International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(9):895-906. doi:10.7150/ijbs.7508 This issue Cite
Research Paper
BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate
Junjun Jing1,2, Yinshi Ren2, Zhaowen Zong2, Chuanju Liu3, Nobuhiro Kamiya4, Yuji Mishina5, Ying Liu2, Xuedong Zhou1, Jian Q. Feng2 ![]()
1. State Key Laboratory of Oral Diseases, West China Hospital of Stomatology, Sichuan University, China.
2. Department of Biomedical Sciences, Texas A&M Baylor College of Dentistry, Dallas, TX, USA.
3. Department of Orthopaedic Surgery and Cell Biology, New York University School of Medicine, New York, NY, USA.
4. Center for Excellence in Hip Disorders, Texas Scottish Rite Hospital for Children, Dallas, TX, USA.
5. Department of Biologic and Materials Sciences, School of Dentistry, University of Michigan, Ann Arbor, MI, USA.
Received 2013-8-27; Accepted 2013-9-16; Published 2013-9-18
Citation:
Jing J, Ren Y, Zong Z, Liu C, Kamiya N, Mishina Y, Liu Y, Zhou X, Feng JQ. BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate. Int J Biol Sci 2013; 9(9):895-906. doi:10.7150/ijbs.7508. https://www.ijbs.com/v09p0895.htm
Other stylesAbstract
Bone morphogenic proteins (BMPs) are critical for both chondrogenesis and osteogenesis. Previous studies reported that embryos deficient in Bmp receptor (Bmpr)1a or Bmpr1b in cartilage display subtle skeletal defects; however, double mutant embryos develop severe skeletal defects, suggesting a functional redundancy that is essential for early chondrogenesis. In this study, we examined the postnatal role of Bmpr1a in cartilage. In the Bmpr1a conditional knockout (cKO, a cross between Bmpr1a flox and aggrecan-CreERT2 induced by a one-time-tamoxifen injection at birth and harvested at ages of 2, 4, 8 and 20 weeks), there was essentially no long bone growth with little expression of cartilage markers such as SOX9, IHH and glycoproteins. Unexpectedly, the null growth plate was replaced by bone-like tissues, supporting the notions that the progenitor cells in the growth plate, which normally form cartilage, can form other tissues such as bone and fibrous; and that BMPR1A determines the cell fate. A working hypothesis is proposed to explain the vital role of BMPR1A in postnatal chondrogenesis.
Keywords: BMPR1A, Growth plate, Cell fate, Chondrogenesis, Endochondral Bone.
Introduction
During endochondral ossification, the progenitor cells committed act as the stem cells that replenish the pool of proliferative chondrocytes in the resting zone of the growth plate. The resulting daughter cells exit the cycle and undergo maturation via prehypertrophic, hypertrophic and terminal hypertrophic processes, followed by calcified cartilage formation. The vascular invasion then leads to the removal of cartilage, formation of bone marrow, and new bone formation and growth. An impressive body of studies has documented that the transition from cartilage to bone is highly regulated by numerous transcriptional factors and local growth factors such as Indian hedgehog (Ihh), Sox9, and PTHrP [9, 14, 16, 23, 24], as well as BMPs.
These BMPs are vital not only to osteogenesis [3, 12, 13] but to chondrogenesis as well. For example, Bmp2 cKO mice (a cross between Bmp2 fx/fx and Col2a1CreERT2 mice and induced by tamoxifen) displayed severe defects in chondrocyte proliferation, differentiation and apoptosis in the growth plates although Bmp4 cKO exhibited no apparent chondrocyte defects [23]. Studies of their key receptors -1A and -1B (both expressed throughout the growth plate) showed that the removal of Bmpr1a in the cartilage by the Col 2-Cre line or conventional Bmpr1b KO has little effect. However, double-mutant mice displayed a severe chondrodysplasia phenotype during embryonic development, indicating redundancy but functionally essential for BMP signaling in chondrocyte proliferation, survival and differentiation [26, 27]. Further reports showed a similar early chondrodysplasia of Smad1 and Smad5 double cKO mice [21], confirming the vital role of the canonical Smads 1/5 signaling pathway controlling embryonic chondrogenesis.
