Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(8):811-817. doi:10.7150/ijbs.6734 This issue Cite
Research Paper
The Neuroprotective Effects of Coccomyxa Gloeobotrydiformis on the Ischemic Stroke in a Rat Model
Luning Sun1,*, Ying Jin2,*, Liming Dong3, Ryo Sumi4, Rabita Jahan5, Zhi Li6, ![]()
1. Department of Pathophysiology, College of Basic Medical Science, China Medical University;
2. Department of Pharmacology, Liaoning Medical University;
3. Department of Urology, the Forth Affiliated Hospital, China Medical University;
4. Tanaka Memorial Laboratory, Nikken Sohonsha Corporation;
5. International Education School, China Medical University;
6. Department of ethnopharmacology, School of Pharmaceutical Sciences, China Medical University
* Luning Sun and Ying Jin equally contributed at this work.
Received 2013-5-21; Accepted 2013-7-13; Published 2013-8-20
Abstract
Stroke is a major cause of mortality and the leading cause of permanent disability. In this study, we adopted the classic middle cerebral artery occlusion(MCAO) stroke model to observe the therapeutic effects of coccomyxa gloeobotrydiformis(CGD) on ischemic stroke, and discuss the underlying mechanisms. Low dose (50 mg/kg.day) and high dose (100 mg/kg.day) concentrations of the drug CGD were intragastrically administrated separately for 8 weeks. Infarct volumes, neurologic deficits and degree of stroke-induced brain edema were measured 24 hours after reperfusion. Furthermore, oxidative stress related factors (SOD and MDA), mitochondrial membrane potential, and apoptosis regulatory factors (mitochondrial Cyt-C, Bcl-2, Bax, and caspase-3) were all investigated in this research. We found that CGD attenuated cerebral infarction, brain edema and neurologic deficits; CGD maintained the mitochondrial membrane potential and decreased mitochondrial swelling. It also prevented oxidative damage by reducing MDA and increasing SOD. In addition, CGD could effectively attenuate apoptosis by restoring the level of mitochondrial Cyt C and regulating the expression of Bcl-2, Bax and caspase 3. These results revealed that CGD has a therapeutic effect on ischemic stroke, possibly by inducing mitochondrial protection and anti-apoptotic mechanisms.
Keywords: Coccomyxa gloeobotrydiformis (CGD), ischemic stroke, apoptosis, mitochondrion
Introduction
Stroke is a significant etiologic factor for mortality and permanent disability on a worldwide scale. Studies conducted in the United States, China and Kuwait all provided significant statistics suggesting that ischemic stroke accounts for 85-90% of all stroke cases clinically observed. [1],[2],[3] In addition, there is still no “silver bullet” to reverse acute ischemic stroke at this point. The best outcome in stroke patients is associated with fast and early arterial recanalization, followed by prevention strategies against arterial re-occlusion, and to promote neuroprotection [4]. Systemic recombinant tissue plasminogen activator (r-tPA) remains the only approved treatment plan and the fastest way to initiate thrombolysis for acute ischemic stroke within the first three hours after stroke onset.[5] Unfortunately, only a small number of patients are candidates for r-tPA therapy. Furthermore, the reperfusion injury following the arterial recanalization often complicates ischemic stroke management. Therefore, an effective neuroprotective medication with resistance to the ischemia-reperfusion injury is urgently needed.
Coccomyxa gloeobotrydiformis (CGD) is a newly found microalga, which can efficiently relieve the stroke risk factors including hypertension, diabetes mellitus and hyperlipermia.[6] The data obtained showed a copious amount of α-linolenic acid(ALA), approximately 24%. The increased ALA intake has demonstrated improvements towards reperfusion and reduced the risk of both MCAO-induced mortality rate and infarct volumes.[7] Moreover, our previous study has proved that CGD could prevent neurodegeneration and improve learning and memory ability of aged rats ( See the supplementary material), which suggested the neuroprotective effects of this algae. Hence, this might be a favorable drug worthy of further investigation for its properties against ischemic stroke, considering its anti-oxidant and neuroprotective effects.
