to confirm the inhibition of DOC-1R on G1/S transition. HeLa cells were transfected with pFLAG-DOC-1R or control vector and plus-labeled with BrdU at 24 h post-transfection as described above. Incorporation of BrdU into DNA and ectopic expression of DOC-1R were identified by immunofluorescence assays, respectively. The percentage of BrdU-positive cells was 2% in DOC-1R overexpressing cells (11 in 449 transfected cells) and 11% in control cells (47 in 419 transfected cells) (Fig. 1B). Cells overexpressing DOC-1R showed a statistically lower incorporation of BrdU than control (P<0.001), suggesting that ectopic expression of DOC-1R could significantly inhibit DNA synthesis in cultured cells.
Since CDK2 is one of the critical kinases controlling G1/S transition and DNA replication, DOC-1R-mediated CDK2 activity was investigated to confirm CDK2 involvement. Our data showed that DOC-1R overexpression suppressed CDK2 enzymatic activity in HeLa cells compared to the control cells (P<0.01; Fig. 1C). Thus, DOC-1R inhibited G1/S transition and decreased CDK2 activity.
Overexpression of DOC-1R inhibited CDK2 expression at the transcriptional level
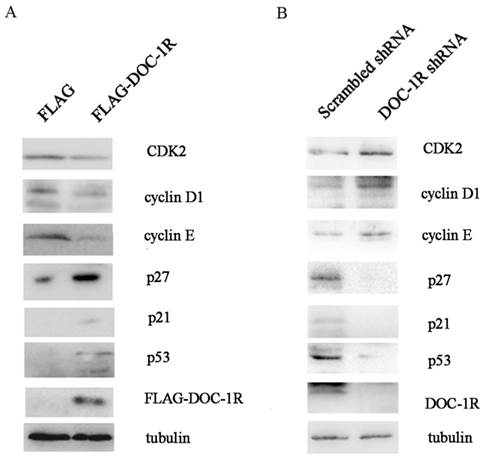
We further investigated the molecular mechanisms responsible for the effects of DOC-1R antiproliferative activity. We first overexpressed DOC-1R protein using transient transfection in HeLa cells, and then detected the expression of different cell cycle-related proteins. We found that DOC-1R overexpression decreased the level of CDK2 protein compared to the control cells, as well as decreased the levels of G1-phase cyclins, cyclin D1 and E, and elevated the levels of CDK2 inhibitors, p21 and p27. Furthermore, p53 levels were also induced by DOC-1R overexpression (Fig. 2A). Silencing DOC-1R expression with a shRNA plasmid confirmed the molecular regulations in HEK-293 cells (Fig. 2B).
Effects of DOC-1R expression on the regulation of G1 phase-related gene expressions. (A) Overexpression of DOC-1R. pFLAG-DOC-1R was transfected into HeLa cells and 48 h after transfection, cells were extracted and subjected to Western blot analysis of CDK2, cyclin D1, cyclin E, p21, p27 and p53 protein, respectively. (B) Knockdown of endogenous DOC-1R. DOC-1R shRNA plasmid was transfected into HEK-293 cells and 48 h post-transfection, cells were extracted and subjected to Western blot analysis of related proteins. A scrambled shRNA plasmid was used as a control.
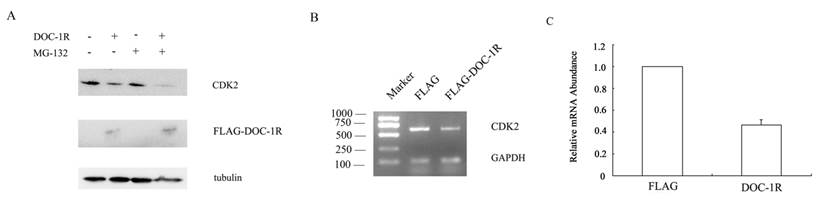
DOC-1, the homolog of DOC-1R, is reported to target CDK2 proteolysis in an ubiquitin-dependent manner. To determine whether DOC-1R negatively regulated CDK2 expression in a similar manner, we transfected pFLAG-DOC-1R or control vectors into HeLa cells and incubated it in the presence or absence of the proteosome inhibitor MG-132 for 8 h. Figure 3A shows that the reduced CDK2 expression was not averted when the transfected cells were grown in presence of the proteosome inhibitor, suggesting that the DOC-1R-mediated reduction of CDK2 protein levels was independent of proteosome. Indeed, CDK2 mRNA levels were significantly down-regulated in DOC-1R overexpressing cells compared to the control cells (Fig. 3B, 3C), suggesting that DOC-1R inhibited CDK2 expression by decreasing CDK2 mRNA levels.
