Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(8):778-791. doi:10.7150/ijbs.6459 This issue Cite
Research Paper
Interaction of Crk with Myosin-1c Participates in Fibronectin-Induced Cell Spreading
Hyejin Oh1, Hwan Kim1, Baehyun Shin2, Kun Ho Lee3, Myeong Gu Yeo4 ![]() , Woo Keun Song1
, Woo Keun Song1 ![]()
1. Bio Imaging and Cell Dynamics Research Center,
2. Bio Remodeling and Gene Therapy Lab., School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, Korea,
3. Department of Marine Biology, Chosun University, Gwangju, 501-759, Korea
4. Department of Alternative Medicine, Nambu University, Gwangju 506-706, Korea
Received 2013-4-12; Accepted 2013-7-26; Published 2013-8-15
Abstract
We previously reported a novel interaction between v-Crk and myosin-1c, and demonstrated that this interaction is essential for cell migration, even in the absence of p130CAS. We here demonstrate a role for Crk-myosin-1c interaction in cell adhesion and spreading. Crk-knockout (Crk‑/‑) mouse embryo fibroblasts (MEFs) exhibited significantly decreased cell spreading and reduced Rac1 activity. A stroboscopic analysis of cell dynamics during cell spreading revealed that the cell-spreading deficiency in Crk‑/‑ MEFs was due to the short protrusion/retraction distances and long persistence times of membrane extensions. The low activity of Rac1 in Crk‑/‑ MEFs, which led to delayed cell spreading in these cells, is consistent with the observed defects in membrane dynamics. Reintroduction of v-Crk into Crk‑/‑ MEFs rescued these defects, restoring cell-spreading activity and membrane dynamics to Crk+/+ MEF levels, and normalizing Rac1 activity. Knockdown of myosin-1c by introduction of small interfering RNA resulted in a delay in cell spreading and reduced Rac1 activity to low levels, suggesting that myosin-1c also plays an essential role in cell adhesion and spreading. In addition, deletion of the v-Crk SH3 domain, which interacts with the myosin-1c tail, led to defects in cell spreading. Overexpression of the GFP-myosin-1c tail domain effectively inhibited the v-Crk-myosin-1c interaction and led to a slight decrease in cell spreading and cell surface area. Collectively, these findings suggest that the v-Crk-myosin-1c interaction, which modulates membrane dynamics by regulating Rac1 activity, is crucial for cell adhesion and spreading.
Keywords: v-Crk, myosin-1c, cell adhesion, cell spreading
Introduction
The cell membrane, which serves as a platform for the localization of various components that actively participate in membrane transport and organelle structure, undergoes dramatic changes through structural assembly and disassembly [1]. Dynamic changes in the cell membrane are prominent during cell adhesion and spreading in response to extracellular stimuli [2, 3]. Cell adhesion, accomplished by an interaction between the cell and the extracellular matrix (ECM), is followed by cell spreading; this is associated with extensive membrane deformation and leads to formation of a membrane protrusion called the lamellipodium [4]. Cell adhesion and spreading also activate cell adhesion components and cytoskeletal proteins, including FAK (focal adhesion kinase), Src, paxillin, p130CAS (Crk-associated substrate), vinculin, and other signaling molecules [5]. Activation of these molecules modulates the membrane dynamics that occur during cell adhesion and spreading [6, 7]. Thus, it is of interest to understand the mechanism by which these proteins are activated as well as the membrane dynamics that occur during cell adhesion and spreading.
Crk is an adaptor protein containing Src homology 2 (SH2) and 3 (SH3) domains. A number of proteins have been identified as Crk-binding proteins, including C3G (RapGEF), SOS1 (son of sevenless homolog 1), DOCK180 (180 kDa protein downstream of CRK), and growth factor receptors [8-13]. Association of these proteins with Crk enables membrane targeting of Crk and transduces various cellular signals. C3G, a Rap1 GEF (guanine nucleotide exchange factor) that contains a Crk-SH3-binding proline-rich motif, transduces signals from receptors to the nucleus via the Ras/Raf/MAPK (mitogen-activated protein kinase) pathway [14, 15]. The role of C3G in the cell-spreading process has been well characterized through rescue studies in C3G-knockout mouse embryo fibroblasts (MEFs). SOS1, a Ras GEF, binds through its PxxP motif (amino acids [a.a.] 1021-1034) to Crk/CrkL, and knockdown of SOS1 inhibits Crk-mediated transformation of NIH3T3 cells and migration of HEY (ovarian carcinoma) cells [16, 17]. DOCK180 enhances Crk/p130CAS complex formation and also positively regulates membrane spreading through Rac1 activation [18, 19]. Among Crk-binding proteins, p130CAS is a tyrosine-phosphorylated protein which is recognized by the Crk SH2 domain. This interaction enables Crk/p130CAS to move to the membrane edge to form focal adhesion complexes, which leads to membrane protrusion and cell migration [20, 21]. Loss of p130CAS interaction with Crk causes defects in the phosphorylation of focal adhesion proteins and leads to delays in cell spreading and migration [22, 23]. Crk knockdown by small (interfering) hairpin RNA (shRNA) significantly decreases cell movement in wound-healing and invasion assays [24]. Notably, Crk ablation in mice is embryonic lethal because of developmental defects, such as edema, hemorrhage, and cardiac defects. It has been suggested that these developmental defects are attributable to the loss of specific interactions, especially with p130CAS, that play a role in the regulation of actin dynamics during cell spreading/migration [25]. These reports suggest that Crk may modulate signals initiated by cell adhesion to the ECM and transfer them to adhesion components and/or cytoskeletal proteins involved in cell spreading.
In a previous study, we identified myosin-1c as a novel binding protein of v-Crk and showed that myosin-1c rescued the cell-migration-defective phenotype of p130CAS-knockout MEFs [26]. Myosin family proteins are actin-based motor proteins that have been well studied in muscle contraction, cellular motility processes, and actin-cytoskeleton regulation processes [27-29]. Many studies have demonstrated that myosin plays essential roles in regulating the formation and dynamics of actin-rich structures [30, 31]. Myosin-1c, in particular, is widely expressed in vertebrates and is enriched in perinuclear regions, membrane ruffles, and membrane marginal regions [32, 33]. Myosin-1c participates in the retention of lamellipodia and membrane protrusions in neuronal growth cone dynamics [34] and has been reported to function in cell-spreading processes, which require membrane expansion [35]. However, the precise mechanism by which myosin-1c contributes to cell adhesion and spreading is largely unknown.
In the current study, we confirmed that myosin-1c is a v-Crk binding protein and demonstrated that v-Crk is able to rescue the cell-spreading defects of Crk‑/‑ MEFs through interactions with myosin-1c. To further analyze the functional consequences of v-Crk-myosin-1c interactions, we investigated how these interactions affect fibronectin-induced cell adhesion and spreading as well as membrane dynamics. Here, we provide evidence that the interaction of v-Crk with myosin-1c is essential in cell adhesion and spreading and modulates the membrane dynamics that occur during these cellular processes.
Materials and Methods
Materials and chemicals
Rabbit anti-myosin-1c polyclonal antibody and mouse anti-Flag M2 monoclonal antibody were purchased from Sigma-Aldrich (St. Louis, MO). Mouse anti-Crk monoclonal antibody was purchased from BD Biosciences (Franklin Lakes, NJ). Alexa 488- and Alexa 594-conjugated secondary antibodies were purchased from Molecular Probes (Eugene, OR). Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich.