In this study, we adopted the well-established method of MCAO model to observe therapeutic effects of CGD on ischemic stroke in rats. Furthermore, we discuss the underlying mechanisms of the drug and its effects on the specimen.
Materials and methods
This animal study was approved by the Institutional Animal Care and Use Committee. All procedures in this study were carried out in accordance with the recommendations in the guide for the care and use of laboratory animals of China Medical University. All surgery was performed under anesthesia, and all efforts were made to minimize pain and suffering for the animals.
Animals
The rat MCAO model was used as a stroke model. Adult male Sprague-Dawley rats weighing 250 to 280 g (Experimental animal center of Liaoning Medical University) were anesthetized with 10% chloralhydrate (300 mg/kg, i.p.) and induced to MCAO by using a previously described method [8]. During surgery, the rectal temperature was maintained at normal values by using a heating pad throughout the procedure. After awakening from anesthesia, the rats were kept in cages and allowed free access to food and water. After 60 minutes of MCAO, the blood flow was restored for 24 hours. Animals in the control group underwent surgical brain microdissection without ligating the arteries. Animals were divided into four groups: Sham operated group (n=12); MCAO model control group (n=16); low-dose CGD (50 mg/kg.day) treated MCAO model group (n=16) and high-dose CGD (100 mg/kg.day) treated MCAO model group (n=16). The CGD was administered intragastrically for a total duration of 8 weeks. In the control group, a similar volume of saline solution was administrated.
Neurological evaluations
Neurological evaluations were performed 24 hours after reperfusion by using neurological deficit scoring scale adapted from Longa et al [9]. The scale rates the presence or absence of neurological signs in rats, as follows: 0'-no neurological deficit; 1'-retracts left forepaw when lifted by the tail; 2'-circles to the left; 3'-falls while walking; 4'-does not walk spontaneously; 5'-dead after surgery. Only those animals showing a value greater than or equal to 1' in the Longa scale 30 min after recovery from anesthesia were considered stroke-affected and included in the study. The above behavioral observations were carried out in a blinded manner.
Infarct size
Animals were decapitated 24 hours after reperfusion under anesthesia, and the infarct volumes were measured as described previously [10]. Coronal sections of the brain (n=6) were cut into 2mm slices and immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, USA) at 37℃ for 20 min, followed by 10% formaldehyde solution. The infarcted tissue remained unstained (white), whereas normal tissue was stained red by the TTC. The infarct zone was demarcated and analyzed by Image J software. Infarct areas of all the sections were added to ascertain the total infarct area, which was multiplied by the thickness of the brain sections to obtain the total infarct volume.
Brain edema
Degree of stroke-induced brain edema was assessed 24 hours after reperfusion using the wet-dry method as described previously [11]. The ipsilateral MCAO brain hemisphere (n=4) was dissected in a humidity chamber and the wet weight was recorded immediately. The ischemic hemisphere was then incubated at 100℃ until a constant dry weight was obtained. Brain edema was analyzed by calculating tissue water content based on the formula: percentage of brain water content = (1-dry weight/wet weight)×100%.
Oxidative stress
Hippocampal tissues from 6 individual rats from each group were collected 24 hours after reperfusion and then placed into an ice-cold RIPA lysis buffer (Beyotime institute of biotechnology, Shanghai, China). The samples were then homogenized by a polytron homogenizer, followed by centrifugation at 3000 rpm/minute for 15 minutes. The supernatant was collected for superoxide dismutase (SOD) and malonaldehyde (MDA) measurement. The total superoxide dismutase assay kit with WST-1 (Beyotime institute of biotechnology, Shanghai, China) and lipid peroxidation MDA assay kit (Beyotime institute of biotechnology, Shanghai, China) were selected for SOD and MDA measurement. The assay was conducted according to the manufacturer's instructions.