Inhibition of CDK2 expression at the transcriptional level by overexpression of DOC-1R. (A) DOC-1R overexpression down-regulated CDK2 expression via an ubiquitin-independent manner. HeLa cells were transfected with DOC-1R or control plasmid in presence or absence of the proteosome inhibitor MG-132 (0.01 mM) for 8 h and then subjected to Western blot analysis of CDK2 or FLAG-DOC-1R. (B, C) DOC-1R overexpression reduced CDK2 mRNA levels. Cells cultured in the same conditions were subjected to RNA isolation and then RT-PCR (B) and qRT-PCR (C) to detect CDK2 expression.
DOC-1R reduced CDK2 activation by inhibiting the interaction of CDK2 with cyclin E and A
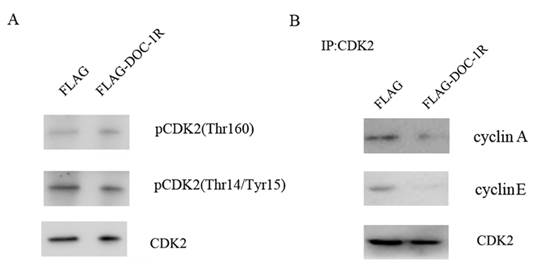
Previous studies have shown that for cells to activate CDK2, CDK2 protein has to bind to its regulatory subunit cyclin E or A, the Thr160 site has to be phosphorylated by CDK activator (CAK), and CDC25 has to dephosphorylate its Thr14 and Tyr15 sites [14, 15]. Our data showed that the phosphorylation levels of CDK2 Thr160 and Thr14/Tyr15 were not affected by DOC-1R overexpression (Fig. 4A). In contrast, DOC-1R overexpression significantly decreased the formation of cyclin E/A-CDK2 complexes (Fig. 4B), suggesting that DOC-1R inhibits the associations of cyclin E/A-CDK2 complexes, resulting in CDK2 inactivation.
DOC-1R-mediated inhibition of CDK2 activation through repressing cyclin E/A-CDK2 interactions. (A) Western blot. HeLa cells were transfected with pFLAG-DOC-1R and then subjected to protein extraction and western blot analysis for total CDK2, pCDK2 (Thr160) and pCDK2 (Thr14/Tyr15) proteins, respectively. (B) Immunoprecipitation-Western blot assay. The cells with the same conditions as A were subjected to immunoprecipitation with an anti-CDK2 antibody, as well as Western blot analysis for cyclin E and A protein, respectively.
DOC-1R interacted with CDK2 protein
According to the previous report, DOC-1R was suggested to associate with CDK2 protein. To explore the association, we further performed GST pull-down and Western blot assays. Our data showed that DOC-1R protein was able to pull-down CDK2 protein, but not cyclin E or A protein (Fig. 5A), suggesting that DOC-1R binds to a monomeric form of CDK2 in vitro.
Interaction between DOC-1R and CDK2 proteins. (A) GST pull-down assay. GST or GST-DOC-1R proteins were expressed in Escherichia coli BL21, extracted, incubated with total lysates of HeLa cells and MagneGST particles, and then subjected to Western blot analysis of CDK2, cyclin A and cyclin E, respectively. (B) Immunoprecipitation-Western blot assay. Recombinant retrovirus carrying DOC-1R cDNA was produced and used to infect HeLa cells. Immunoprecipitation was performed as described above following with Western blot analysis. (C) Fluorescent assay. pEGFP-DOC-1R or control was transfected into CHO cells with pDsRED-CDK2. Forty-eight hours later, cell images were viewed and analyzed with a fluorescent microscope.
