ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2013; 9(8):766-777. doi:10.7150/ijbs.5711 This issue Cite
Research Paper
B19, a Novel Monocarbonyl Analogue of Curcumin, Induces Human Ovarian Cancer Cell Apoptosis via Activation of Endoplasmic Reticulum Stress and the Autophagy Signaling Pathway
Wanglei Qu 1*, Jian Xiao 2*, Hongyu Zhang2*, Qiong Chen 1, Zhouguang Wang 2, Hongxue Shi 2, Liang Gong3, Jianqiang Chen3, Yanlong Liu2, Risheng Cao4, Jieqiang Lv 1 ![]()
1. Department of Gynecology and Obstetrics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, China;
2. School of Pharmacy, Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, China;
3. Department of otolaryngology, Cixi Hospital, Wenzhou Medical University, Ningbo, China;
4. Department of Gastroenterology, The First Affiliated Hospital, Nanjing Medical University, Nanjing, China;
* The first three authors contributed equally to this study.
Abstract
Background: The unfolded protein response, autophagy and endoplasmic reticulum (ER) stress-induced apoptosis regulate tumor cell fate and have become novel signaling targets for the development of cancer therapeutic drugs. Curcumin has been used to treat several different cancers, including ovarian cancer, in clinical trials and research; however, the role of ER stress and autophagy in the therapeutic effects of curcumin and new curcumin analogues remains unclear.
Methods: Cell viability was determined using the MTT assay. Apoptosis was detected using flow cytometry with PI/Annexin V-FITC staining. The expression levels of ER stress- and autophagy-related proteins were analyzed by western blotting. The activation of autophagy was detected using immunofluorescence staining.
Results: We demonstrated that B19 induced HO8910 cell apoptosis in a dose-responsive manner. We also determined and that this effect was associated with corresponding increases in a series of key components in the UPR and ER stress-mediated apoptosis pathways, followed by caspase 3 cleavage and activation. We also observed that B19 treatment induced autophagy in HO8910 cells. The inhibition of autophagy using 3-methyladenine (3-MA) increased levels of intracellular misfolded proteins, which enhanced ovarian cancer apoptosis.
Conclusions: Our data indicate that ER stress and autophagy may play a role in the apoptosis that is induced by the curcumin analogue B19 in an epithelial ovarian cancer cell line and that autophagy inhibition can increase curcumin analogue-induced apoptosis by inducing severe ER stress.
Keywords: Curcumin analogues, B19, ovarian cancer, apoptosis, ER stress, autophagy.
Background
Epithelial ovarian cancer (EOC) is the sixth most common cancer and the fifth leading cause of cancer mortality in women worldwide [1]. This lethal gynecological malignancy is commonly diagnosed at a late stage due to a silent early stage and ease of metastasis. It is typically treated in its advanced stages with cytoreductive surgery and platinum-based chemotherapy. Unfortunately, despite recent developments in treatment techniques, the five-year survival rate for ovarian patients with cancer is only 20-40% [2]. Therefore, the development of novel therapies for the management of ovarian cancer is especially urgent. This development is the long-term objective of the current project.
Endoplasmic reticulum (ER) is an essential cellular compartment for protein synthesis and maturation. Various physiological and pathological conditions may affect ER homeostasis and interfere with proper protein folding, ultimately causing an imbalance between ER protein folding load and capacity, leading to the accumulation of unfolded or misfolded proteins in the ER lumen [3]. This cellular condition is referred to as ER stress. When misfolded proteins accumulate in the ER, the resulting stress activates the unfolded protein response (UPR) [4]. The UPR has three primary outputs: a transient reduction in protein translation to decrease ER protein load, the upregulation of ER regulators to augment the folding and export capacity of the ER, and the activation of ER-associated protein degradation (ERAD) [5]. The UPR can also activate cell death programs under severe or prolonged ER stress, when adaptive responses are exceeded and/or a dysfunctional UPR is unable to correct ER stress [6].
When misfolded proteins cannot be degraded by the proteasome, the UPR may upregulate autophagy machinery [7]. Autophagy is an evolutionarily conserved protein degradation pathway in eukaryotes which plays important roles in regulating protein homeostasis and which is essential for cell survival under metabolic stress. Ubiquitinated proteins are degraded by the proteasome through the ER-associated degradation pathway and by autophagy through the ER-activated autophagy pathway [5]. In addition to its role in cellular homeostasis, autophagy can be a form of programmed cell death or may play a cytoprotective role in situations of nutrient starvation [8]. Recent studies suggest that UPR signaling also affects interactions within the tumor microenvironment. The UPR, autophagy and ER stress-induced apoptosis regulate tumor cell fate. Moreover, different anticancer treatments activate UPR signaling in tumor cells, a process that has been proposed to either enhance cancer cell death or to act as a mechanism of resistance to chemotherapy [9, 10].
Curcumin, a well-known constituent of traditional medicine, has been used to treat several different cancers in clinical trials, including ovarian cancer [11]. However, many studies indicate that its poor bioavailability and pharmacokinetic profiles limit its application in anti-cancer therapies [12,13]. The chemical modification of curcumin is an effective way to obtain potential analogues with superior bioavailability and more effective anti-tumor activities. In our earlier study, we designed and synthesized a series of mono-carbonyl analogues of curcumin with greater stability and superior pharmacokinetic profiles. We also demonstrated that certain analogues exhibited more effective anti-tumor activity compared with curcumin [14-16]. (1E, 4E)-1, 5-bis (2-methoxyphenyl) penta-1, 4-dien-3-one (referred to as B19) is one of the novel curcumin analogues we synthesized and has been demonstrated to cause ER stress-related apoptosis in human non-small cell lung cancer H640 cells [15]. However, it has not been determined whether ER stress plays a role in curcumin analogue-induced apoptosis in epithelial ovarian cancer cell lines.