DNA constructs
v-Crk cDNA was cloned from pMEXneo/v-Crk into the pLHCX retroviral vector (Clontech Laboratories, Inc., Mountain View, CA). The following individual v-Crk domains were subcloned into the pLHCX vector using the indicated primer pairs: gag, 5'-GGC CGC GGC CGC ACC GAA GCC GTC AT-3' (sense) and 5'-CCT ATC GAT TAG GTT GTC GAA TGC CTT GTA GTC CCC CCG GTC CTC GGA GTC GAA CTG -3' (antisense); SH2-SH3 domain, 5'-GAG CGG CCG CTG GTA CTG GGG GCG G-3' (sense) and 5'-CCA TCG ATT AGG TTG TCG AAT GCC TTG TAG TCT TCA ACT TCC TCC TGC CTG AGG ATA ACG-3' (antisense); and SH3 domain, 5'-AGG CGG CCG CTA TGT GCG AGC TCT C-3' (sense) and 5'-CCA TCG ATT AGG TTG TCG AAT GCC TTG TAG TCT TCA ACT TCC TCC TGC CTG AGG ATA ACG-3' (antisense). The Flag sequence was inserted into the C-terminal region for tagging individual domains with the Flag epitope. For GST pull-down assays, the myosin-1c motor domain, IQ/tail domain, and tail domain only were subcloned into the pGEX4T-1 vector. All plasmid constructs were sequenced to confirm the fidelity of cloning steps.
Generation of stable cell lines
Crk-knockout (Crk‑/‑) and wild-type (Crk+/+) MEFs, kindly provided by Dr. Curran (The Children's Hospital of Philadelphia, Philadelphia, PA), were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg/ml penicillin/streptomycin. pLHCX expression plasmids for full-length v-Crk and its domains were transfected into the retroviral packaging cell line, Phoenix-ECO (Nolan Laboratory, Stanford University, Stanford, CA), using calcium phosphate precipitation. Retrovirus-containing cell supernatants were then used to infect the Crk‑/‑ cell line. Beginning 2 days after viral infection, cells were selected by growing for 1 week in the presence of 300 μg/ml of hygromycin B (Calbiochem, San Diego, CA), after which clones were isolated and confirmed by immunoblot analysis.
Myosin-1c siRNA design and transfection
The following myosin-1c siRNAs, synthesized by Dharmacon (Thermo Scientific, Cheshire, UK) were designed to target the indicated cDNA sequences [36]: siRNA #1, 5'-AAG GCG TTG TAC AGC CGG ACA TT-3'; and siRNA #2, 5'-AAG CTT CCA GAC AGG GAT CCA TG-3'. A scrambled sequence (5'-CAG TCG CGT TTG CGA CTG G-3') was used as a control. Crk‑/‑ and Crk+/+ MEFs (2.0 × 105 cells/ml) were plated and transfected with either 5 nmol of scrambled or 20 nmol of siRNA #1 and siRNA #2 using the Dharmafect 3 reagent (Dharmacon RNA Technologies). siRNA-transfected cells were visualized by cotransfecting with siGLO-TRITC. After transfection, cells were incubated for 72 hours and analyzed for myosin-1c expression by immunoblotting and immunocytochemistry.
Immunoprecipitation and immunoblotting
Crk‑/‑, Crk+/+, and v-Crk-transfected Crk‑/‑ (Crk‑/‑/vCrk) MEFs were plated on fibronectin (10 μg/ml)-coated coverslips, incubated for 1 hour, and lysed by incubating on ice for 10 minutes in lysis buffer (1% Triton X-100, 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 10 mM NaF, 1 mM sodium pyrophosphate, 2 mM Na3VO4) supplemented with a protease-inhibitor cocktail. The protein concentration was determined using the Bradford method (Bio-Rad Laboratories, Berkeley, CA). For immunoprecipitation assays, cell lysates were incubated with anti-Crk antibody and protein-G Sepharose (GE Healthcare, Uppsala, Sweden) for 2 hours at 4°C and then resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After transferring proteins to PVDF (polyvinylidene difluoride) membranes and blocking with 5% non-fat dry milk, blots were incubated with the indicated primary and secondary antibodies. Immunoreactive proteins were detected by enhanced chemiluminescence (Pierce Chemical, Rockford, IL) using a LAS4000 mini biomolecular imager (GE Healthcare).
Stroboscopic analysis of cell dynamics assay
For kymography analyses, time-lapse images were obtained with an Olympus IX81 using a 40X (0.60 NA) objective. Cells in microscopic recording media (containing 5 μM mitomycin C in phenol red-free DMEM; Gibco (Invitrogen, Carlsbad, CA, USA) were plated onto fibronectin-coated coverslips and allowed 10 minutes to attach. Thereafter, images were acquired every 10 seconds for 5 minutes using an iXon EM DU-897 illuminated EMCCD camera mounted on an Olympus IX81 microscope. Kymographs were generated from 10-pixel-wide (1.06 μm) regions in the direction of individual lamellipodia protrusions. Kymographs were produced using Metamorph (Universal Imaging Corp., Downingtown, PA) and analyzed using SigmaPlot software.
Immunocytochemistry
For immunocytochemistry assays, cells were plated on fibronectin-coated coverslips and incubated for the indicated time at 37°C. Thereafter, cells were fixed and permeabilized by incubating sequentially in 4% paraformaldehyde/phosphate-buffered saline (PBS) and 0.5% Triton X-100/PBS for 10 and 5 minutes, respectively. The actin cytoskeleton was visualized with Alexa 555- or 594-conjugated phalloidin (Invitrogen). The coverslips were then mounted with PermaFluor Aqueous Mounting Medium (Thermo Scientific, Cheshire, UK). Images were acquired with an Olympus FV1000 confocal microscope and analyzed with Metamorph software.
GST pull-down assay/Rac1 activation assay
Myosin-1c domains subcloned into pGEX4T-1 were transformed into the Escherichia coli strain BL21, and expression of recombinant proteins was induced by incubating at 18°C overnight with 0.5 mM IPTG (isopropylthio-β-galactoside). The cells were sonicated in lysis buffer containing sarkosyl and neutralized with Triton X-100. After centrifugation, soluble fractions were incubated with GST-Sepharose 4B (Incospharm Kribbs, Daejeon, Korea) and washed with lysis buffer. Recombinant myosin-1c variants-bound beads were incubated with Crk+/+ MEF lysates for 30 minutes at 4°C, after which samples were separated by SDS-PAGE. Rac1 activation assays were performed using a Rac1 activation assay kit (Millipore, Billerica, MA) as described by the manufacturer.
In vitro binding assay
Recombinant proteins of GST-tagged myosin-1c variants were expressed in E. Coli (BL21) by IPTG induction. Bacteria pellets were suspended in sarkosyl-containing STE buffer (150 mM NaCl, 20 mM Tris-HCl pH 7.4, 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and lysed using a French press. Sarkosyl in the supernatant was neutralized by adding Triton X-100, and proteins were immobilized on glutathione sepharose beads. His-tagged v-Crk-SH3 was induced with IPTG and immobilized on Ni-NTA beads according to the manufacturer's procedure. His-v-Crk-SH3 was eluted from Ni-NTA beads using 200 mM imidazole and dialyzed against 20 mM Tris-HCl (pH 7.4) and 150 mM NaCl. One nanomole of v-Crk-SH3 (35 μg) was reacted with 1 nmol of glutathione-bead-immobilized myosin-1c variants. After extensive washing with binding buffer, the resulting products were separated by SDS-PAGE.