Yet, one of the key questions remaining to be addressed is whether there is a molecule that initiates and guides the progenitor cell in the resting zone of the growth plate, which solely differentiates chondrogenesis. Would the growth plate form non-cartilaginous tissues such as bones if such a critical factor were removed? In this study, we attempted to answer this question using BMPR1A as the target molecule. We took advantage of an inducible Aggrecan-CreERT2 knock-in allele mouse line (Agc1CreERT2) in which the Cre recombinase is expressed in all the cartilage layers from the resting zone to the hypertrophic zone when tamoxifen is injected [20]. We crossed this line to the homozygous Bmpr1a flox/flox line [19], thus generating Bmpr1a cKO mice with a one-time tamoxifen injection at birth and then harvested the bones separately at age of 2, 4, 8 and 20 weeks. Our data indicated that there is essentially no long bone growth after Bmpr1a is removed postnatally from the progenitor cells in the growth plate. Surprisingly, the null growth plate is gradually replaced by bone and fiber-like tissues. These findings lead to the novel concepts that the progenitor cell in the growth plate is plastic and BMPR1A determines the chondrogenic cell fate.
Materials and Methods
Mice and tamoxifen administration
To avoid neonatal death, mice harboring a floxed allele of Bmpr1a (Bmpr1a Osxflox) [19] were crossed with Agc1CreERT2 mice, permitting a temporally regulated Cre-mediated recombination by TM (Sigma-Aldrich) [1]. The offspring were further intercrossed to obtain mice homozygous for floxed Bmpr1a and heterozygous for the Agc1CreERT2 transgene. The mice were then injected intraperitoneally with TM 1.5-3.0 mg/10 g body weight or vehicle (corn oil containing 10% ethanol) at newborns and sacrificed at ages of 2-wks, 4-wks and 8-wks separately. All animal protocols were approved by the Animal Welfare Committee at Texas A&M Baylor College of Dentistry.
X-ray radiography and high-resolution microcomputed tomography [µ-CT]
Femurs from one-month-old Bmpr1a cKO mice and control mice were dissected of free skeletal muscles for radiography with a Faxitron MX-20 specimen radiography system (Faxitron X-ray Corp., Buffalo Grove, IL) as described previously [25]. For high-resolution µ-CT analyses, the femurs were scanned at 3.5-μm resolution using a µ-CT35 imaging system (Scanco Medical, Basserdorf, Switzerland), and the trabecular bone was analyzed from 20 sections underneath the growth plate at a threshold of 160, as previously described [7].
Histology
The bones were fixed in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (pH 7.4), decalcified, and embedded in paraffin using standard histological procedures as previously described [6]. The tissue blocks were cut into 4-µm thick mesio-distal serial sections to compare the tissue samples from the experimental and control groups and were then mounted on glass slides. The sections were used for H&E stain, Safranine O stain, toluidine blue stain, and immunohistochemistry, as described previously [6, 7]. For immunohistochemistry analyses, the following antibodies were used, anti-Osterix (Santa Cruz Biotechnology, USA; 1:400), rabbit anti-aggrecan antibody (A kind gift from Dr. Koji Kimata, Institute for Molecular Science of Medicine, Aichi Medical University, Japan; 1:200) [11], rabbit anti-IHH polyclonal antibody (Abcam,USA;1:1000), rabbit anti-fibronectin polyclonal antibody (Abcam,USA;1:100), rabbit anti-periostin polyclonal antibody (Innovative Research,USA;1:1000), rabbit anti-DMP1 polyclonal antibody ( generously provided by Dr. Chunlin Qin, Baylor College of Dentistry, USA;1:400), or rabbit anti-SOX9 polyclonal antibody (Santa Cruz Biotechnology, USA, 1:100). All the immunohistochemistry experiments were detected with a 3, 3-diaminobenzidine kit (Vector Laboratories, Burlingame, CA) according to the instructions of the manufacturer.
Backscattered scanning electron microscopy [SEM], acid etching SEM, and FITC imaging
The long bones were dissected and fixed in 70% ethanol at room temperature for 24 h. The tissue specimens were dehydrated in ascending concentrations of ethanol (from 70% to 100%), embedded in MMA without decalcification and sectioned using a water-cooled diamond-impregnated circular saw (Isomet, Buehler). The cut surface was polished using 1, 0.3 and 0.05 μm alumina alpha micropolish II solutions (Buehler) on a soft cloth rotating wheel [18]. Each sample was placed in an ultrasonic bath between steps and immediately following the polishing steps. The dehydrated specimens were then sputter-coated with carbon and scanned with a backscattered electron detector in a JEOL JSM-6300 scanning electron microscope (JEOL, Japan). The parameters were kept constant while the backscattered SEM images were taken under the scanning electron microscope, as described previously [7, 17].