In our present study, we investigated the anticancer effects of B19 on the epithelial ovarian cancer cell line HO8910. We demonstrated that B19 may induce ER stress by promoting the formation of ubiquitinated misfolded proteins and that both autophagy and apoptosis were activated. The autophagy inhibitor 3-methyladenine (3-MA) increased the level of ubiquitinated proteins, which elevated ER stress and resulted in a higher apoptotic rate in these cells when they were treated with B19. This study was designed to test our hypotheses that ER stress plays a role in curcumin analogue-induced apoptosis in epithelial ovarian cancer cell lines and that autophagy inhibition can increase curcumin analogue-induced apoptosis by inducing severe ER stress.
Materials and methods
Materials
The human ovarian cancer HO8910 cell line was purchased from the Institutes of Cell Biology in Shanghai, China. Dulbecco's modified eagle's medium (DMEM), Fetal bovine serum (FBS), propidium iodide (PI) and Annexin V-FITC were obtained from Invitrogen (Carlsbad, CA, U.S.A.). Anti-Grp78 (sc-13968), anti-CHOP (sc-7351), anti-Ub (sc-8017) and anti-PDI (sc-136230) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Anti-caspase-3 (ab90437), anti-LC3 (ab58610) and anti-ATF6β (ab103673) antibodies were obtained from Abcam (Abcam, PLC, Hong Kong).
The synthesis of B19
The general procedure of synthesis of B19 was as follows [14]: 7.5 mmol acetone was added to 10 ml of a solution of 15 mmol aryl-aldehyde in MeOH. The solution was stirred at room temperature for 20 min, followed by the dropwise addition of 1.5 ml of 7.5 mmol NaOCH3/CH3OH. The mixture was stirred at room temperature and monitored using TLC. When the reaction was completed, the residue was poured into a saturated NH4Cl solution and filtered. The precipitate was washed with water and cold ethanol and dried in a vacuum. The solid was purified using chromatography over silica gel using CH2Cl2/CH3OH as the fluent to yield compounds.
Cell culture
The human ovarian cancer HO8910 cell line was cultured at 37°C in 5% CO2 and 95% air atmosphere in DMEM, supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 U/ml streptomycin.
Cell viability assays
Cell viability was determined using MTT assay. We seeded exponentially growing HO8910 cells into 96-well culture plates in 100 µl of medium at a density of 1×104 cells/well. The following day, varying concentrations (5, 10, 15 μM) of B19 were added to the wells in quadruplicate and incubated for 12 h and 24 h. The MTT assays (Beyotime, Nanjing, China) were performed by adding 20 µl MTT (3-(4,5- dimethylthiazol-2-yl)-2,5-diphe-nyltetrazolium bromide (5 mg/ml in PBS) to the cells for 4 h. Next, 150 μl of dimethyl sulfoxide (Beijing Chemical Industry Limited Company, Beijing, China) was added to each well. After shaking for 10 min, absorbance was measured at a wavelength of 570 nm using a microplate reader (Biotek Instruments, Inc., USA). Each treatment was performed in triplicate. The mean of the three values was determined, and the results were expressed as a percent of the control.
Electron microscopy
Electron microscopy and morphometric analysis were performed as described previously [34]. The cells were fixed for 30 min with ice-cold 2.5% glutaraldehyde in 0.1 M cacodylate buffer, embedded in Epon, and processed for transmission electron microscopy using standard procedures. Representative areas were chosen for ultra-thin sectioning and examined on a transmission electron microscope at a magnification of 5,000 × or 10,000 ×.
Flow cytometry analysis
Propidium iodide (PI, 1 μg/ml) and Annexin V-FITC (1 μg/ml) were used for the determination of cell death. After exposure to the different experimental conditions, the cells were trypsinized and incubated with PI and Annexin V-FITC for 15 min at 37°C. The samples were then analyzed using a FACScan flow cytometer. All of the experiments reported in the present study were performed in triplicate.
Immunofluorescence staining
The cells were seeded onto coverslips in 24-well plates at a density of 5×104 cells/well and allowed to recover overnight. After treatment with 10 μM of B19 for 0 h and 12 h, the cells were washed with phosphate-buffered saline (PBS) twice. The cells were then fixed with 4% paraformaldehyde and washed twice more with PBS. The cells were then stained with the nuclear stain Hoechst 33258 (100 μg/ml, Sigma) for 5 min, washed with PBS, and examined using a Nikon 80i microscope to reveal apoptosis.
Microtubule-associated protein light chain 3 (MAPLC3) was examined by indirect immunofluorescence. The cells were cultured on coverslips overnight, treated with 10 μM of B19 for different time periods and rinsed 3 times with PBS. After incubation, the cells were fixed with 4% paraformaldehyde for 20 min, incubated in H2O2 for 20 min to remove endogenous enzyme, blocked with bovine serum albumen (BSA), and incubated with the primary antibody (anti-LC3, used at 1:100 dilution; Santa Cruz Biotechnology, CA) overnight at 4°C. The next day, the slides were incubated with FITC/Texas Red-conjugated secondary antibodies (1:400 dilution) (Santa Cruz Biotechnology, CA) for 1 h, stained with Hoechst 33258 (100 μg/ml) for 5 min, and washed 3 times with PBS. After mounting, the cells were examined using a Nikon 80i microscope.
Western blotting
Whole cellular protein was extracted from HO8910 cells prepared with lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EDTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, 1 μg/ml leupeptin, and 1 mM PMSF) for western blotting. The protein extracts were quantified using a ProteoExtract® Protein Extraction Kits (Merck). For the extraction of total cell proteins, the cells were harvested and washed with cold PBS. The cells were centrifuged for 5 min at 600 g at 4 °C then incubated in cell lysis buffer (150 mM NaCl, 1 mM EDTA, 10 mM HEPES, 1 mM PMSF, and 0.6% NP-40). The lysates were sonicated and incubated for 15 min on ice and then centrifuged at 700 g for 10 min at 4 °C. The supernatant was centrifuged at 14,000 g for another 30 min at 4 °C; cell proteins were contained within the resultant supernatant. For the western blot analysis, lysate proteins (30-50 μg) were resolved using 8%, 10%, and 15% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose transfer membranes (Whatman, Maidstone, UK). The membranes were blocked with 5% nonfat dry milk in buffer (10 mM Tris-HCl, 100 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature and then incubated with the relevant primary antibody overnight at 4°C. Anti-PDI, anti-p62, anti-Ub, anti-Grp78, and anti-CHOP antibodies were each used at a 1:200 dilution. Anti-caspase-3 and anti-LC3 antibodies were used at a 1:500 dilution. The next day, membranes were incubated with horseradish peroxidase-conjugated secondary antibody at a 1:2000 dilution for 1 h at room temperature. The immunoreactive bands were visualized using the diaminobenzidine (Sigma-Aldrich, St. Louis, MO, USA) coloration method. The representative bands were measured using a Bio-Rad GIS gel imaging system. The protein levels were normalized to those of GAPDH, and the ratios of normalized protein to GAPDH were presented as the mean ± SD from three independent experiments. The protein levels were quantified by densitometry using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Experiments were performed at least three times and the data are presented as the mean ± SD. The statistical data analysis was performed using one-way ANOVA. P < 0.05 was considered to represent a statistically significant difference.