Mitochondrial swelling and membrane Potential
Once more, Hippocampal tissues from 6 rats from each group were collected 24 hours after reperfusion. Mitochondria isolation was then carried out as per the manufacturer's instructions (Mitochondria isolation kit for tissue, Abcam). Measurment of mitochondrial swelling was done by the method of Halestrap et al [12]. Two-hundred microliters of isotonic buffer was added to mitochondria (50mg protein) at 30℃ for 5 min. The absorbance rate was measured at 520nm. And then, Rhodamin 123, the specific fluorescent dyes for mitochondria were added to 2.5ml reaction buffer containing 10mg mitochondria protein suspension and incubated for 5 min at room temperature. Mitochondria were pelleted by centrifugation and the fluorescence of the supernatant was read with a spectrophotometer. The parameters included an excitation wavelength of 503nm and an emission wavelength of 527nm. When mitochondrial membrane potential is high, it retains more dye and the fluorescence intensity of the supernatant will decrease. If there is a loss of membrane potential, the dye is free to leak out of mitochondria and the fluorescence intensity in the supernatant will increase.
Immunoassay of Cyt-C
Cyt-C dissociates from the mitochondrial inner membrane, moves into the intermembrane space, then ultimately released into the cytosol. Therefore, it is recognized as a key marker of oxidative stress-induced apoptosis in the intrinsic pathway. Cyt-C was derived from supernatant of mitochondria isolation. Cyt-C content was analyzed using the Cyt-C immunoassay kit (R&D system) according to the manufacturer's instructions.
Western Blot Analysis for Bcl-2, Bax, and caspase-3 protein levels
Hippocampus protein lysates were subjected to 12% SDS-PAGE and transferred onto PVDF membrane (Millipore Corp.). After blocking with 5% skim milk, the membranes were incubated overnight at 4℃ with their respective primary antibodies (Bcl-2, 1:400, Bax, 1:400, Santa Cruz, U.S.A; caspase-3, 1:1000, Cell signaling technology, USA.). The membranes were then incubated with the appropriate amount of secondary horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) antibodies at a 1:2000 dilution. Immunoreactive proteins were then visualized using enhanced chemiluminescence (ECL). The signals were quantified by densitometry using a Western blotting detection system where β-actin served as the loading control.
Statistical Analysis
The data obtained was represented as mean±S.E.M. (Standard error of the mean) of at least three independent preparations. Statistical analysis was performed by the one-way analysis of variance (ANOVA) with Tukeys' post hoc test. A difference with p<0.05 was considered statistically significant.
Results
CGD attenuated cerebral infarction, brain edema, and neurologic deficits
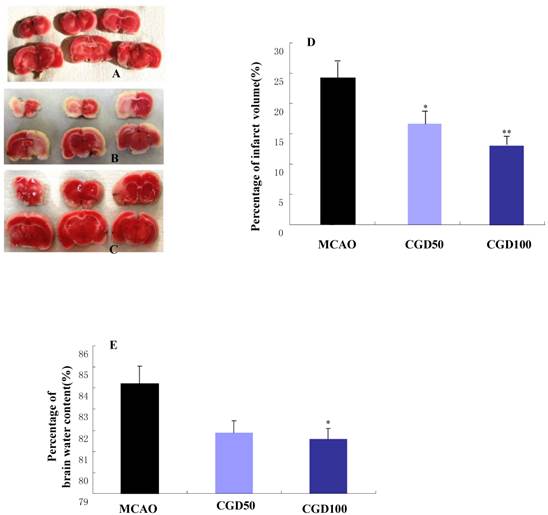
There was no cerebral injury in the sham-operated group (Fig 1A); In the MCAO group, the infarct area was observed in the left cortex and striatum (Fig 1B). As expected, CGD significantly decreased infarct volumes (Fig 1C and D). In comparison to MCAO model group, CGD treated animals had a significantly lower occurrence of edema in their ipsilateral brain hemispheres (Fig1E). The neuroprotective effects of CGD were assessed based on their neurological deficit score. The higher the neurological deficit score, the greater the severity of neurological impairment. In agreement with infarct volume measurement, CGD reduced the neurological deficit score in a dose-dependent manner (Table 1).
A. Infarct area of sham group. B. Infarct area of MCAO group. C. Infarct area of CGD100 group. D. Percentage of infarct volume in each group. E. Percentage of brain water content in each group. *P<0.05, **P<0.01, compared with MCAO group.