Results
Crk plays an essential role in cell spreading on fibronectin
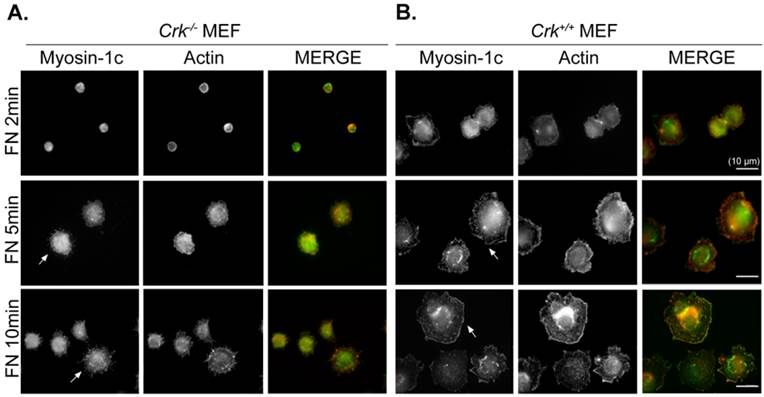
We previously suggested that the interaction of v-Crk with myosin-1c is involved in fibronectin-induced cell migration in p130Cas-knockout cells [26]. To verify the role of Crk-myosin-1c interactions in cell spreading, we examined cell spreading at early times. Cells were incubated on fibronectin (10 μg/ml)-coated coverslips, and then the cell boundary was visualized with phalloidin and myosin-1c staining. As shown in Fig. 1, Crk+/+ MEFs began to spread within 2 minutes and displayed numerous ruffles at the cell boundary after 5 minutes (Fig. 1A). However, Crk‑/‑ MEFs had not spread at all by 5 minutes, and spreading was barely detectable after 10 minutes (Fig. 1B). Instead, filopodia, but no ruffle-like structures, were detected at the cell boundary of Crk‑/‑ MEFs. Myosin-1c was detected at both the membrane edge (especially at membrane ruffles) and in the cytosol of Crk+/+ MEFs, but was only detected in the cytosol of Crk‑/‑ MEFs.
Crk is essential for cell spreading and myosin-1c localization at the membrane edge. Crk‑/‑ and Crk+/+ MEFs were suspended in serum-free DMEM for 1 hour, then plated on fibronectin (FN; 10 μg/ml)-coated coverslips for the indicated times. Cells were stained with anti-myosin-1c antibody and then with Alexa-488 secondary antibody and Alexa-594-phalloidin. Arrows indicate myosin-1c localization at membrane edge areas. Scale bar: 10 μm
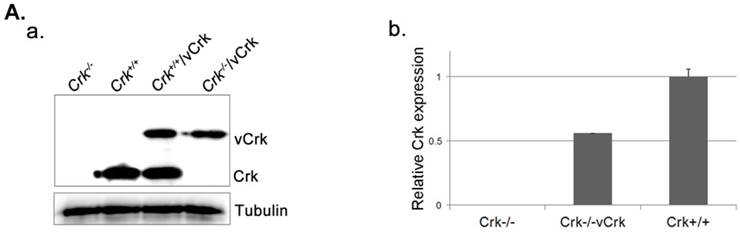
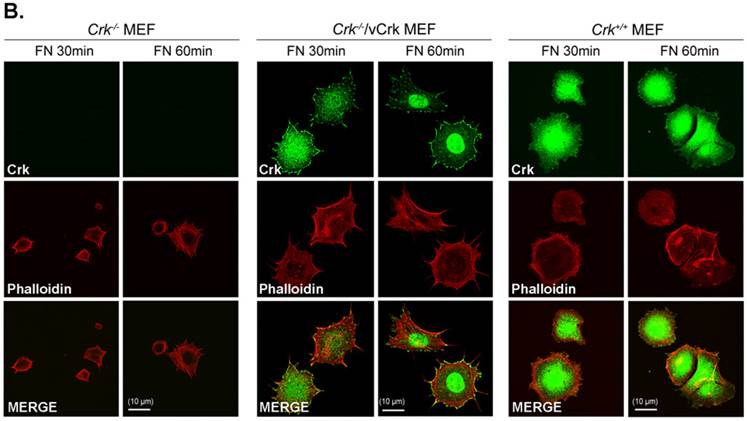
To better understand the role of Crk in cell spreading, we generated cells stably expressing v-Crk. v‑Crk expression was confirmed by immunoblot analysis using an anti-Crk antibody (Fig. 2A). As shown in Fig. 2B, Crk+/+ MEFs exhibited a well-spread, dish-like morphology with smooth curvatures at the cell periphery. In contrast, Crk‑/‑ MEFs cultured on fibronectin-coated coverslips exhibited spherical and irregular shapes until 30 minutes after plating, and showed an atypical morphology with more numerous membrane filopodia after 60 minutes. Notably, introduction of v-Crk into Crk‑/‑ MEFs (Crk‑/‑/vCrk MEFs) rescued this defective cell-spreading phenotype, restoring the dish-like spreading pattern containing filopodia observed in Crk+/+ MEFs. Crk staining was observed in the membrane boundary of both Crk+/+ and Crk‑/‑/vCrk MEFs. These results indicate that Crk plays an essential role in cell spreading on fibronectin.
Expression of v-Crk rescues the cell-spreading defect in Crk-/- MEFs. A. a) Crk‑/‑ and Crk+/+ MEFs were stably transfected with v-Crk plasmids using a retroviral transfer system with subsequent hygromycin B (300 μg/ml) selection. Cells expressing v-Crk and cellular Crk were verified by immunoblotting using an anti-Crk antibody. b) Histogram indicates v-Crk expression in Crk‑/‑/vCrk MEFs and cellular Crk in Crk+/+ MEFs for comparison of relative Crk and v-Crk expression levels. Data represent means ± SEM of four separate experiments. B. Crk‑/‑, Crk‑/‑/vCrk, or Crk+/+ MEFs were plated on fibronectin (FN; 10 μg/ml)-coated coverslips and incubated for 30 or 60 minutes. Cells were first stained with mouse anti-Crk antibody and then with Alexa-488 conjugated mouse secondary antibody and Alexa-555-phalloidin. Scale bar: 10 μm.