FITC staining [2, 5]- Fluorescein isothiocyanate, a small molecular dye, fills in all non-mineralized cells/fibers. Thus, the dye provides a visual representation of the organization of the growth plate and perichondrium under the confocal microscope. Immediately after harvesting and dissection, the long bones were placed in fresh 4% paraformaldehyde fixative at 4C for two days followed by embedded in MMA bone blocks. A cross section (1-2 mm thick) was cut with a diamond-bladed saw (Buehler, Lake Bluff, IL), and the cortical sections were then sanded and ground to a final thickness of approximately 100 μm for confocal imaging.
Statistical analysis
Statistical significance was determined by an independent-sample t-test using SPSS 12.0. A P value of < 0.05 was considered statistically significant.
Results
Bmpr1a cKO long bone growth is arrested after one-time tamoxifen injection at birth
If BMPR1A controls endochondral bone formation starting from chondrogenesis, we reasoned that its removal from the chondrogenic progenitor cells by Agc1CreERT2, a Cre line expressed in all chondrocyte zones [20] [also see Supplementary Material: Figure S1], would lead to the arrest of longitudinal bone growth. To test this hypothesis, we initially injected tamoxifen one time at birth and sacrificed the mice at the age of 2 weeks. BrdU stains showed no cell proliferation, and the safranin O stain revealed little production of glycoproteins and a lack of endochondral bone formation (Fig. 1). We then proceeded with a more thorough study at the age of 1 month. The photograph showed an extremely short status, and the μCT images revealed a sharp reduction in bone length with a small metaphysis bone mass in the null femur (Fig. 2a). The quantitative data demonstrated that these changes are highly significant (Fig. 2b).
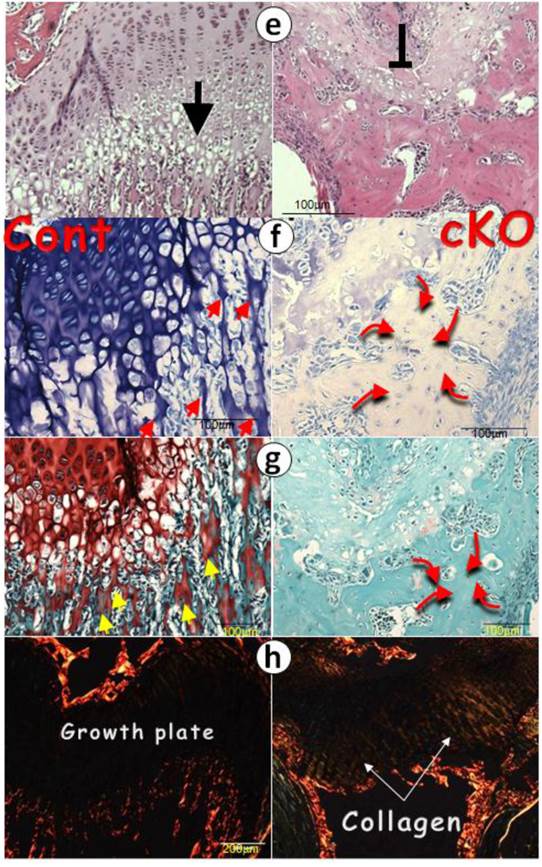
At the electron microscope (EM) level, we observed numerous calcified trabeculae with no signs of osteocytes in the control femur metaphyses, which are connected to the chondrocyte columns (fig 2c, left panels). In contrast, in the null metaphysis (Fig. 2c, right panels), there were no chondrocyte columns but numerous mineral spheres (red arrowheads) that directly formed a few large clusters of bone. The FITC image (Fig. 2d) further confirmed this observation, in which there were mature osteocytes in the null bone (arrows) but not in the control metaphysis. To better understand the nature of the null bone formation in the metaphysis, we examined the null growth plate using H&E stain (Fig. 2e), toluidine blue stain (Fig. 2f), and safranin O stain (Fig. 2g). These histological images disclosed a lack of mature chondrocytes and proteoglycan production. Furthermore, there were no trabeculae in the cKO metaphyses. Instead, there were a few pieces of a “large and fused bone mass” that appeared to have been formed from the surrounding mesenchymal cells with no sign of the cartilage residue (red curved arrows), plus ectopically expressed type I collagen in the cKO growth plate revealed by polarized light (Fig. 2h).
Importantly, the arrest of long bone growth cannot be compensated for according to time, as the Bmpr1a-null mice harvested at all stages (from 2, 4, 8 and 20 weeks with 3-5 animals a stage) displayed the identical phenotype of short long bones by radiographs (Fig. 3). The failure of long bone growth indicates that there is no new progenitor cells added after deletions of Bmpr1a gene by one time injection of tamoxifen at newborn stage.