Results
B19 inhibits HO8910 cell viability and induces apoptosis
The synthesis of the novel curcumin analogue B19 was performed as shown in Figure 1A and as described above. To evaluate the effect of B19 on ovarian cancer cells, we treated HO8910 cells with increasing concentrations of B19 for 12 h and 24 h and then examined cell viability using the MTT assay. We observed that B19 significantly suppressed the viability of HO8910 cells in a dose-dependent manner within 24 h (Figure 1B). We then treated HO8910 cells with 10 μM of B19 for increasing amounts of time. The results showed that at certain concentrations, B19 exhibited greater levels of inhibition of HO8910 cell growth in a time-dependent manner (Figure 1C). Based on the results of the MTT assay, we treated HO8910 cells with varying concentrations (5, 10, 15 μM) of B19 for 12 h, and examined apoptotic chromatin condensation by Hoechst 33258 staining. Compared with the control group, B19-induced apoptotic chromatin condensation was clearly observed in HO8910 cells (Figure 1D). We observed ultrastructural changes in HO8910 cells that were treated with B19 for 12 h using electron microscopy. Compared with the controls, the most prominent features of the B19-treated cells were an extensive distension of the ER and apoptotic morphology, with condensation and margination of nuclear chromatin and disrupted cell membranes (Figure 1E).
B19 activates the UPR signaling pathway and induces ER stress-associated apoptosis
We next asked if ER stress-induced cell death was involved in the effects of B19. We therefore measured the levels of ER stress-associated chaperone proteins to determine UPR activation. The most abundant and well-characterized ER chaperone proteins include protein disulfide isomerase (PDI) as detected using western blotting in the present analysis. The ER luminal marker PDI is considered to be an indicator of ER stress activation [17]. As shown in Figure 2A and 2B, after treatment with 10 μM B19, PDI expression was up-regulated at 1 h, 4 h and 8 h and then decreased when treatment was prolonged to 24 h.
GRP78 is reported to be the gatekeeper to the activation of ER stress. Upon ER stress, GRP78 releases the UPR sensors, allowing for the activation and transduction of UPR signals across the ER membrane to the cytosol and the nucleus [4]. We thus measured the protein expression levels of GRP78 and CHOP after B19 treatment using western blot analysis. The time course results indicate that the expression of GRP78 and CHOP are significantly up-regulated following treatment with 10 μM B19 from 1 h to 24 h (Figure 2A). In addition, as shown in Figure 2C and 2D, protein expression of GRP78 and CHOP increased in a dose-dependent manner after 12 h of B19 treatment.
To further confirm the effects of B19 on ovarian cancer cells, HO8910 cells were treated with various concentrations (5, 10, 15 μM) of B19 for 12 h. As shown in Figure 3A, 3B, 3C and 3D, the expression of the ER stress-related proteins PDI, GRP78 and CHOP increased significantly compared with the control and the vehicle (DMSO) groups. During ER stress, the unfolded protein response components, including activating transcription factor 6 (ATF-6) and X-box binding protein 1 (XBP-1), induce the transcription of CHOP and ultimately lead to caspase cascade activation to complete the execution of ER stress-induced apoptosis [18]. We next determined the levels of ATF-6 and XBP-1 upon treatment with B19. As shown in Figure 3E, 3F and 3G, the protein expression of ATF-6 and XBP-1 was significantly increased after the cells were incubated with B19 for 12 h, especially at concentrations of 10 and 15 μM. As B19 induced ER stress-mediated apoptotic downstream events after commitment to the UPR pathway, the activation of caspase-3 was analyzed by western blotting. It was observed that the cleavage of procaspase-3 increased significantly after treatment with B19 for 12 h (Figure 3E and 3H).
B19 inhibits HO8910 cell viability and induces apoptosis. A: Schematic representation of the synthesis of B19. B: The cells were treated with varying doses of B19 for 12 h and 24 h. Cell viability was determined using the MTT assay. The data are presented as the mean ± SD, n = 5. *P < 0.05 vs. control group, **P < 0.01 vs. control group. C: The cells were treated with 10 μM of B19 for varying times. Cell viability was determined using MTT assay. The data are presented as the mean ± SD, n = 5. *P < 0.05 vs. control group, **P < 0.01 vs. control group. D: The cells were treated with varying concentrations (5, 10, 15 μM) of B19 for 12 h and stained with Hoechst 33258. Cell morphology was observed by microscopy (200 ×). (a) Control, (b) 5 μM, (c) 10 μM, (d) 15 μM. E: Representative transmission electron microscopy photomicrographs of HO8910 cells that were treated with 10 μM B19 for 12 h. The nuclear and cellular profiles and ER morphologies are normal in the control cells (small panel a, 5,000 ×). Exposure to 10 μM B19 for 12 h resulted in apoptosis (small panel b, 3,000 ×, arrows indicate distended ER), (small panel c, 5,000 ×, arrows indicate apoptotic bodies) (bar, 2 μm).