Crk modulates membrane dynamics
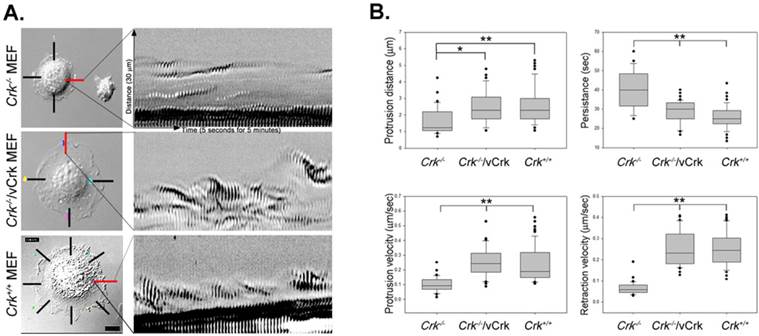
We next analyzed the role of Crk in membrane dynamics during cell spreading. These kinetic analyses employed kymography, which extracts intensity values along a defined line region in each image of a time-lapse series. Images were then pasted side-by-side to generate a composite image of object movement over time. The resulting kymographs represent the dynamics of membrane activity at a single point along the cell perimeter. A series of repeated patterns was detected in kymographs, representing a cycle of protrusion and retraction (Fig. 3A). A regular repeated pattern was observed in Crk+/+ MEFs, but not in Crk‑/‑ MEFs. Stable expression of v-Crk in Crk‑/‑ MEFs largely restored the pattern of Crk+/+ MEFs, replacing the irregular pattern observed in Crk‑/‑ MEFs. This analysis also yielded quantitative measurements of membrane dynamic parameters during cell spreading (Fig. 3B). Expressed as means ± SD, the values for persistence time were 40.30 ± 1.7, 28.62 ± 0.88 and 25.82 ± 0.66 seconds for Crk‑/‑, Crk‑/‑/vCrk and Crk+/+ MEFs, respectively; the corresponding values for velocity of protrusion were 0.10 ± 0.05, 0.26 ± 0.11 and 0.24 ± 0.01 μm/s, and those for velocity of retraction were 0.07 ± 0.03, 0.27 ± 0.01 and 0.27 ± 0.01 μm/s. Kymographs of Crk‑/‑ MEFs appeared as “dunes”, reflecting slow protrusion over time and infrequent withdrawal. The short protrusion/retraction distance and long persistence time of Crk‑/‑ MEFs led to a low velocity of protrusion and retraction (Fig. 3B). Over time, this behavior resulted in less membrane extension in Crk‑/‑ MEFs than in Crk+/+ MEFs, consistent with the cell-spreading defect observed in Crk‑/‑ MEFs. Interestingly, kymographs of Crk‑/‑/vCrk MEFs displayed long protrusion distances and short persistence times, indicating that restoration of Crk function significant increased protrusion velocity and led to frequent lamellipodia protrusion and ruffle formation. In fact, the membrane dynamics and overall cell-spreading behavior of Crk‑/‑/vCrk MEFs were comparable to those of control, wild-type MEFs. Taken together, these results indicate that Crk modulates membrane dynamics and thereby induces cell spreading.
Membrane dynamics and Rac1 activity is restored by v-Crk expression. A. MEFs expressing various Crk variants were incubated on fibronectin (FN; 10 μg/ml)-coated coverslips for 5 minutes and imaged every 5 seconds for 5 minutes. A 1.06-μm-wide box (region of interest) was drawn in the direction of cell protrusion; the montage was created by spreading out 61 images of the region of interest. At least 10 kymographs were generated for each MEF. The red line indicates the portion used to create kymographs displayed in the figure. B. Persistence, protrusion/retraction distance, and protrusion/retraction time were measured and calculated from kymographs. Graphs were generated using SigmaPlot software. *P < 0.001; **P < 0.005. C. For Rac1 activation assays, lysates from Crk variant-expressing MEFs were incubated with GST-PBD beads at 4°C. Bead-bound proteins were eluted with SDS buffer and boiled prior to SDS-PAGE. D. Active Rac1 was quantified using Multi-Gauge software.
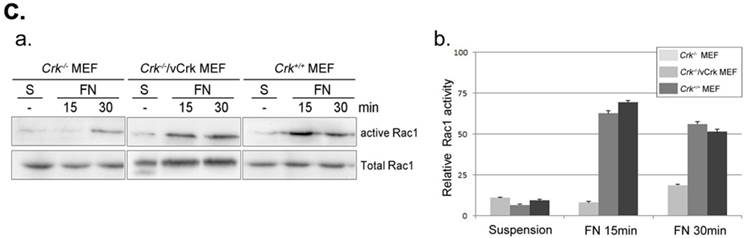
The small GTPase Rac1 is a predominant effector of cellular responses, including cytoskeletal rearrangement and cell spreading and migration, through regulation of membrane dynamics [37, 38]. We previously reported that v-Crk enhances membrane dynamics through Rac1 activation, leading to recovery of cell migration in p130CAS-knockout MEFs [39, 26]. To examine whether Crk induces ruffle formation via Rac1 activation, we performed Rac1 activation assays (Fig. 3C). Activated Rac1 was observed within 15 minutes of fibronectin stimulation in Crk+/+ MEFs, whereas Rac1 in Crk‑/‑ MEFs was not fully activated even after 30 minutes. Interestingly, Rac1 activation was restored almost to Crk+/+ levels in Crk‑/‑ MEFs stably expressing v‑Crk. This observation is consistent with our previous kymography results, in which cell spreading and membrane dynamics were restored by v-Crk expression.
Myosin-1c knockdown affects cell spreading and Rac1 activity
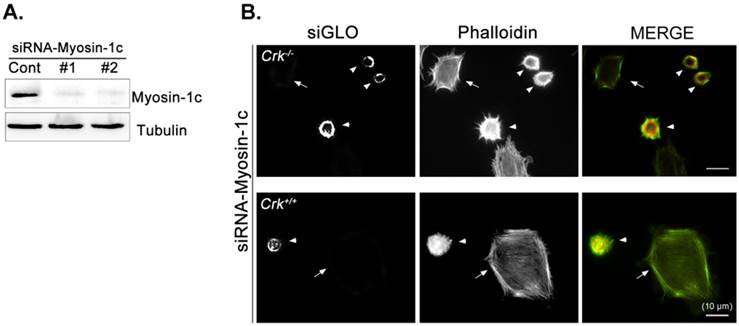
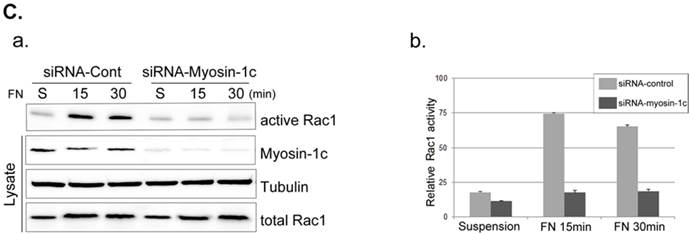
Next, we focused on the roles of myosin-1c in cell spreading, examining the effect of myosin-1c knockdown on fibronectin-stimulated cells. Cells were transfected with siRNA targeting myosin-1c (or control siRNA) together with TRITC-siGLO (to visualize siRNA-transfected cells); knockdown of myosin-1c was confirmed by immunoblot analysis (Fig. 4A). Cell spreading was clearly detected in Crk+/+ MEFs, but was less evident in Crk‑/‑ MEFs. However, Crk+/+ MEFs transfected with siRNA-myosin-1c (Fig. 4B, arrowhead) displayed dramatically reduced cell spreading compared to Crk+/+ MEFs (Fig. 4B, arrow), indicating that myosin-1c is essential for cell spreading. To test the involvement of myosin-1c in Rac1 activation, we performed Rac1 activation assays in wild-type and myosin-1c-knockdown MEFs (Fig. 4C). Wild-type MEFs exhibited full activation of Rac1 within 15 minutes of fibronectin stimulation, whereas Rac1 was not activated even after 30 minutes in myosin-1c-knockdown MEFs. The loss of Rac1 activity in myosin-1c-knockdown MEFs led to a significant cell-spreading defect, suggesting that both myosin-1c and Crk are required for cell spreading.