Taken together, the above data demonstrate the indispensable role of BMPR1A in postnatal endochondral bone formation, and a lack of new progenitor cells added in the growth plate after birth.
There is a change from cartilage to a mixture of bone and fibers in the Bmpr1a cKO growth plate
It is known that SOX9 [4, 9] and IHH [10, 15] are critical for cartilage formation. We reasoned that the failure of chondrogenesis in the Bmpr1a null growth plate could be the consequence of reductions in these two factors. Next we examined their expression levels in the null growth plate and detected little expression (Fig. 4a-b). In addition, there were only patched expressions of aggrecan in the null growth plate, which likely reflects the remains of this extracellular matrix protein from the existing cartilage prior to Bmpr1a removal (Fig. 4c).
Perhaps the most surprising finding is the ectopic expression of bone and fibrous markers in the null growth plate, including OSX, DMP1, periostin (highly expressed in periosteum), and fibronectin (Fig. 4d-g). As a result, the null growth plate stops normal chondrogenesis and instead ectopically forms a mixture of bone and fibrous tissue as shown by H&E stain, and FITC stain. In addition to this mixture, there were also numerous blood vessels (arrows) and TRAP (Tartrate-Resistant Acid Phosphatase) positive cells in the null growth plate (Fig. 4h-j).
Fig 1
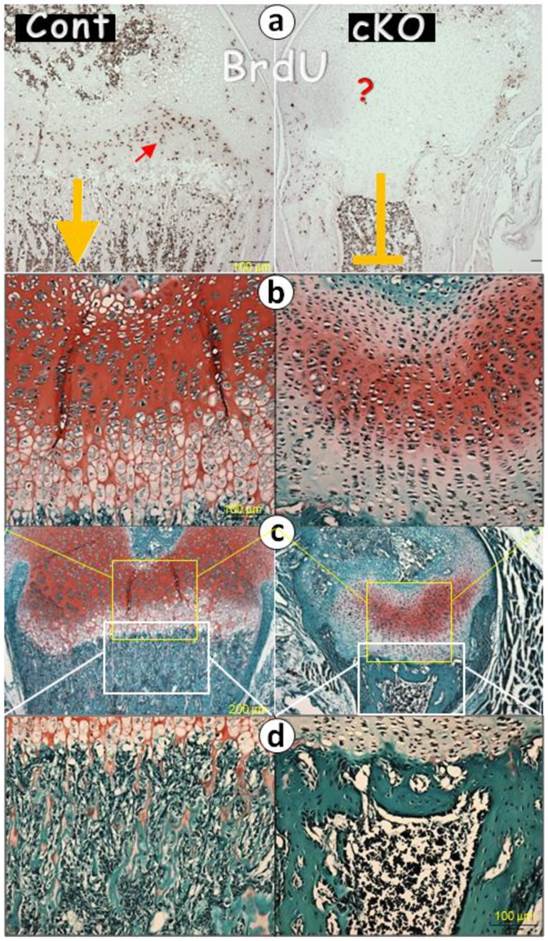
Deletion of Bmpr1a leads to a lack of chondrogenesis at age of 2-weeks (right panels). (a) Representative BrdU stained images of the Bmpr1a cKO (conditional knockout) mice (crossing between Bmpr1a flox and aggrecan CreER with one time tamoxifen injection at newborn and harvesting at age of 2-weeks, right) and the Cre negative littermate (left) showed no visible BrdU positive cells in the null growth plate; and (b-d). The safranin O stained images revealed lacks of hypertrophic chondrocytes and chondrocyte columns (b-c); and in the cKO metaphyses there were no trabeculae but a few large amounts of well-formed bone clusters, in which there was no signs of safranin O stains (the red color reflecting glycoproteins) in the cKO bone.