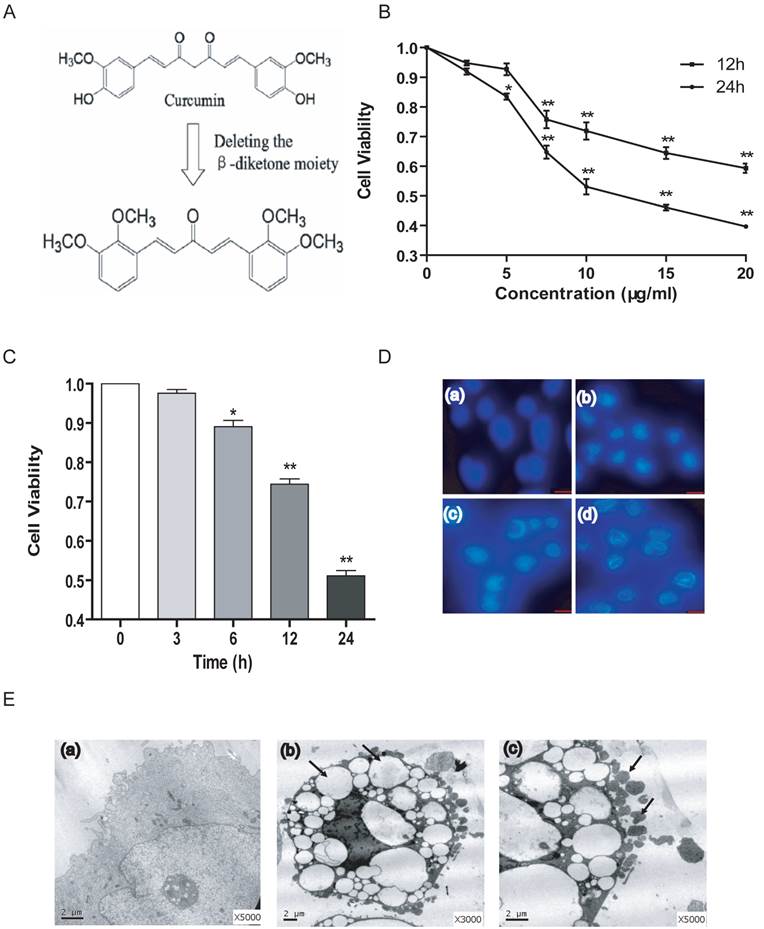
B19 activates the ER stress-associated proteins PDI, GRP78 and CHOP in HO8910 cells. A: HO8910 cells were treated with 10 μM B19 for 0 h, 1 h, 4 h, 8 h, 12 h, or 24 h. Western blot analysis for the protein expression of PDI, GRP78 and CHOP. B, C and D: Quantitation of PDI, GRP78 and CHOP levels. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group. GAPDH was used as a protein loading control and for band density normalization.
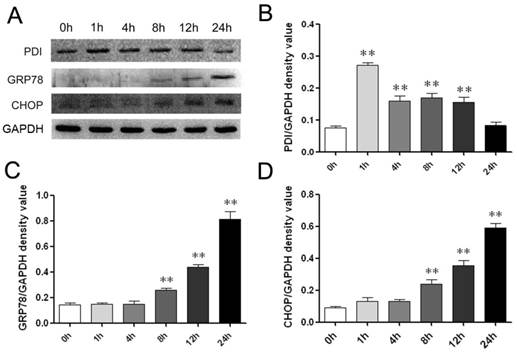
B19 activates the ER stress-associated proteins GRP78, CHOP, ATF-6 and XBP-1 and caspase-3 cleavage in HO8910 cells. A: HO8910 cells were treated with 5, 10 or 15 μM B19 for 12 h. The expression levels of PDI, GRP78 and CHOP were determined by western blot. B, C and D: Quantitation of protein expression of PDI, GRP78 and CHOP levels. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group. GAPDH was used as a protein loading control and for band density normalization. E: HO8910 cells were treated with 5, 10 or 15 μM B19 for 12 h. The expression of ATF-6, XBP-1 and caspase-3 were determined by western blot. F, G and H: The quantitation of ATF-6 and XBP-1 levels and the cleavage of caspase-3. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group. GAPDH was used as a protein loading control and for band density normalization.
The activation of autophagy is involved in the effects of B19 on HO8910 cells
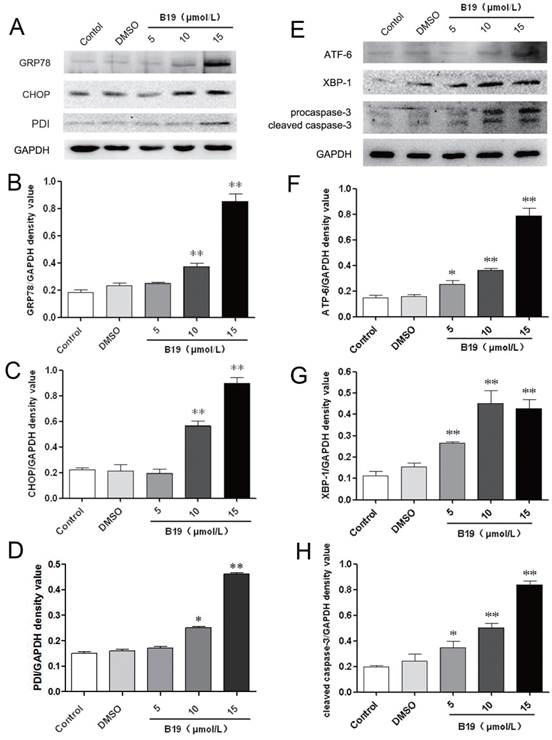
It has been suggested that the activation of ER stress and autophagy have a complex relationship in the process of cell death. To further investigate whether the activation of autophagy is also involved in the effects of B19, we examined HO8910 cells by electron microscopy for autophagosome-like structures after 4 h of B19 treatment. As shown in Figure 4A, classic autophagic vacuoles with double membranes were observed in the cytoplasm of HO8910 cells that were treated with 10 μM B19 but not in the control group (Figure 1Ea). Furthermore, we detected the activation of autophagy proteins in B19-treated HO8910 cells by western blotting. The autophagy protein microtubule-associated light chain protein 3 (LC3) is the mammalian equivalent of yeast Atg8. When autophagy is activated, LC3-I is cleaved to the proteolytically derived LC3-II and aggregates in autophagosomal membranes, an that is action essential for the formation of the autophagosome [19]. We used indirect immunofluorescence staining to detect the localization of LC3. As shown in Figure 4B, the green signals of LC3 accumulated significantly in HO8910 cells that were treated with B19 for 4 h. The transformation of LC3-I to LC3-II was also determined using western blot analysis. The ratio of LC3-II to LC3-I in HO8910 cells that were treated with B19 for 1 h and 4 h increased significantly compared with the control group (Figure 4C and 4D).