Neurological Deficit Grading System
| Groups | Sham | MCAO | CGD50 | CGD100 |
|---|---|---|---|---|
| Score | 0 | 2.67±0.36 | 1.92±0.23 | 1.17±0.29** |
**P<0.01; compared with MCAO group
CGD defend against oxidative damage
The activitity of SOD in the sham operated group was 13.8±1.16 U/mg protein. MCAO caused a significant reduction of SOD to 9.63±20.88U/mg protein. High dose of CGD could restore the activity of SOD to its normal level (12.9±0.94 U/mg protein). MDA is an index for lipid peroxidation. As shown in Table 2, MDA level increased significantly in the MCAO group, and CGD is capable of defending oxidative damage by reducing MDA.
Level of hippocampal SOD and MDA
| SOD, U/mg protein | MDA, μmol/mg protein | |
|---|---|---|
| Sham | 13.80±1.16 | 2.89±0.33 |
| MCAO | 9.63±0.88** | 4.53±0.41** |
| CGD50 | 11.89±0.95 | 3.29±0.39# |
| CGD100 | 12.90±0.94# | 2.94±0.32## |
**P<0.01, compared with sham group; #P<0.05, ##P<0.01, compared with MCAO group.
CGD ameliorated mitochondrial membrane swelling and restore membrane potential
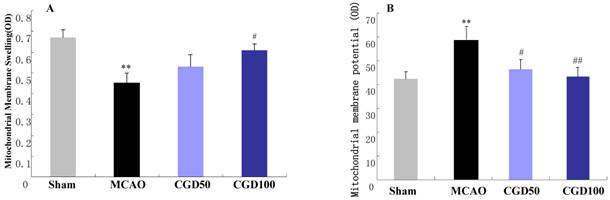
Membrane swelling was measured after MCAO for 60 min followed by reperfusion for 24 hours. Six animals from each group were randomly selected for the measurement of mitochondrial membrane swelling. The absorbance of the mitochondria from the MCAO group decreased significantly compared with sham group, which can be restored by CGD (Fig 2A). In the MCAO group, the fluorescence intensity in the supernatant increased remarkablly in comparison to the sham group. And CGD significantly decreased the fluorescence intensity in the supernatant (Fig 2B). This indicated a loss of mitochondrial membrane potential during MCAO/reperfusion and the restoration of the membrane potential by CGD treatment.
A. Effect of CGD on the mitochondrial membrane swelling; B. Mitochondria membrane potential. Means±SEM n=6. **p<0.01, compared with sham group; # p<0.05, ##P<0.01, compared with MCAO group compared with MCAO group.
CGD restored the level of mitochondrial Cyt C
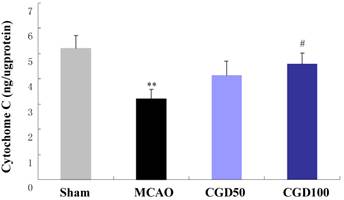
As shown in Fig 3, concentration of mitochondrial Cyt-C in MCAO group decreased in comparison to the sham group. CGD could restore the level of mitochondrial Cyt-C, which also indicated that CGD inhibited the oxidative stress-induced apoptosis in the intrinsic pathway.
Content of Mitochondrial Cyt-C by immunoassay. Means±S.E.M; **p<0.01, compared with sham group; # p<0.05, compared with MCAO group.