Knockdown of myosin-1c causes a cell-spreading defect. A. Crk+/+ MEFs were transfected with siRNA-control (Cont; Scrambled) and two-types of siRNA-myosin-1c (#1 and #2). Cell lysates were blotted with an anti-myosin-1c antibody to determine myosin-1c expression levels. B. Crk+/+ cells transfected with siRNA-control or siRNA-myosin-1c together with siGLO-TRITC (to visualize transfected cells) were plated on fibronectin (FN; 10 μg/ml)-coated coverslips, incubated for 10 minutes, and stained with Alexa-647 phalloidin (to detect actin). Scale bar: 10 μm. C. Crk+/+ cells were transfected with siRNA-myosin-1c #1, plated on FN-coated coverslips, and incubated for the indicated times. Cell lysates were then incubated with GST-PBD beads at 4°C. Active Rac1 bound to GST-PBD beads was eluted and detected with an anti-Rac1 antibody. Active Rac1 was quantified using Multi-Gauge software. Arrowhead: cells transfected with siRNA-myosin-1c; Arrow, non-transfected cells.
Crk and myosin-1c interaction participates in cell spreading
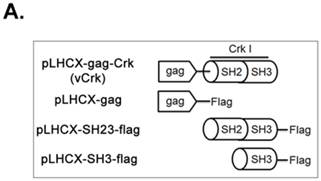
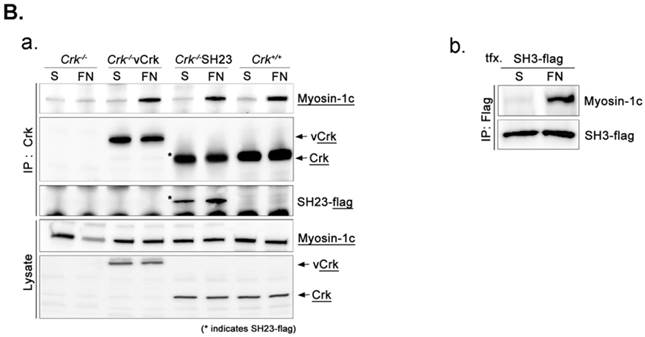
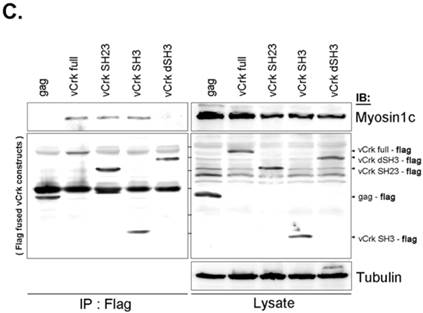
To assess the implications of Crk-myosin-1c interactions during cell spreading, we sought to determine which domains of Crk and myosin-1c are responsible for their interaction. Cells were transfected with the v-Crk variants, gag-Crk (v-Crk), SH2SH3-Flag or SH3-Flag (constructed from v‑Crk cDNA and cloned into the expression vector pLHCX; Fig. 5A), and subjected to co-immunoprecipitation and immunoblot analysis. Endogenous myosin-1c co-immunoprecipitated with transfected v-Crk, SH2SH3-Flag or SH3-Flag, indicating an interaction between myosin-1c and these Crk variants; notably, cells expressing SH3-Flag strongly interacted with myosin-1c (Fig. 5B), and deletion of v-Crk-SH3 abolished the interaction with myosin-1c (Fig. 5C), indicating that the v-Crk SH3 domain is essential for this interaction.
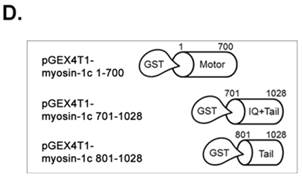
SH3 domain of v-Crk binds to the myosin-1c tail domain (a.a. 801-1028). A. Diagram shows Crk variant constructs used for domain mapping. Four variants were cloned into the pLHCX expression vector for retroviral packaging. B. Identification of the myosin-1c binding domain of v-Crk. Cells were suspended in serum-free media for 1 hour and plated on fibronectin (FN; 10 μg/ml)-coated coverslips for 1 hour. Lysates of transfected cells were immunoprecipitated with anti-Crk (a) or anti-flag (b) antibodies and blotted with an anti-myosin-1c antibody. C. The SH3 domain is required for v-Crk and myosin-1c interaction. Crk‑/‑ MEFs were transfected with Flag-tagged constructs of v-Crk variants, including v-Crk dSH3, and lysates were immunoprecipitated with anti-flag antibody. Anti-myosin-1c antibody was used for immunoblot analysis. D. GST-myosin-1c recombinant plasmids were constructed. E. Three GST-myosin-1c domains were expressed and purified in vitro and incubated with lysates of Crk+/+ MEFs for GST pull-down assays. Crk bound to GST-myosin-1c domains was detected with an anti-Crk antibody. The bottom panel displays GST-myosin-1c variant proteins immobilized on beads. Cell lysate control is shown at left in the upper panel. F. Recombinant proteins of GST-myosin-1c variants (full-length, a.a. 1-1028; [dTail], a.a. 1-802; [dPxxP], a.a. 1-764 linked to 803-1028) or vector control were expressed and purified using glutathione-Sepharose beads. The protein expression of the constructs was verified by Coomassie Blue staining. Immobilized GST-myosin-1c variants were mixed with recombinant His6-v-Crk-SH3 protein for 2 hours at 4°C. Bound v-Crk-SH3 was detected with anti-His monoclonal antibody.
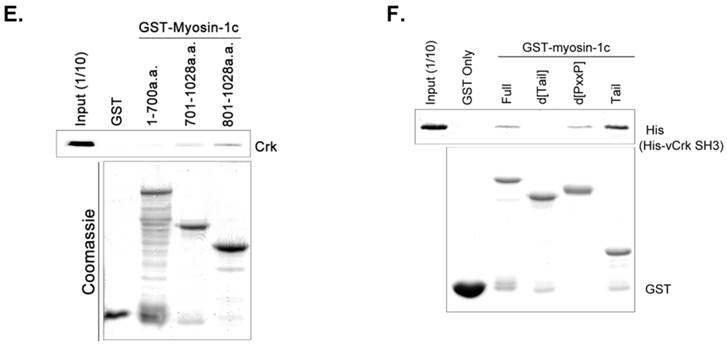
To further investigate the Crk-SH3-myosin-1c interaction, we carried out GST pull-down assays and in vitro binding assays with various myosin-1c constructs (Fig. 5E and F). As shown in Fig. 5E, recombinant GST-myosin-1c (a.a. 701-1028), containing the IQ and tail domains, interacted with Crk. Moreover, GST-myosin-1c (a.a. 801-1028), containing only the tail domain, strongly interacted with Crk. Interestingly, the tail domain (a.a. 801-1028) of myosin-1c (1028 a.a.; Uniprot ID: Q9WTI7-2) lacks well-known SH3-binding motifs, such as PxxP, RxxK, RPLPVAP, PPPALPPKKR, RKGDYASY and WxxQF; in contrast, the IQ domain contains two PxxP motifs (765-PRCP-768 and 799-PTPP-802). To determine whether these two PxxP motifs are responsible for Crk binding, we constructed a myosin-1c deletion mutant lacking these motifs (d[PxxP]; deletion of a.a. 765-802) and tested its binding with Crk. Myosin-1c-d[PxxP] retained Crk binding, suggesting that these PxxP motifs do not participate in binding to Crk (Fig. 5F). Collectively, these results suggest that myosin-1c interacts primarily through its tail domain (a.a. 803-1028) with the SH3 domain of Crk.