The inhibition of autophagy by a specific inhibitor enhances ER stress-induced apoptosis
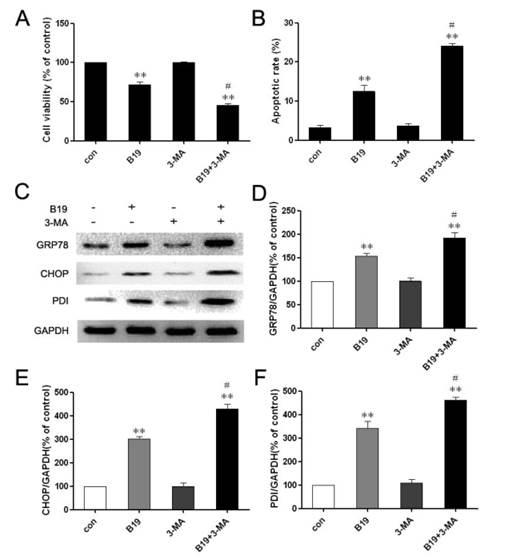
The above results demonstrated that B19 may induce both autophagy and apoptosis in HO8910 cells and that these events occurred in a defined temporal sequence. We detected autophagy at 4 h (an earlier time point) and apoptosis at 12 h (a later time point). The autophagy-specific inhibitor 3-MA, which by itself has no toxic effects according to previous studies [20], was used to elucidate the role of autophagy in HO8910 cells that were treated with B19. As shown in Figure 5A, MTT assays indicated that 3-MA alone has no significant effects on cell viability. When combined with B19, 3-MA enhanced the cytotoxic effect of B19 in HO8910 cells. We next observed the effects of autophagy inhibition on B19-induced apoptosis in HO8910 cells by flow cytometry of cells stained with PI/Annexin V-FITC. It was observed that apoptosis increased markedly in HO8910 cells that were treated with B19 in combination with 3-MA compared with the group treated with B19 alone (Figure 5B).
To further investigate the relationship between autophagy and ER stress-induced apoptosis, the expression levels of PDI, GRP78 and CHOP were determined by western blotting. As shown in Figure 5C, 5D and 5E, HO8910 cells that were treated with B19 in combination with 3-MA exhibited up-regulation of PDI, GRP78 and CHOP compared with the group treated with B19 alone. Collectively, these results indicate that the apoptosis induced by the curcumin analogue B19 was related to the activation of autophagy and that the inhibition of autophagy by the autophagy-specific inhibitor 3-MA aggravated the ER stress-induced apoptosis induced by B19.
B19 activates autophagy in HO8910 cells. A: Representative transmission electron microscopy photomicrographs of HO8910 cells that were treated with 10 μM B19 for 4 h (10,000 ×, arrows indicate autophagosome-like structures with double membranes). B: The localization of LC3 in HO8910 cells that were treated with 10 μM B19 for 4 h. The cells were observed by microscopy (400 ×). C: HO8910 cells were treated with 10 μM B19 for 0 h, 1 h, 4 h, 8 h, 12 h or 24 h. Western blot analysis for LC3 expression. D: The quantitation of the ratio of LC3-II to LC3-I. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group. GAPDH was used as a protein loading control and for band density normalization.
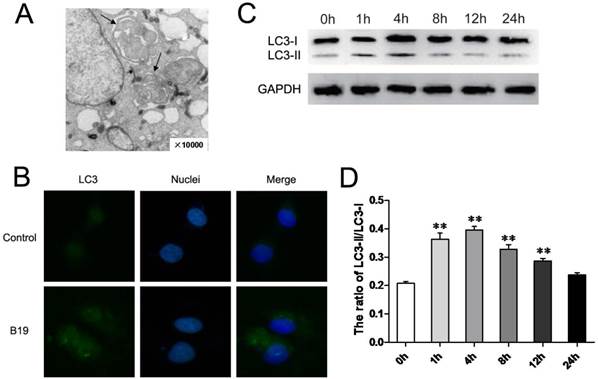
Combination treatment with the autophagy inhibitor 3-MA enhances B19-induced apoptosis. A: HO8910 cells were treated with B19 (10 μM) and/or 3-MA (10 mM) for 12 h. Cell viability was determined using MTT assay. The data are presented as the mean ± SD, n = 5. **P < 0.01 vs. control; #P < 0.05 vs. B19 group. B: HO8910 cells were treated with B19 and/or 3-MA for 12 h and then stained with PI and Annexin V-FITC. Positively stained cells were counted using FACScan. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control; #P < 0.05 vs. B19 group. C: Western blot analysis for the expression of GRP78, CHOP and PDI in cells that were treated with B19 and/or 3-MA for 12 h. D, E and F: The quantitation of GRP78, CHOP and PDI protein levels. The data are presented as the mean ± SD, n =3. *P < 0.01 vs. control group, #P < 0.05 vs. B19 group.