Fig 2
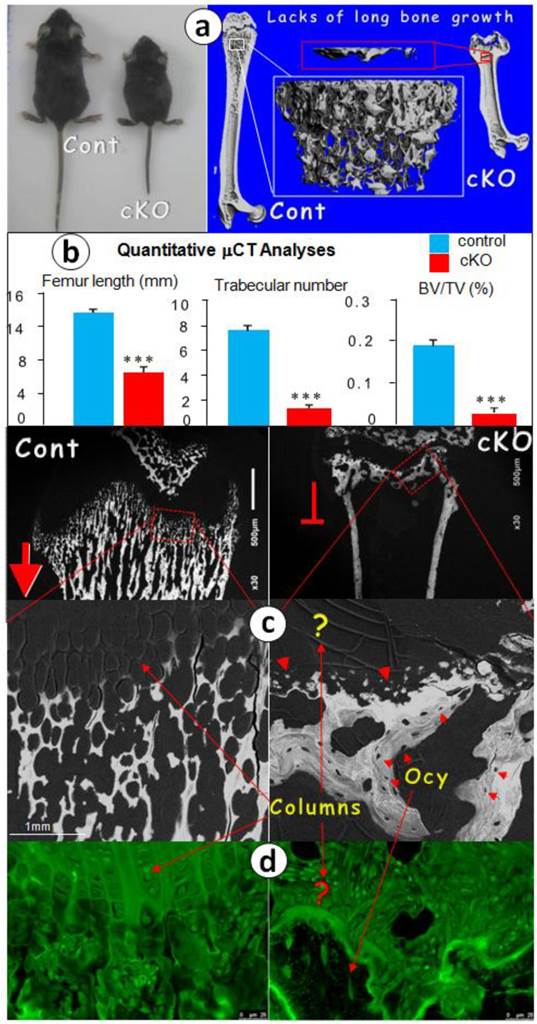
Postnatal long bone growth is arrested in the 1-mon-old Bmpr1a cKO mice (right panels) after a one-time tamoxifen injection at birth. (a) Representative photographic and μCT images showed the arrest of long bone growth in the Bmpr1a cKO mice with few signs of bone in the cKO metaphysis (upper panel); (b) The quantitative data revealed a significant reduction of femur length, trabecular number and bone volume in the cKO metaphysis; (c-d) The representative backscattered SEM (c) and FITC (d) images showed a lack of chondrocyte columns and trabeculae in the cKO femur. Instead, there were numerous spherical mineral bodies adjacent to a few large clusters of mature bones, in which the osteocytes (Ocy) were well-formed; (e) The H&E stain displayed similar bone clusters in the cKO metaphysis, which appeared from the surrounding mesenchymal cells; (f) The toluidine blue stain images showed no signs of the cartilage residues in the metaphysis bones that are directly formed from the mesenchymal cells (red half-circle arrows) compared to the control (red arrows, left); (g) The safranine O stain images revealed no cartilage extracellular matrices in the cKO bone compared to the control (yellow arrows); and (h) The polarized images disclosed numerous collagen fibers in the cKO growth plate compared to the age-matched control, in which there was no type I collagen.
Fig 3
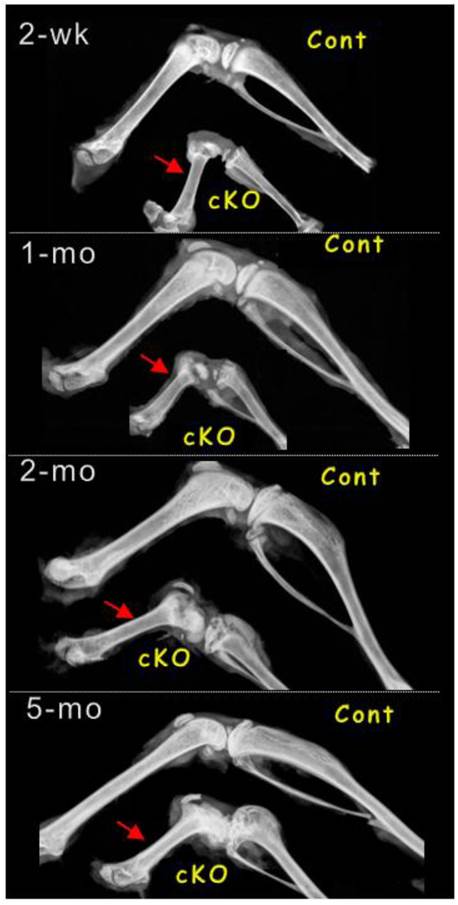
Removal of Bmpr1a leads to lacks of bone growth at newborns by one-time tamoxifen injection. Representative radiograph images showed a stop in lone bone growth at the following stages: 2-weeks, 1-month, 2-months and 5-months. The minor difference among different null long bone could be due to the variations of tamoxifen injections, as the injections occurred at newborn and some solution injected could not be leaked out.