CGD regulates the expression of Bcl-2, Bax and caspase 3 proteins
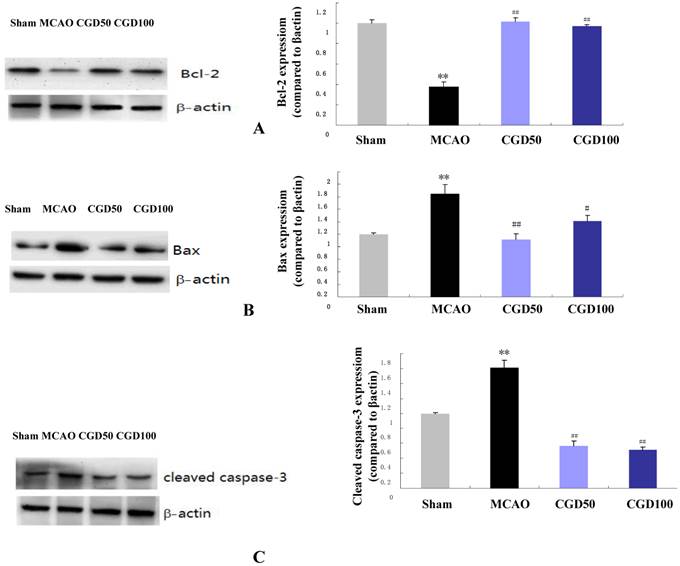
The anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax are crucial determinants of the apoptotic response and also control the release of Cyt-C. We found that expression of Bcl-2 protein was significantly higher in the CGD treated group compared to MCAO group (Fig 4A), and the expression of Bax was to the contrary played an opposing factor (Fig 4B). As expected, cleaved caspase-3 the apoptotic cell death marker, decreased significantly in the CGD treated group (Fig 4C).
Expression of Bcl-2, Bax and caspase 3 proteins by Western Blot. Means±S.E.M, **p<0.01vs sham group. ## p<0.01 vs MCAO group.
Discussion
The most effective intervention for acute ischemic stroke is reperfusion of ischemic penumbra. However, the accompanied ischemia-reperfusion injury often causes death of neurocytes within this region, which may induce the ultimate infarction. Therefore, anti-reperfusion injury and neuroprotection are two key problems faced during stroke management. It's well understood that oxidative stress is the major mechanism of ischemia-reperfusion injury. The mitochondrion is the fundamental site for reactive oxygen species (ROS) production. It also plays a major role in regulating neurocyte apoptotic pathway during ischemic-reperfusion. This process involves pro- and anti-apoptotic protein binding and the releasing of cytochrome c (Cyt-C). As mentioned earlier, CGD can effectively alleviate stroke risk factors, and also contains a higher concentration of antioxidant-ALA. These factors can improve reperfusion and reduce both MCAO-induced mortality rate and infarct volumes. Our previous study showed that CGD increased the level of endothelial nitric oxide synthase (eNOS), whose expression remarkably decreased ischemic stroke in animals [13]. Prior reports have demonstrated that eNOS is a source of NO• involved in mitochondrial biogenesis [14]. Therefore, its convincing that CGD plays the fundamental roles as an antioxdant and a neuroprotector through its positive impact on the mitochondrion.
This study showed that CGD could protect the brain from ischemic injury as evidenced by the reduction in infarct size and improvement in neurological function. Since neurons are highly susceptible to ischemia-reperfusion injury, maintenance of mitochondrial integrity is, without question, a critical determinant of cells fate. Mitochondrial transmembrane potential is a sensitive indicator reflecting the mitochondrial function. The decline of mitochondrial transmembrane potential was correlated with the opening of the permeability transition pore, which leads to the release of caspase-activating proteins and swelling of mitochondria. Consequently, it disrupts the outer mitochondrial membrane. Therefore, mitochondrial swelling also reflects the opening of the permeability transition pore. In this study, MCAO group showed a significant increase in mitochondrial swelling and a prominent decrease in mitochondrial membrane potential. Its apperant that CGD treatment could both maintain mitochondrial membrane potential and decrease mitochondrial swelling. In addition, CGD also prevents oxidative damage by reducing MDA and increasing SOD. As a result, CGD has the ability to relieve the ischemia- reperfusion injury and reduce the infarct volumes during stroke through its mitochondrial protective effects.
Apoptosis is one of the main forms of neurocyte death in the ischemic penumbra during the progression of ischemic stroke, and mitochondrion is the essential organelle involved in cell apoptosis. Interactions between the pro-apoptotic Bax and anti-apoptotic Bcl-2 family proteins on the mitochondrial outer membrane are believed to play an important role in cell survival [15]. With apoptotic stimuli, Bax is post-transcriptionally activated, and oligomerizes and translocates to mitochondria, then it triggers Cyt-C releasing from mitochondria. Cyt-C is a post mithochondrial activator of apoptosis. Cyt-C releasing activates downstream caspases of the intrinsic pathway through the formation of the apoptosome, a complex of dATP, cytochrome, procaspase 9 and Apaf1[16]. Our study showed a significant reduction of mitochondrial Cyt-C in MCAO group, which may be due to the up-regulated Bax level and down regulated Bcl-2 level. And in the CGD treated group, mitochondrial Cyt-C was almost restored to the normal level. So CGD could effectively attenuate apoptosis through inhibiting Bax-induced Cyt-C releasing from mitochondria. It might be the mechanism of the neuroprotetive effects of CGD on ischemic stroke.