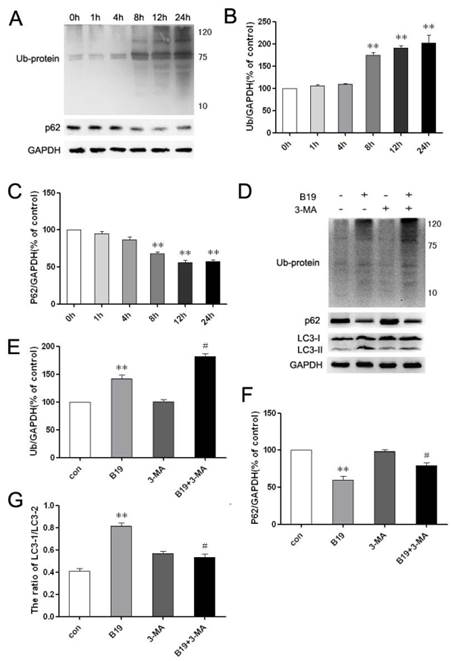
Blocking autophagy results in the accumulation of ubiquitinated proteins in HO8910 cells treated with B19
The ubiquitin-binding protein p62/SQSTM1 (sequestosome 1) is multifunctional, as it promotes survival-critical signals, acts as a receptor/adaptor for autophagy of ubiquitinated substrates and recruits ubiquitinated proteins (Ub-proteins) to autophagosomes for degradation [21, 22]. To investigate whether the clearance of accumulated proteins and the related process of autophagic degradation are involved in the effects of B19, we examined the levels of p62 and Ub-proteins by western blot. The level of Ub-proteins increased noticeably after treatment with 10 μM B19 for 8 h, 12 h and 24 h, and the level of p62 decreased noticeably at these time points (Figure 6 A and 6B). Upon combination of B19 treatment with 10 mM 3-MA for 8 h, the transformation of LC3-I to LC3-II and p62 degradation were inhibited and Ub-protein accumulation was noticeably enhanced compared with cells that were treated with B19 alone (Figure 6C and 6D).
Discussion
Epithelial ovarian cancer has been the leading cause of mortality among gynecologic malignancies worldwide during the last twenty years [23]. To a certain extent, the low survival rate is due to the side effects of chemotherapy and the acquired resistance to chemotherapy. The development of a novel therapy is urgently needed to improve the survival rate of this condition. Recent studies have suggested that during tumorigenesis, cancer cells are often exposed to hypoxia, nutrient starvation, oxidative stress and other metabolic dysregulations that cause the accumulation of unfolded or misfolded proteins in the ER, a condition that is referred to as “ER stress” [12]. To handle the accumulation of the unfolded or misfolded proteins, organisms evolved a group of ER signal transduction pathways, collectively termed the unfolded protein response [24], to alter transcriptional and translational programs to maintain ER homeostasis [25]. Most normal cells do not undergo an active “stress” response, and the UPR pathways remain quiescent. This discrepancy between tumor cells and normal cells allows for the agents that target ER stress to achieve specificity in cancer therapy. Clinical data support the hypothesis that the manipulation of ER stress may enhance the efficacy of chemotherapeutic drugs [26, 27] and that ER stress-mediated apoptotic pathways may provide novel targets for the development of antitumor drugs [28].
The inhibition of autophagy enhances Ub-protein accumulation in HO8910 cells that were treated with B19. A: HO8910 cells were treated with 10 μM B19 for 0 h, 1 h, 4 h, 8 h, 12 h or 24 h. Western blot analysis for the level of Ub-proteins and p62. B and C: The quantitation of Ub-proteins and p62 levels. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group. GAPDH was used as a protein loading control and for band density normalization. D: HO8910 cells were treated with B19 (10 μM) and/or 3-MA (10 mM) for 8 h. Western blot analysis for the level of Ub-proteins, p62 and LC3 levels in HO8910 cells that were treated with B19 and B19 in combination with 3-MA for 8 h. E, F and G: Quantitation of Ub-proteins, p62 and the ratio of LC3 II to LC3 I protein levels. The data are presented as the mean ± SD, n = 3. **P < 0.01 vs. control group, #P < 0.05 vs. B19 group. GAPDH was used as a protein loading control and for band density normalization.
Many studies have demonstrated that curcumin protects against ovarian cancer. Curcumin has been shown to activate multiple pathways, including the ER stress and UPR pathways [29, 30]. This compound has been determined to be safe in clinical trials, even at high concentrations. However, because of its instability in vitro and poor metabolic properties in vivo, the clinical application of curcumin has been significantly limited. More potent and soluble curcumin analogues have been developed. In our previous study, B19, a novel monocarbonyl analogue of curcumin, exerted cytotoxicity on H460 cells through the induction of apoptosis, which involves the ER stress signaling pathway [15]. In the present study, we tested B19 cytotoxicity on human ovarian cancer HO8910 cells. The MTT assay results indicated that B19 exhibited powerful cytotoxic effects against HO8910 cells in a time and dose-dependent manner.
In addition to apoptotic morphological changes, the most prominent ultrastructural change after B19 treatment was an extensively dilated ER. We hypothesized that B19 treatment interferes with proper protein folding in the ER, leading to misfolded protein accumulation in the ER lumen. Therefore, the possibility of whether B19 activates the UPR signaling pathway and induces ER stress-associated apoptosis was examined. Using electron microscopy, we observed extensive distension in the ER and an apoptotic morphology in ovarian cancer cells that were treated with B19. These processes are assisted and monitored by several resident chaperones and Ca2+-binding proteins, including glucose-regulated proteins such as GRP78, the immunoglobulin heavy chain binding protein BiP, calreticulin and calnexin, as well as several folding enzymes, such as the thioredoxin-like protein PDI [31]. Studies of the role of PDI in cancer have demonstrated a pro-oncogenic, pro-survival function for PDI in cancer and therapeutic resistance. PDI up-regulation in response to ER stress helps to ameliorate misfolded protein and stress-induced apoptosis. Based on these morphological results, we next analyzed the levels of ER stress-associated proteins using western blot analysis. Our data indicate that B19 increases PDI protein expression after treatment of at least 1 h. PDI up-regulation may be the earliest sign of ER stress activation upon treatment with B19.