Fig 4
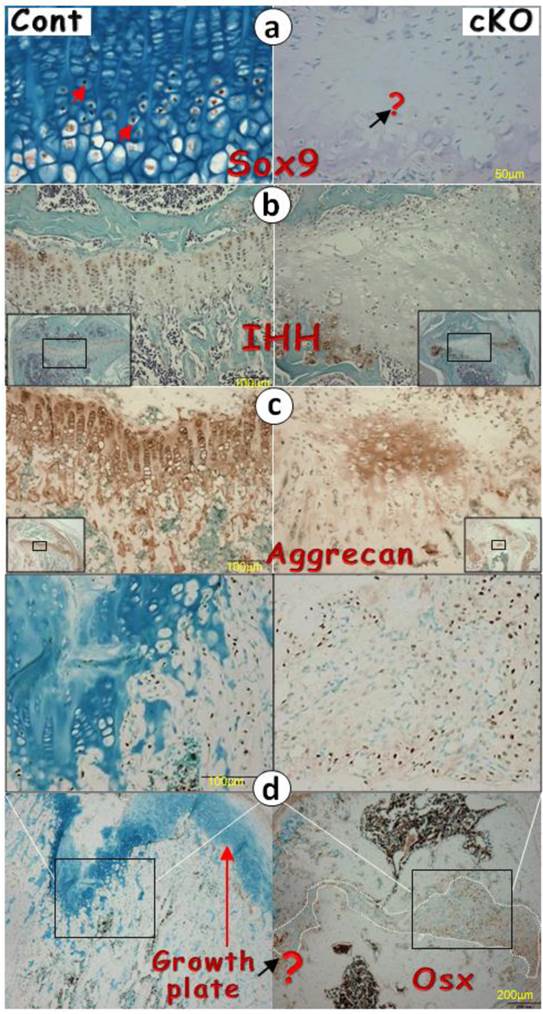
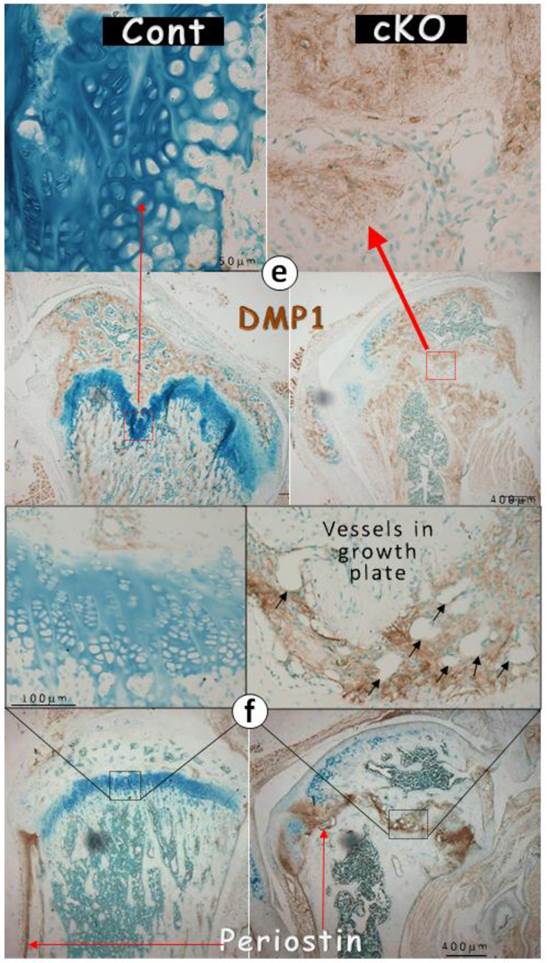
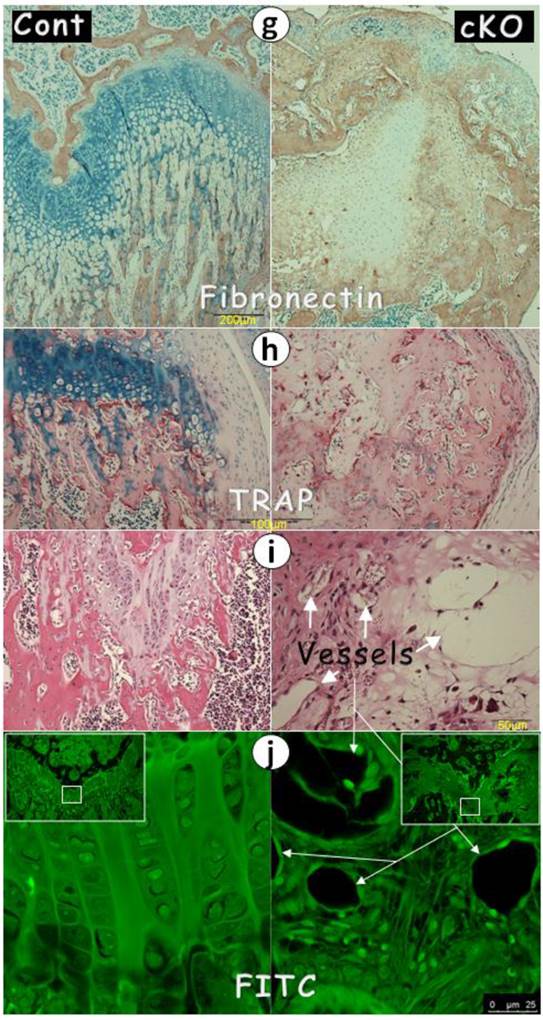
The lack of chondrogenic markers in the Bmpr1a null (cKO) growth plate (a-g) IHC stain images in the cKO growth plate indicated either a lack of signals of SOX9 (a) and IHH (b) separately, or faint signals of aggrecan (c), or ectopic expressions of OSX (d), DMP1 (e), periostin (f), or fibronectin (g); (h-i) The H&E stain images displayed numerous blood vessels and TRAP (Tartrate-Resistant Acid Phosphatase) positive cells; (j) The FITC confocal images revealed blood vessels and fibrous-like tissues in the cKO growth plate; and (k) The entire Bmpr1a-null growth plate was replaced by an ectopic bone-like structure.