In conclusion, this study is the first to demonstrate the roles of CGD on ischemic stroke. Although the underlying mechanisms are still to be verified, our results suggest CGD is an effective neuroprotectant against cell apoptosis through inhibiting Bax-induced Cyt-C released from mitochondria. CGD is a naturally produced drug with minimal side effects. It can mitigate ischemic reperfusion injury and reduce the infarct volumes during stroke through its mitochondrial protective effects. Even though our present study was exclusively based on animal testing, it proved our initial theory of the drug. Post-treatment studies will be performed in the near future to further advance our knowledge of the clinical application of CGD.
Supplementary Material
Supplementary information.
Acknowledgements
This research was funded by Nikken Sohonsha Corporation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Seshadri S, Wolf PA, Beiser A. et al. Elevated midlife blood pressure increases stroke risk in elderly persons: the Framingham Study. Arch Intern Med. 2001;161(19):2343- 50
2. Ashkanani A, Hassan KA, Lamdhade S. Risk factors of stroke patients admitted to a general hospital in Kuwait. Int J Neurosci. 2013;123(2):89-92
3. Zhao D, Liu J, Wang W. et al. Epidemiological transition of stroke in China: twenty-one-year observational study from the Sino-MONICA-Beijing Project. Stroke. 2008;39(6):1668-74
4. Adams HP Jr, del Zoppo G, Alberts MJ. et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38(5):1655-1711
5. Rha JH, Saver JL. The impact of recanalization on ischemic stroke outcome: a meta-analysis. Stroke. 2007;38(3):967-73
6. Ishida Y, Honda T, Mabuchi S. et al. Validation of thermally assisted hydrolysis and methylation-gas chromatography using a vertical microfurnace pyrolyzer for the compositional analysis of fatty acid components in microalgae. J Agric Food Chem. 2012;60(17):4222-6
7. Nguemeni C, Delplanque B, Rovère C. et al. Dietary supplementation of alpha-linolenic acid in an enriched rapeseed oil diet protects from stroke. Pharmacol Res. 2010;61(3):226-33
8. Goldmacher GV, Nasser R, Lee DY. et al. Tracking transplanted bone marrow stem cells and their effects in the rat MCAO stroke model. PLoS One. 2013;8(3):e60049
9. Longa EZ, Weinstein PR, Carlson S. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84-91
10. Tsubokawa T, Jadhav V, Solaroglu I. Lecithinized superoxide dismutase improves outcomes and attenuates focal cerebral ischemic injury via antiapoptotic mechanisms in rats. Stroke. 2007;38(3):1057-62
11. Ahmed SH, He YY, Nassief A. et al. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke. 2000;31(1):193-9
12. Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268(1):153-60
13. Niu PP, Yang G, Zheng BK. et al. Relationship between endothelial nitric oxide synthase gene polymorphisms and ischemic stroke: a meta-analysis. Acta Neurol Scand. 2013 doi: 10.1111/ane.12120
14. Nisoli E, Tonello C, Cardile A. et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310(5746):314-7
15. Vela L, Gonzalo O, Naval J. et al. Direct interaction of Bax and Bak with Bcl-2 Homology domain-3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J Biol Chem. 2013;288(7):4935-46
16. Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55(3):310-8
Author contact
![]() Corresponding author: Zhi Li, Department of Ethnopharmacology, School of Pharmaceutical Sciences, China Medical University Shenyang, China. E-mail address:lizhilelecom.
Corresponding author: Zhi Li, Department of Ethnopharmacology, School of Pharmaceutical Sciences, China Medical University Shenyang, China. E-mail address:lizhilelecom.