GRP78, a member of glucose-regulated protein family (GRPs), is one of the most abundant ER luminal chaperone proteins and is upregulated by ER stress. Our data demonstrated that B19 could increase GRP78 mRNA levels and protein expression, especially after 8 h treatment. Once GRP78 is activated, it binds to unfolded proteins and dissociates three major ER stress sensors: pancreatic ER kinase (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) [32]. ATF6 is activated by proteolysis, translocates to the nucleus and regulates the expression of ER chaperones and XBP1 to facilitate protein folding, secretion, and degradation in the ER [6]. The other ER stress-associated protein, CHOP, is a member of the C/EBP family of bZIP transcription factors, and its expression is induced to high levels by ER stress. ATF-6 and PERK activation have been reported to increase CHOP gene expression, triggering an ER stress-specific cascade for the implementation of apoptosis. We observed up-regulation of CHOP protein after HO8910 cells were treated with B19, indicating that the B19 may induced ER stress developed into the commitment phase of apoptosis. All of the upstream signals ultimately lead to caspase activation, resulting in the ordered and sequential dismantling of the cell, completing the execution of ER stress-induced apoptosis [33, 34]. In the present study, we also detected the B19-induced activation of two of the unfolded protein response pathways via the monitoring of AFT-6 and XBP-1 levels and found that both GRP78 and CHOP were activated by B19 treatment. Overall, our data demonstrate the capacity of B19 to activate all of the key proteins in the initiation, commitment, and execution phases of ER stress. These data preliminary indicate that the compound B19 may trigger the ER stress-dependent apoptotic pathway. However, we really do not know whether B19-induced cell apoptosis is ER stress-dependent or not. In non-small cancer cell line H460, knockdown of CHOP by specific siRNAs attenuated B19-induced ER stress-mediated apoptotic cascade [15]. We supposed that B19-induced apoptosis also is, at least partially, ER stress-dependent in HO8910 cells.
Using electron microscopy, we also observed autophagosome-like structures in B19-treated HO8910 cells. Autophagy is essentially a protein degradation system of the cell's own lysosomes. A variety of stress signals, such as nutrient starvation or treatment with different anticancer agents, stimulate the autophagy process. That is, conditions that induce ER stress also lead to the induction of autophagy [35,36]. Autophagy is generally characterized by the presence of cytoplasmic vacuoles and autophagosomes, the absence of marginated nuclear chromatin [37,38] and an increase in cleavage of LC3. The role of autophagy in the process of cell death is controversial, but it has been confirmed that crosstalk between apoptosis and autophagy is essential in programmed cell death. In cancer cells treated with anticancer agents, autophagy appears to play a protective role. Suppression the autophagy with the autophagy inhibitor wortmannin and knockdown of Atg5 triggered cucurbitacin-induced cell death to shift from autophagic cell death to caspase-dependent apoptosis [39,40]. The simultaneous knockdown of the autophagic gene Beclin-1 synergistically restored antiestrogen sensitivity in resistant human breast cancer cells [5]. In this study, we detected the expression of LC3 protein. Both immunofluorescence staining results and western blot analysis indicated that B19 treatment results in the activation of an early stage of autophagy induction. Autophagy inhibition by the inhibitor 3-MA aggravated the apoptosis induced by B19.
Proteasomal degradation and autophagy have been identified as the two primary mechanisms that regulate protein clearance in stressed cells. The protein p62 reportedly conveys ubiquitinated proteins to both the proteasomal and autophagic degradation systems. Ubiquitinated proteins are degraded by proteasomes through the ER-associated degradation pathway and by autophagy through the ER-activated autophagy pathway [41, 42]. In particular, p62 was the first protein shown to bind directly with LC3 to facilitate autophagic degradation of ubiquitinated protein aggregates [43]. In the present study, we demonstrated that ubiquitinated proteins significantly accumulated after B19 treatment, in parallel with an increase in the cleavage of LC3 and a reduction in p62 protein levels. When we treated HO8910 cells with B19 in combination with 3-MA to block autophagy, the levels of LC3II decreased accordingly. Meanwhile, ubiquitinated proteins accumulated and p62 levels significantly increased. All of these facts indicate that B19 activates both apoptosis and autophagy maybe in response to ER stress caused by ubiquitinated proteins. Autophagy helps to remove ubiquitinated proteins, protects the ER and mitigates ER stress. The activation of autophagy after ER stress can either be cell-protective or cytotoxic. Studies have confirmed that autophagy eliminates misfolded proteins and plays a protective role in cell survival against ER stress in certain conditions [20]. Certain researchers have reported that activation of autophagy plays a protective role or is involved in chemoresistance in tumor cells [44]. Other researchers have documented that autophagy acts as programmed cell death type II and efficiently suppresses the growth of malignant glioma cells after curcumin treatment [45]. However, the role of autophagy-related clearance in HO8910 cells treated with B19 had not been examined. In the present study, we demonstrated that when autophagy was blocked by the specific inhibitor 3-MA, ubiquitinated proteins accumulated and ER stress-mediated apoptosis was enhanced in HO8910 cells that were treated with B19. These data indicate that the activation of autophagy clears ubiquitinated proteins as a self-defense mechanism, which alleviates ER stress and subsequently apoptosis. The clearance of these misfolded proteins by autophagy may be crucial to alleviate ER stress and maintain homeostasis. That is, autophagy may help HO8910 cells avoid B19-induced cell death.
However, there is no doubt that the limitations of B19 in ovarian cancer therapy still need for further study and investigation. For example, to confirm ER-stress is required for B19-induced apoptosis, ER stress inhibitors or specific siRNA should be used. In this study, only in vitro outcome is not enough, which needs to be further investigated on the human ovarian cancer nude mice model outcome. Moreover, misfolded proteins are usually turned over quickly or form aggregation, to give more evidences about the regulation of misfolded proteins by B19, SDS-soluble and insoluble fractions determination, or 35S-pulse-chase of newly syntheses proteins, should be performed. Nevertheless, the anti-cancer effect of B19 in ovarian cancer is feasible, to demonstrate the underlying mechanisms is necessary in the further studies.
In summary, we showed for the first time that treatment with a novel monocarbonyl analogue of curcumin, B19, can inhibit the growth of human ovarian cancer HO8910 cells in vitro. Our results indicated that B19 may induce ER stress, apoptosis and autophagy in human ovarian cancer HO8910 cells. The B19-induced activation of ER stress-mediated apoptosis may open up new therapeutic options in ovarian cancer treatment. Autophagy efficiently transports B19-induced misfolded proteins for degradation, allowing cells to avoid ER stress-mediated apoptosis. In addition, autophagy blockage increased the sensitivity of HO8910 cells to B19. We recommend that the use of B19 as an anticancer agent for ovarian tumors should be pursued because of its prominent effects and anticancer mechanisms, including the induction of ER stress, autophagy and apoptosis.