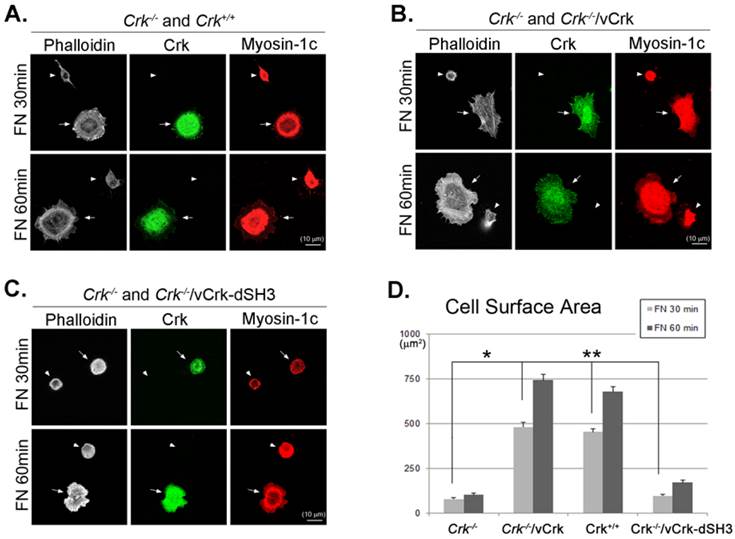
To further verify the importance of Crk-myosin-1c interaction during fibronectin-induced cell spreading, we constructed a v-Crk mutant lacking the SH3 domain responsible for myosin-1c binding (Crk‑/‑/vCrk-dSH3) and examined its effects on cell spreading. Crk‑/‑ MEFs were mixed and co-incubated with Crk+/+, Crk‑/‑/vCrk or Crk‑/‑/vCrk-dSH3 MEFs on fibronectin-coated coverslips for the indicated times, and cell-spreading area was measured. As shown in Fig. 6, the cell-spreading area of Crk+/+ MEFs was much larger than that of Crk‑/‑ MEFs (Fig. 6A). As expected, Crk‑/‑/vCrk MEFs displayed a level of cell spreading similar to that of Crk+/+ MEFs (Fig. 6B). However, Crk‑/‑/vCrk-dSH3 MEFs, lacking the myosin-1c binding domain of v-Crk, exhibited a defect in cell spreading that was comparable to that of Crk‑/‑ MEFs (Fig. 6C).
The interaction between v-Crk and myosin-1c is important for cell spreading. Crk‑/‑ MEFs (arrowheads) co-cultured with Crk+/+ MEFs (arrows) (A), Crk‑/‑ MEFs (arrowheads) transfected with v-Crk (arrows) (B), or Crk‑/‑ MEFs (arrowheads) transfected with v-Crk-dSH3 (arrows) (C) were trypsinized, suspended in serum-free media for 1 hour, plated on fibronectin (FN; 10 μg/ml)-coated coverslips, and incubated for 30 minutes (FN 30) or 60 minutes (FN 60). Cells were stained with anti-Crk and anti-myosin-1c antibodies and Alexa-647 phalloidin. Scale bar: 10 μm. D. Cell surface areas of Crk‑/‑, Crk‑/‑/vCrk, and Crk+/+ MEFs were analyzed and compared with those of Crk‑/‑/vCrk-dSH3 MEFs using Metamorph software. *P < 0.001, **P < 0.0005. E-G. Representative experiment (of four) showing Crk+/+ MEFs transfected with the indicated GFP-tagged myosin-1c constructs. Arrows: MEFs transfected with GFP-myosin-1c constructs. Arrowheads: non-transfected MEFs. Scale bar: 10 μm H. Cell surface area was measured in more than 60 cells. Results are presented as means ± SEM (error bars). I. Crk+/+ MEFs were transfected with different amounts of GFP-myosin-1c tail construct, as indicated in the figure. Endogenous Crk was immunoprecipitated with anti-Crk antibody, and bound myosin-1c was detected with anti-myosin-1c antibody. *P < 0.001, **P < 0.0005.
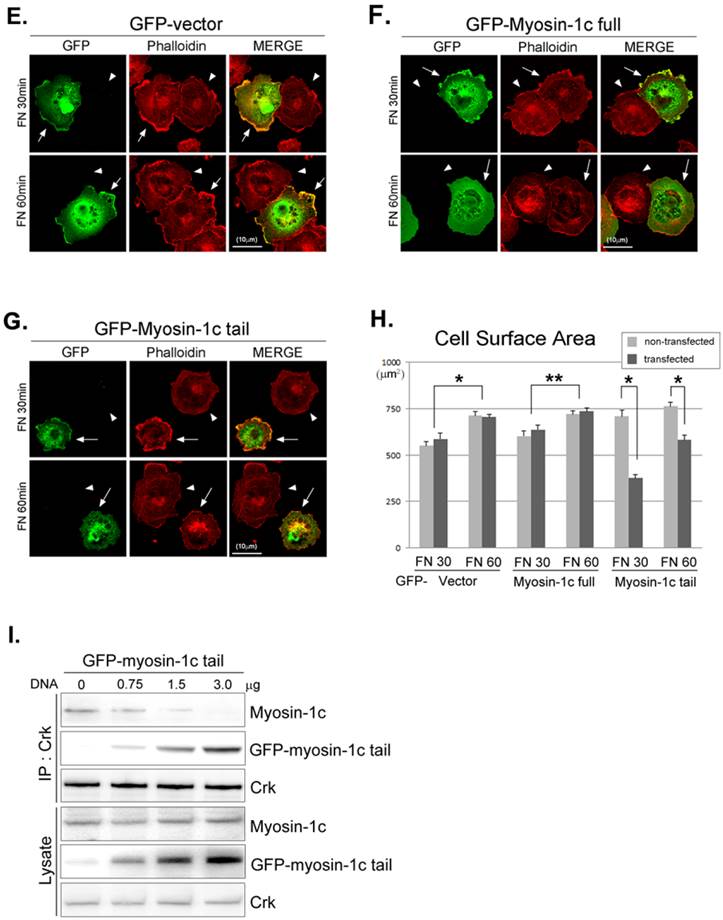
There are many known Crk-SH3-interacting proteins that mediate various cellular events. The SH3-deletion mutant of Crk is unable to bind Crk-SH3-interacting proteins, including DOCK180, C3G and SOS1, and the inability to bind these proteins may in turn cause the cell-spreading defect. To test this possibility, we ectopically expressed the GFP-myosin-1c tail domain to specifically inhibit the interaction between v-Crk and myosin-1c (Fig. 6E-H) and examined cell spreading. Cell spreading in Crk+/+MEFs transfected with GFP vector or full-length GFP-myosin-1c was not significantly different compared with that in non-transfected cells (Fig. 6E and F). In contrast, cells transfected with GFP-myosin-1c tail domain exhibited slightly diminished spreading and decreased cell surface area compared with non-transfected MEFs (Fig. 6G and H). In a parallel experiment, we confirmed that overexpression of the GFP-myosin-1c tail effectively inhibited endogenous Crk-myosin-1c interactions (Fig. 6I). These results indicate that the interaction between the v-Crk SH3 domain and myosin-1c tail participates in fibronectin-induced MEF cell spreading.
Discussion
In a previous study, we demonstrated that the interaction of v-Crk with the novel binding partner, myosin-1c, was essential for cell migration and showed that this interaction was able to recover the cell-migration defect of p130CAS-knockout MEFs [26]. We here investigated the role of v-Crk-myosin-1c interactions in the ECM-mediated processes of cell adhesion and spreading.