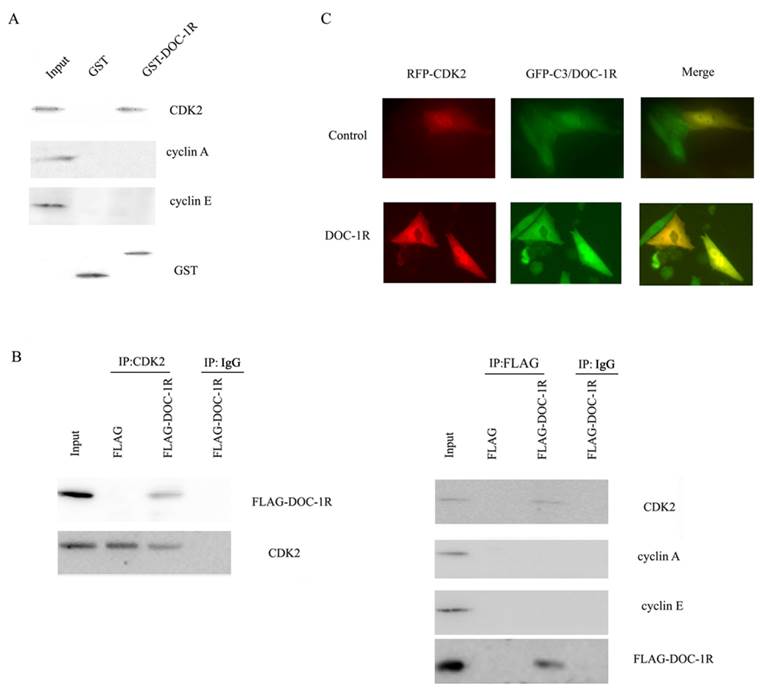
Similarly, viral supernatants from DOC-1R-transfected GP2-293 cells were incubated with HeLa cells and then immunoprecipitated and blotted with anti-FLAG antibody or anti-CDK2 antibody, respectively. The data showed that consistent with the pull-down data, DOC-1R associates with monomeric CDK2 in mammalian cells (Fig. 5B). The fluorescent assay data also confirmed the association (Fig. 5C). Taken together, these data demonstrate that DOC-1R binds to CDK2 protein.
Next, we further investigated the region of DOC-1R protein responsible for interacting with CDK2 protein. A previous study revealed that amino acid residues 120 to 122 (TER) of DOC-1R protein was its potential CDK binding site, while amino acid residues 110 to 112 (RAL) was as its putative cyclin-CDK binding site [3]. To verify the potential binding sites, four plasmids carrying GST-DOC-1R mutated cDNA were constructed (Fig. 6A). Truncated proteins were then expressed in Escherichia coli BL-21 and pull-down assays were performed. As shown in Figure 6B, the GST-DOC-1R mutant (1-106) retained CDK2, while the GST-DOC-1R mutant (1-63) did not, suggesting that amino acid residues between 64 and 106 could be the region of DOC-1R protein binding to CDK2 protein.
Identification of the amino acid region of DOC-1R protein that interacted with CDK2 protein. (A) Schematic representation of the truncated DOC-1R proteins. (B) GST pull-down assay. Truncated DOC-1R proteins were expressed in Escherichia coli BL-21 cells, pulled down with total lysates of HeLa cells and MagneGST particles, and then immunoblotted with anti-CDK2 antibody. (C) Schematic representation of the truncated CDK2 protein. (D) GST pull-down assay. Truncated FLAG-CDK2 proteins were subjected to pull-down assays with recombinant GST-tagged DOC-1R protein and then immunoblotted with anti-FLAG antibody.