Acknowledgements
This work was supported by the National Natural Science Funding of China (30901819), Natural Science Funding of Zhejiang province (LY12H13002), and funds from the Zhejiang Provincial Project of Key Group (2010R50042), Key project of the Zhejiang traditional Chinese medicine administration (2012 ZZ012), the Science Foundation of Ninbo city (2012A610206).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Permuth-Wey J, Sellers TA. Epidemiology of ovarian cancer. Methods Mol Biol. 2009;472:413-37
2. McGuire WP, Hoskins WJ, Brady MF. et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334:1-6
3. Clarke R, Cook KL, Hu R. et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321-31
4. Duprez J, Jonas JC. Role of activating transcription factor 3 in low glucose- and thapsigargin-induced apoptosis in cultured mouse islets. Biochem Biophys Res Commun. 2011;415:294-9
5. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455-62
6. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133-49
7. Kouroku Y, Fujita E, Tanida I. et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230-9
8. Criollo A, Maiuri MC, Tasdemir E. et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029-39
9. Carew JS, Nawrocki ST, Cleveland JL. Modulating autophagy for therapeutic benefit. Autophagy. 2007;3:464-7
10. Dalby KN, Tekedereli I, Lopez-Berestein G. et al. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322-9
11. Strimpakos AS, Sharma RA. Curcumin: preventive and therapeutic properties in laboratory studies and clinical trials. Antioxid Redox Signal. 2008;10:511-45
12. Appiah-Opong R, de Esch I, Commandeur JN. et al. Structure-activity relationships for the inhibition of recombinant human cytochromes P450 by curcumin analogues. Eur J Med Chem. 2008;43:1621-31
13. Anand P, Kunnumakkara AB, Newman RA. et al. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807-18
14. Xiao J, Wang Y, Peng J. et al. A synthetic compound, 1,5-bis(2-methoxyphenyl)penta-1,4-dien-3-one (B63), induces apoptosis and activates endoplasmic reticulum stress in non-small cell lung cancer cells. Int J Cancer. 2012;131:1455-65
15. Wang Y, Xiao J, Zhou H. et al. A novel monocarbonyl analogue of curcumin, (1E,4E)-1,5-bis(2,3-dimethoxyphenyl)penta-1,4-dien-3-one, induced cancer cell H460 apoptosis via activation of endoplasmic reticulum stress signaling pathway. J Med Chem. 2011;54:3768-78
16. Zhao C, Yang J, Wang Y. et al. Synthesis of mono-carbonyl analogues of curcumin and their effects on inhibition of cytokine release in LPS-stimulated RAW 264.7 macrophages. Bioorg Med Chem. 2010;18:2388-93
17. Zhong J, Kong X, Zhang H. et al. Inhibition of CLIC4 enhances autophagy and triggers mitochondrial and ER stress-induced apoptosis in human glioma U251 cells under starvation. PLoS One. 2012;7:e39378
18. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381-9
19. Gonzalez-Polo RA, Niso-Santano M, Ortiz-Ortiz MA. et al. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol Sci. 2007;97:448-58
20. Yu H, Su J, Xu Y. et al. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer. 2011;47:1585-94
21. Pankiv S, Clausen TH, Lamark T. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
22. Bjorkoy G, Lamark T, Brech A. et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603-14
23. Singh AP, Senapati S, Ponnusamy MP. et al. Clinical potential of mucins in diagnosis, prognosis, and therapy of ovarian cancer. Lancet Oncol. 2008;9:1076-85
24. Perlmutter DH. Misfolded proteins in the endoplasmic reticulum. Lab Invest. 1999;79:623-38
25. Wang G, Yang ZQ, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. American journal of translational research. 2010;2:65-74
26. Liao PC, Tan SK, Lieu CH. et al. Involvement of endoplasmic reticulum in paclitaxel-induced apoptosis. J Cell Biochem. 2008;104:1509-23
27. Tiwary R, Yu W, Li J. et al. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS One. 2010;5:e11865
28. Hill DS, Martin S, Armstrong JL. et al. Combining the endoplasmic reticulum stress-inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin Cancer Res. 2009;15:1192-8
29. Pae HO, Jeong SO, Jeong GS. et al. Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem Biophys Res Commun. 2007;353:1040-5
30. Wu SH, Hang LW, Yang JS. et al. Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria-dependent pathways. Anticancer Res. 2010;30:2125-33
31. Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28-32
32. Armstrong JL, Flockhart R, Veal GJ. et al. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285:6091-100
33. Szegezdi E, Logue SE, Gorman AM. et al. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880-5
34. Schleicher SM, Moretti L, Varki V. et al. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist Updat. 2010;13:79-86
35. Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA. et al. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine A-induced cell death. Oncogene. 2012.
36. Yorimitsu T, Nair U, Yang Z. et al. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299-304
37. Selimovic D, Porzig BB, El-Khattouti A. et al. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal. 2013;25:308-18
38. Joubert PE, Werneke SW, de la Calle C. et al. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med. 2012;209:1029-47
39. Shrivastava A, Kuzontkoski PM, Groopman JE. et al. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther. 2011;10:1161-72
40. Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278:403-13
41. Wang Z, Zhang H, Xu X. et al. bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol Lett. 2012;212:137-46
42. Zhang HY, Zhang X, Wang ZG. et al. Exogenous Basic Fibroblast Growth Factor Inhibits ER Stress-Induced Apoptosis and Improves Recovery from Spinal Cord Injury. CNS Neurosci Ther. 2012.
43. Hundeshagen P, Hamacher-Brady A, Eils R. et al. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011;9:38
44. Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T. et al. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med. 2009;13:3644-54
45. Aoki H, Takada Y, Kondo S. et al. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72:29-39
Author contact
![]() Corresponding author: Department of Gynecology and Obstetrics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, 325035, China. Tel: +86 577 86699350; Fax: +86 577 86689983. E-mail:23813766com (JQ. Lv),
Corresponding author: Department of Gynecology and Obstetrics, The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, 325035, China. Tel: +86 577 86699350; Fax: +86 577 86689983. E-mail:23813766com (JQ. Lv),
Received 2012-12-14
Accepted 2013-8-2
Published 2013-8-14