Fig 5
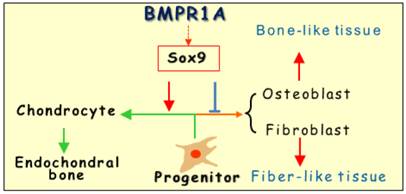
The working hypothesis is that BMPR1A determines the cell fate of the growth plate via SOX9: The progenitor cells proliferate and exit the cell cycle and undergo maturation away from the resting zone to form chondrocytes followed by endochondral bone formation. In the absence of Bmpr1a, these progenitor cells will form either osteoblast cell or fibrous like cells, leading to a mixture structure of bone (dominant) and fibrous tissue in the growth plate.
Discussion
The early embryonic studies, which showed the critical but redundant role of Bmpr1a in endochondral bone formation [26], motivated us to investigate the postnatal roles of Bmpr1a in cartilage development. Our key findings were: (1) There is a lack of long bone growth after the removal of Bmpr1a in the progenitor cells, suggesting an essential role of BMPR1A in early chondrogenesis; and (2) There is a replacement of chondrogenic markers (SOX9, IHH and glycan proteins) by osteogenic markers (such as OSX, DMP1 and periostin) in the Bmpr1a-null growth plates, causing a lack of cartilage and ectopic bone formation. Based on these findings, we propose that the default program in the growth plate is the formation of chondrocytes, in which BMPR1A determines the cell fate from two aspects: progenitor cell proliferation and exit the cell cycle and undergo maturation away from the resting zone. In the absence of Bmpr1a, these progenitor cells form either osteoblast cells (dominant) or fibrous-like cells that lead to ecotopic bone formation (Fig. 5). Concurrent with this observation, a similar role of the specification of cell fate during the development of the dorsal spinal cord by type I BMPR-A and -B was reported by Hazen et al. [8], in which the cananical Smad signaling pathway is required.
It is not currently clear why the removal of Bmpr1a alone from cartilage by Col 2 Cre results in no apparent phenotype during embryonic development [26], whereas the deletion of the same gene in cartilage by Agc1CreERT2 completely halts long bone growth. One of approaches to solving this puzzle is to induce Bmpr1a KO (a cross between Bmpr1a flox and Agc1CreERT2) in the embryonic stage, which is now being carried out in the lab. Finishing this work will distinguish the role of Bmpr1a in different developmental stages.
The work with a one-time injection of tamoxifen to remove Bmpr1a in cartilage at birth, which leads to a lack of long bone growth postnatally (Fig. 3), raises the possibility that the progenitor cell number might already be present in the growth plate at birth with no more new progenitor cells added. This observation may explain why the rodent growth plate remains unfused during the animal's lifetime but there is essentially no long bone lengthening after 28-30 weeks [22]. In fact, in the 5-month-old control mouse, there is very limited cell proliferation in the growth plate compared to the early stage (Supplementary Material: Figure S2a). On the other hand, the cell proliferation remains active in the Bmpr1a-null mice, in which the cell fate is already transferred from the chondrogenic to the osteogenic, resulting in a non-stop expansion of bone mass identical to intramembrous bone formation (Supplementary Material: Figure S2b).
Overall, the removal of Bmpr1a in early chondrogenic cells resulting in a lack of long bone growth, plus a switch of the cell fate, lend support to these views: (1) All the progenitor cells in the growth plate seem to be already stored with no more new progenitor cells added during late development; (2) the progenitor cells in the growth plate are plastic; and (3) BMPR1A plays an essential role in both the control of cell fate and postnatal cartilage formation partly through SOX9 (Fig. 5).