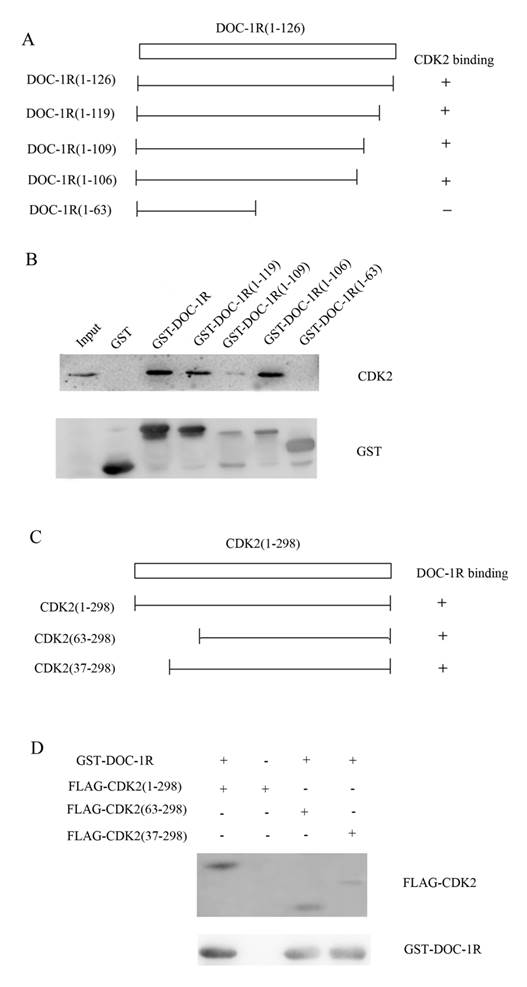
Furthermore, since PSTAIRE helix of CDK2, which is located between the 37 and 63 amino acid residues of CDK2, is necessary for CDK2 to interact with cyclin A/E [16-18], we hypothesized that DOC-1R occupied the CDK2 PSTAIRE helix and thus disabled CDK2 to interact with cyclin E or A. To map the DOC-1R interactive domains in CDK2, two deleted mutants, pFLAG-CDK2 (37-298) and FLAG-CDK2 (63-298), were obtained (Fig. 6C) [17]. Pull-down assays revealed that both of the mutants retained DOC-1R protein (Fig. 6D), suggesting that the PSTAIRE helix was not responsible for the DOC-1R-CDK2 association. Given that DOC-1R interacted with the monomeric form of CDK2, the pull-down data demonstrated that the interaction of DOC-1R with CDK2 might have shielded CDK2 PSTAIRE helix from interacting with cyclin E or A, resulting in DOC-1R-mediated inactivation of CDK2.
Discussion
In this study, we demonstrated the antitumor activity of DOC-1R via the inhibition of cell cycle G1/S phase transition in HeLa cell line. To the best of our knowledge, this is the first study to demonstrate DOC-1R-regulation of CDK2 expression and activity. Our data indicated that DOC-1R overexpression decreased CDK2 expression and inhibited CDK2 activation. Furthermore, the association of DOC-1R with CDK2 was found to be independent of the PSTAIRE helix of CDK2, and the amino acid region between 64 and 106 of DOC-1R protein was identified to be responsible for their interaction.
Indeed, as an important modulator in the cell cycle, CDK2 regulates cell cycle G1/S and G2/M transitions and modulates events in S phase, such as DNA replication and centrosome duplication, in coordination with cyclin E/A [5-11]. Moreover, CDK2 protein also participates in regulation of cell recombination, differentiation and apoptosis, and plays essential roles in senescence and meiosis [19-25]. In addition, altered CDK2 activity has been reported to have a close link with human carcinogenesis [5,6,12,19,24].
In the current study, we showed that DOC-1R expression in HeLa cells inhibited CDK2 expression and activation, which confirmed the data from our previous study that overexpression of DOC-1R could significantly inhibit the growth and colony forming ability of NIH3T3 cells. However, unlike DOC-1, the homolog of DOC-1R, which binds with CDK2 and induces its degradation in an ubiquitin-dependent manner, DOC-1R did not target CDK2 for ubiquitin-dependent proteolysis [26, 27]. Rather DOC-1R repressed CDK2 expression at the mRNA level. This finding may have an impact on our future studies of DOC-1R function and antitumor activity as a tumor suppressor gene.