Because Crk ablation in mice is embryonic lethal at late stages of development [25], we used MEFs derived from Crk‑/‑ mice as an experimental model. Notably, the cellular behavior of Crk‑/‑ MEFs had not been previously studied. Crk‑/‑ MEFs exhibited defects in cell spreading and membrane ruffle formation. In kymograph analyses, Crk‑/‑ MEFs exhibited distinctive alterations in membrane dynamic parameters, showing shortened protrusion/retraction distances and a longer persistence time compared to Crk+/+ MEFs. The defects in spreading and membrane ruffle formation of Crk‑/‑ MEFs appeared as a “dunes-like” pattern in kymograph analyses reflecting overall membrane dynamics, suggesting that Crk plays a crucial role in modulating membrane dynamics.
Crk, a signaling adaptor protein, integrates signals from a wide variety of sources, including growth factors and ECM molecules [40, 41]. Crk, which lacks catalytic domains, has SH2 and SH3 domains that interact with many phosphotyrosine-containing proteins; these interactions enable Crk to modulate a variety of signaling pathways [42-44]. We previously reported that Crk coupling to p130CAS, and assembly of a p130CAS/Crk complex serves as a molecular switch for cell migration. Upon cell adhesion or stimulation with growth factors, Crk/p130CAS complex formation [26] and/or interaction of Crk with tyrosine-phosphorylated paxillin is necessary for phosphorylation of FAK (Tyr-397), which in turn facilitates cellular processes such as cell adhesion, spreading, and migration. [45, 46, 43]. Crk‑/‑ MEFs exhibit a defect in integrin-mediated cellular events, including haptotactic cell motility and FAK autophosphorylation during cell migration [47]. Thus, Crk interactions with cellular proteins enable Crk to transmit extracellular stimuli to intracellular proteins and play important roles during cell adhesion, spreading, and migration.
We demonstrated that the Crk SH3 domain specifically interacts with the myosin-1c tail domain. The SH3 domain serves as an adaptor to link tyrosine kinases to specific target proteins. Several reports have demonstrated that myosin-1c localizes to the plasma membrane and participates in the regulation of membrane dynamics. For example, myosin-1c serves vesicular trafficking functions, such as transportation of glucose transporter (GLUT) to lipid rafts, where cytoskeletal proteins are activated [48]. Myosin-1c is also able to propel the F-actin filament at the leading edge of motile cells to induce membrane protrusion through binding to phosphoinositides [49]. In accord with these reports, we found that deletion of the Crk SH3 domain, which directly interacts with myosin-1c, led to a defect in cell spreading in which both Crk and myosin-1c failed to translocate to membrane edges. This suggests that Crk is essential for myosin-1c translocation at the leading edge of motile cells, and v-Crk-myosin-1c interactions at the membrane edge are involved in membrane dynamics that lead to cell spreading. Notably, Crk is able to interact with several proteins through its SH3 domain. For example, DOCK180 (dedicator of cytokinesis) activates Rac1 and stimulates cell spreading through formation of a complex with CrkII and p130CAS during integrin signaling [12, 18]. c-Abl in association with Crk stimulates lamellipodia formation, which, in turn, leads to cell spreading [50]. Thus, the interaction of Crk with these proteins as well as myosin-1c may be essential processes in cell spreading.
Actin cytoskeletal organization in Crk‑/‑ MEFs was quite different from that in Crk+/+ MEFs (Figs. 1 and 2). Crk+/+ MEFs displayed a ring-shaped actin cytoskeleton just beneath membrane boundaries, whereas Crk‑/‑ MEFs possessed an actin cytoskeleton with much more of a stress-fiber pattern. In addition, Crk‑/‑ MEFs displayed numerous filopodia and a small spreading area, and the gaps between filopodia remained unfilled by membrane ruffles, even with long incubation times. In contrast, Crk+/+ MEFs showed both numerous filopodia and ruffles at very early cell spreading times and filling of gaps between filopodia by membrane ruffles. It is well known that actin cytoskeletal organization is regulated by members of the small GTPase family, such as Rac1, RhoA, and Cdc42 [51, 52]. Thus, the low Rac1 activity detected in Crk‑/‑ MEFs may be responsible for the decrease in membrane ruffles in gaps between filopodia, suggesting that both Crk and myosin-1c are essential for Rac1 activation, which, in turn, is required for membrane ruffle formation during cell adhesion and spreading.
In summary, we demonstrated that the v-Crk SH3 domain interacts with the myosin-1c tail, and showed that the Crk/myosin-1c complex regulates membrane dynamics, such as those involving Rac1 activity, that are crucial for initiation of cell adhesion and spreading. Ongoing studies to define the functional roles of the Crk/myosin-1c complex are expected to provide additional insight into the molecular mechanisms that govern cell spreading.
Acknowledgements
This research was supported by the Bio Imaging and Cell Dynamics Research Center (2007-0056492) and the Converging Research Center Program (2012K001361) funded by the Ministry of Education, Science and Technology. Kun Ho Lee (NRF-2011-0017009) was granted by National Research Foundation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ryan G.L, Watanabe N, Vavylonis D. A review of models of fluctuating protrusion and retraction patterns at the leading edge of motile cells. Cytoskeleton (Hoboken). 2012;69:195-206
2. Hagel M, George E.L, Kim A, Tamimi R, Opitz S.L, Turner C.E, Imamoto A, Thomas S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol Cell Biol. 2002;22:901-915
3. Yamazaki D, Fujiwara T, Suetsugu S, Takenawa T. A novel function of WAVE in lamellipodia: WAVE1 is required for stabilization of lamellipodial protrusions during cell spreading. Genes Cells. 2005;10:381-392
4. Huttenlocher A. and Horwitz A.R. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074
5. Ballestrem C, Erez N, Kirchner J, Kam Z, Bershadsky A, Geiger B. Molecular mapping of tyrosine-phosphorylated proteins in focal adhesions using fluorescence resonance energy transfer. Journal of Cell Science. 2006;119:866-875
6. Vuori K. Integrin signaling: tyrosine phosphorylation events in focal adhesions. J Membr Biol. 1998;165:191-199
7. Juliano R.L, Reddig P, Alahari S, Edin M, Howe A, Aplin A. Integrin regulation of cell signalling and motility. Biochem Soc Trans. 2004;32:443-446
8. Mayer B.J, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272-275
9. Birge R.B, Fajardo J.E, Mayer B.J, Hanafusa H. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J Biol Chem. 1992;267:10588-10595
10. Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, Matuoka K, Takenawa T, Kurata T, Nagashima K. et al. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci U S A. 1994;91:3443-3447
11. Feller S.M, Knudsen B, Hanafusa H. Cellular proteins binding to the first Src homology 3 (SH3) domain of the proto-oncogene product c-Crk indicate Crk-specific signaling pathways. Oncogene. 1995;10:1465-1473
12. Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770-1776