The activities of cyclin-CDK complexes are negatively regulated by CDK inhibitors (CKIs). To date, two CKI families, CIP/KIP and INK4, have been identified according to their sequence homology and their mechanisms of cell cycle arrest. INK4 family members, consisting of p16, p15, p18 and p19, share a similar structure dominated by several ankyrin repeats and specifically inhibit the activity of CDK4 and CDK6. In contrast, CIP/KIP family members, including p21, p27 and p57, share a nuclear-localization sequence (NLS) and a conserved amino-terminal domain, causing them to localize into the nuclei and to ubiquitously inactivate CDKs by affecting their phosphorylation status [28]. However, DOC-1R protein is different from CIP/KIP family members in sequence characters, sub-localization in cells, and even the inhibitory mechanism of CDK2 activity. Thus, DOC-1R is suggested to belong to a new kind of CKI.
ERK2 regulates CDK2 activity by affecting CDK2 nuclear localization [29]. Abolishing ERK2 activity by a MEK inhibitor significantly reduces CDK2 activity in mammalian cells [30]. A previous study revealed that DOC-1R had a potential ERK2 phosphorylation site (21 to 24 amino acid residues, PGTP) and that phosphorylation is observed in meiosis of mouse oocytes [3], suggesting an intriguing link among ERK2, CDK2 and DOC-1R protein. However, further study is needed to confirm such a link.
Our current study also detected the down-regulation of CDK2 mRNA levels in DOC-1R overexpressing cells, although the mechanism of such an action remains to be determined. Furthermore, we also observed that overexpression of DOC-1R could reduce β-catenin expression (data not shown). Taken together, our data suggest that DOC-1R might negatively regulate the canonical Wnt pathway, resulting in the depression of cyclin D1 expression and consequently the reduction of CDK2 mRNA levels [31, 32].
In this study, we detected decreased association of CDK2 with cyclin A in DOC-1R overexpressing cells. The cyclin A-CDK2 complex is required for the progression of the S phase of the cell cycle, as well as G2/M transition [10,11]. Our data support the critical role of DOC-1R in G2/M transition. However, although different distributions of G2/M cells were observed between DOC-1R transient transfectant and control, synchronization might be helpful to increase the sensitivity due to the limitation of the G2-M cell population in HeLa cells.
In summary, our current study provided evidence that DOC-1R might play a crucial role in negatively regulating cell cycle G1 phase progression and G1/S transition by repressing CDK2 expression and activation. Future studies will further investigate the clinical significance of DOC-1R expression in different tumor cancer cells.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (No. 81100419 and 81170543).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang X, Tsao H, Tsuji T. et al. Identification and mutation analysis of DOC-1R, a DOC-1 growth suppressor-related gene. Biochem Biophys Res Commun. 1999;225:59-63
2. Sheng XJ, Jiang L, Zhou WQ. et al. Cloning of the mouse Doc-1R gene by genomic walking. Zhong Hua Yi Xue Yi Chuan Xue Za Zhi. 2002;21:240-4
3. Terret ME, Lefebvre C, Djiane A. et al. DOC-1R: A MAP kinase substrate that control microtubule organization of metaphase Ⅱ mouse oocytes. Development. 2003;132:5169-77
4. Zhou WQ, Jiang L, Sheng XJ. et al. Construction and expression of mouse DOC-1R antisense gene vector. Ai Zheng. 2002;21:122-6
5. Li Z, Pützer BM. Spliceosomal protein E regulates neoplastic cell growth by modulating expression of cyclin E/CDK2 and G2/M checkpoint proteins. J Cell Mol Med. 2008;12:2427-38
6. Wright RH, Castellano G, Bonet J. et al. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 2012;26:1972-83
7. Huang X, Summers MK, Pham V. et al. Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol Cell. 2011;42:511-23
8. Neganova I, Vilella F, Atkinson SP. et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells. 2011;29:651-9
9. Baydoun HH, Pancewicz J, Bai X. et al. HTLV-I p30 inhibits multiple S phase entry checkpoints, decreases cyclin E-CDK2 interactions and delays cell cycle progression. Mol Cancer. 2010;9:302
10. De Boer L, Oakes V, Beamish H. et al. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene. 2008;27:4261-8
11. Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genet. 2010;6:e1000863
12. Liu Q, Gao JL, Liu X. et al. Monitoring cyclin-dependent kinase 2 (CDK2) activity with Kinase-Glo, a luminescence-based ATP quantitative reagent. Afr J Biotechnol. 2011;10:1217-20
13. Wang F, Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARγ. Mol Biol Cell. 2009;20:801-8
14. Ovejero S, Ayala P, Bueno A. et al. Human Cdc14A regulates Wee1 stability by counteracting CDK-mediated phosphorylation. Mol Biol Cell. 2012;23:4515-25
15. Tu YS, Kang XL, Zhou JG. et al. Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells. Eur J Pharmacol. 2011;670:356-64
16. Jeffrey PD, Russo AA, Polyak K. et al. Mechanism of CDK activation revealed by the structure of a cyclin A-CDK2 complex. Nature. 2010;376:313-20
17. Prince T, Sun L, Matts RL. Cdk2: a genuine protein kinase client of Hsp90 and Cdc37. Biochemisty. 2005;44:15287-95
18. Futreal A, Otyepka M, Mann DJ. A cancer-derived mutation in the PSTAIRE helix of cyclin-dependent kinase 2 alters the stability of cyclin binding. Biochim Biophys Acta. 2010;803:858-64
19. Garcia HH, Brar GA, Nguyen DH. et al. Indole-3-Carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem. 2005;280:8756-64
20. Campaner S, Doni M, Hydbring P. et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene Myc and a Cdk2 senescence switch. Nat Cell Biol. 2010;12:54-9
21. Viera A, Rufas JS, Martínez I. et al. CDK2 is required for proper homologous pairing, recombination and sex-body formation during male mouse meiosis. J Cell Sci. 2009;122:2149-59
22. Hwang CY, Lee SM, Park SS. et al. CDK2 differentially controls normal cell senescence and cancer cell proliferation upon exposure to reactive oxygen species. Biochem Biophys Res Commun. 2012;425:94-9
23. Hiromura K, Pippin JW, Blonski MJ. et al. The subcellular localization of cyclin-dependent kinase 2 determines the fate of mesangial cells: role in apoptosis and proliferation. Oncogene. 2002;21:1750-8
24. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122-44
25. Fang Y, DeMarco VG, Nicholl MB. Resveratrol enhances radiation sensitivity in prostate cancer by inhibiting cell proliferation and promoting cell senescence and apoptosis. Cancer Sci. 2012;103:1090-8
26. Shintani S, Ohyama H, Zhang X. et al. p12DOC-1 is a novel cyclin-dependent kinase 2-associated protein. Mol Cell Biol. 2000;21:6300-7
27. Buajeeb W, Zhang X, Ohyama H. et al. Interactions of the CDK2-associated protein-1, p12DOC-1/CDK2AP1, with its homolog p14DOC-1R. Biochem Biophys Res Commun. 2004;315:998-1003
28. Romanov VS, Pospelov VA, Pospelova TV. Cyclin-dependent kinase inhibitor p21Waf1: contemporary view on its role in senescence and oncogenesis. Biochemistry. 2012;77:575-84
29. Chiariello M, Gomez E, Gutkind S. Regulation of cyclin-dependent kinase (Cdk) 2 Thr-160 phosphorylation and activity by mitogen-activated protein kinase in late G1 phase. Biochem J. 2000;349:869-76
30. Blanchard DA, Mouhamad S, Auffredou MT. et al. Cdk2 associates with MAP kinase in vivo and its nuclear translocation is dependent on MAP kinase activation in IL-2-dependent Kit 225 T lymphocytes. Oncogene. 2000;19:4184-9
31. Berthet C, Aleem E, Coppola V. et al. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775-85
32. Hanse EA, Nelsen CJ, Goggin MM. et al. Cdk2 plays a critical role in hepatocyte cell cycle progression and survival in the setting of cyclin D1 expression in vivo. Cell Cycle. 2009;8:2802-9
Author contact
![]() Corresponding author: E-mail: luoyangcmu.edu.cn.
Corresponding author: E-mail: luoyangcmu.edu.cn.