13. Smit L, van der Horst G, Borst J. Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, Crk, and Crk-L adaptors. J Biol Chem. 1996;271:8564-8569
14. Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, Kaibuchi K, Matsui H, Hatase O, Takahashi H. et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15:6746-6753
15. Okino K, Nagai H, Nakayama H, Doi D, Yoneyama K, Konishi H, Takeshita T. Inactivation of Crk SH3 domain-binding guanine nucleotide-releasing factor (C3G) in cervical squamous cell carcinoma. Int J Gynecol Cancer. 2006;16:763-771
16. Chen H, Wu X, Pan Z.K, Huang S. Integrity of SOS1/EPS8/ABI1 tri-complex determines ovarian cancer metastasis. Cancer Res. 2010;70:9979-9990
17. Zheng J, Machida K, Antoku S, Ng K.Y, Claffey K.P, Mayer B.J. Proteins that bind the Src homology 3 domain of CrkI have distinct roles in Crk transformation. Oncogene. 2010;29:6378-6389
18. Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12:3331-3336
19. Kiyokawa E, Hashimoto Y, Kurata T, Sugimura H, Matsuda M. Evidence that DOCK180 up-regulates signals from the CrkII-p130(Cas) complex. J Biol Chem. 1998;273:24479-24484
20. Mayer B.J. and Hanafusa H. Association of the v-crk oncogene product with phosphotyrosine-containing proteins and protein kinase activity. Proc Natl Acad Sci U S A. 1990;87:2638-2642
21. Dey T, Mann M.C, Goldmann W.H. Comparing mechano-transduction in fibroblasts deficient of focal adhesion proteins. Biochem Biophys Res Commun. 2011;413:541-544
22. Klemke R.L, Leng J, Molander R, Brooks P.C, Vuori K, Cheresh D.A. CAS/Crk coupling serves as a "molecular switch" for induction of cell migration. J Cell Biol. 1998;140:961-972
23. Meenderink L.M, Ryzhova L.M, Donato D.M, Gochberg D.F, Kaverina I, Hanks S.K. P130Cas Src-binding and substrate domains have distinct roles in sustaining focal adhesion disassembly and promoting cell migration. PLoS One. 2010;5:e13412
24. Rodrigues S.P, Fathers K.E, Chan G, Zuo D, Halwani F, Meterissian S, Park M. CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Mol Cancer Res. 2005;3:183-194
25. Park T.J, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol. 2006;26:6272-6282
26. Yeo M.G, Sung B.H, Oh H.J, Park Z.Y, Marcantonio E.E, Song W.K. Focal adhesion targeting of v-Crk is essential for FAK phosphorylation and cell migration in mouse embryo fibroblasts deficient src family kinases or p130CAS. J Cell Physiol. 2008;214:604-613
27. Perry S.V. and Cotterill J. Interaction of actin and myosin. Nature. 1965;206:161-163
28. Huxley H.E. Muscular contraction and cell motility. Nature. 1973;243:445-449
29. Kneussel M. and Wagner W. Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat Rev Neurosci. 2013;14:233-247
30. Giannone G, Dubin-Thaler B.J, Rossier O, Cai Y, Chaga O, Jiang G, Beaver W, Dobereiner H.G, Freund Y, Borisy G, Sheetz M.P. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561-575
31. Terry S.J, Elbediwy A, Zihni C, Harris A.R, Bailly M, Charras G.T, Balda M.S, Matter K. Stimulation of cortical myosin phosphorylation by p114RhoGEF drives cell migration and tumor cell invasion. PLoS One. 2012;7:e50188
32. Cheney R.E. and Mooseker M.S. Unconventional myosins. Curr Opin Cell Biol. 1992;4:27-35
33. Barylko B, Jung G, Albanesi J.P. Structure, function, and regulation of myosin 1C. Acta Biochim Pol. 2005;52:373-380
34. Diefenbach T.J, Latham V.M, Yimlamai D, Liu C.A, Herman I.M, Jay D.G. Myosin 1c and myosin IIB serve opposing roles in lamellipodial dynamics of the neuronal growth cone. J Cell Biol. 2002;158:1207-1217
35. Brandstaetter H, Kendrick-Jones J, Buss F. Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. J Cell Sci. 2012;125:1991-2003
36. Bose A, Guilherme A, Robida S.I, Nicoloro S.M, Zhou Q.L, Jiang Z.Y, Pomerleau D.P, Czech M.P. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature. 2002;420:821-824
37. Price L.S, Leng J, Schwartz M.A, Bokoch G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell. 1998;9:1863-1871
38. Wells C.M, Walmsley M, Ooi S, Tybulewicz V, Ridley A.J. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J Cell Sci. 2004;117:1259-1268
39. Sung B.H, Yeo M.G, Oh H.J, Song W.K. v-Crk induces Rac-dependent membrane ruffling and cell migration in CAS-deficient embryonic fibroblasts. Mol Cells. 2008;25:131-137
40. Wang L, Tabu K, Kimura T, Tsuda M, Linghu H, Tanino M, Kaneko S, Nishihara H, Tanaka S. Signaling adaptor protein Crk is indispensable for malignant feature of glioblastoma cell line KMG4. Biochem Biophys Res Commun. 2007;362:976- 981
41. Birge R, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Communication and Signaling. 2009;7:13
42. Matsuda M, Mayer B.J, Fukui Y, Hanafusa H. Binding of transforming protein, P47gag-crk, to a broad range of phosphotyrosine-containing proteins. Science. 1990;248:1537-1539
43. Schaller M.D. and Schaefer E.M. Multiple stimuli induce tyrosine phosphorylation of the Crk-binding sites of paxillin. Biochem J. 2001;360:57-66
44. Stam J.C, Geerts W.J, Versteeg H.H, Verkleij A.J, van Bergen en Henegouwen P.M. The v-Crk oncogene enhances cell survival and induces activation of protein kinase B/Akt. J Biol Chem. 2001;276:25176-25183
45. Birge R.B, Fajardo J.E, Reichman C, Shoelson S.E, Songyang Z, Cantley L.C, Hanafusa H. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol Cell Biol. 1993;13:4648-4656
46. Schaller M.D. and Parsons J.T. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635-2645
47. Iwahara T, Akagi T, Fujitsuka Y, Hanafusa H. CrkII regulates focal adhesion kinase activation by making a complex with Crk-associated substrate, p130Cas. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17693-17698
48. Wagner M.C, Barylko B, Albanesi J.P. Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol. 1992;119:163-170
49. Jay D.G. The clutch hypothesis revisited: ascribing the roles of actin-associated proteins in filopodial protrusion in the nerve growth cone. J Neurobiol. 2000;44:114-125
50. Antoku S, Saksela K, Rivera G.M, Mayer B.J. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J Cell Sci. 2008;121:3071-3082
51. Saarikangas J, Hakanen J, Mattila P.K, Grumet M, Salminen M, Lappalainen P. ABBA regulates plasma-membrane and actin dynamics to promote radial glia extension. J Cell Sci. 2008;121:1444-1454
52. Zheng D, Niu S, Yu D, Zhan X.H, Zeng X, Cui B, Chen Y, Yoon J, Martin S.S, Lu X, Zhan X. Abba promotes PDGF-mediated membrane ruffling through activation of the small GTPase Rac1. Biochem Biophys Res Commun. 2010;401:527-532
Author contact
![]() Corresponding author: Myeong Gu Yeo, Department of Alternative Medicine, Nambu University, 864-1 Wolgye-dong, Gwangsan-gu, Gwangju, 506-706, Korea, E-mail: mgy11ac.kr and Woo Keun Song, Bio Imaging Research Center, School of Life Sciences, Gwangju Institute of Science and Technology (GIST); Cheomdan-gwagiro 261, Buk-gu, Gwangju 500-712, Korea, E-mail:wksongac.kr
Corresponding author: Myeong Gu Yeo, Department of Alternative Medicine, Nambu University, 864-1 Wolgye-dong, Gwangsan-gu, Gwangju, 506-706, Korea, E-mail: mgy11ac.kr and Woo Keun Song, Bio Imaging Research Center, School of Life Sciences, Gwangju Institute of Science and Technology (GIST); Cheomdan-gwagiro 261, Buk-gu, Gwangju 500-712, Korea, E-mail:wksongac.kr