In summary, the above information supports the view that the progenitor cells in the postnatal growth plate are highly plastic (i.e., these progenitor cells can form other mesenchymal drived tissues when environment changes such as in Bmpr1a mice), and that BMPR1A determines the cell fate of these progenitor cells postnatally.
Supplementary Material
Fig.S1 - Fig.S2.
Acknowledgements
This study was supported by NIH grant DE018486 and by a China State Key Laboratory of Oral Diseases Open Funding grant (SKLODOF 2010-03) to JQF. Authors' roles: Study design, manuscript writing and editing: CL, XZ and JQF. Data collection and analyses: JJJ, ZZ, YM, YL, and NK. We thank Dr. Benoit de Crombrugghe (UT MD Anderson Cancer Center, USA) for his generous donation of the Aggrecan-CreERT2 knock-in mouse line. The authors gratefully acknowledge Mrs. Jeanne Santa Cruz for her English grammar editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Anderson EJ, Kaliyamoorthy S, Iwan J, Alexander D, Knothe Tate ML. Nano-microscale models of periosteocytic flow show differences in stresses imparted to cell body and processes. Ann Biomed Eng. 2005;33:52-62
2. Ascenzi MG, Roe AK. The osteon: the micromechanical unit of compact bone. Front Biosci. 2012;17:1551-81
3. Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216
4. Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR. et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. 2001;98:6698-703
5. Cao Z, Zhang H, Zhou X, Han X, Ren Y, Gao T. et al. Genetic evidence for the vital function of Osterix in cementogenesis. J Bone Miner Res. 2012;27:1080-92
6. Fen JQ, Zhang J, Dallas SL, Lu Y, Chen S, Tan X. et al. Dentin matrix protein 1, a target molecule for Cbfa1 in bone, is a unique bone marker gene. J Bone Miner Res. 2002;17:1822-31
7. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B. et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310-5
8. Hazen VM, Andrews MG, Umans L, Crenshaw EB 3rd, Zwijsen A, Butler SJ. BMP receptor-activated Smads confer diverse functions during the development of the dorsal spinal cord. Dev Biol. 2012;367:216-27
9. Henry SP, Liang S, Akdemir KC, de Crombrugghe B. The postnatal role of Sox9 in cartilage. J Bone Miner Res. 2012;27:2511-25
10. Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339-51
11. Kamiya N, Jikko A, Kimata K, Damsky C, Shimizu K, Watanabe H. Establishment of a novel chondrocytic cell line N1511 derived from p53-null mice. J Bone Miner Res. 2002;17:1832-42
12. Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M. et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007-17
13. Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM. et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development. 2008;135:3801-11
14. Kronenberg HM. Developmental regulation of the growth plate. NATURE. 2003;423:332-6
15. Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131:1309-18
16. Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harbor perspectives in biology. 2013;5:a008334
17. Martin DM, Hallsworth AS, Buckley T. A method for the study of internal spaces in hard tissue matrices by SEM, with special reference to dentine. Journal of microscopy. 1978;112:345-52
18. Miller SC, Omura TH, Smith LJ. Changes in dentin appositional rates during pregnancy and lactation in rats. Journal of dental research. 1985;64:1062-4
19. Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis. 2002;32:69-72
20. Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G. et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948-59
21. Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093-104
22. Roach HI, Mehta G, Oreffo RO, Clarke NM, Cooper C. Temporal analysis of rat growth plates: cessation of growth with age despite presence of a physis. J Histochem Cytochem. 2003;5:373-83
23. Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W. et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124:3428-40
24. Wuelling M, Vortkamp A. Chondrocyte proliferation and differentiation. Endocr Dev. 2011;21:1-11
25. Ye L, Mishina Y, Chen D, Huang H, Dallas SL, Dallas MR. et al. Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J Biol Chem. 2005;280:6197-203
26. Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:5062-7
27. Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR. et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667-78
Author contact
![]() Corresponding author: Jian Q. Feng, MD, PhD. Department of Biomedical Sciences, Baylor College of Dentistry, Texas A&M Health Science Center, 3302 Gaston Ave.Dallas, TX 75246. 214-370-7235 (phone) 214-370-7298 (fax) jfengtamhsc.edu.
Corresponding author: Jian Q. Feng, MD, PhD. Department of Biomedical Sciences, Baylor College of Dentistry, Texas A&M Health Science Center, 3302 Gaston Ave.Dallas, TX 75246. 214-370-7235 (phone) 214-370-7298 (fax) jfengtamhsc.edu.
Citation styles
APA
Jing, J., Ren, Y., Zong, Z., Liu, C., Kamiya, N., Mishina, Y., Liu, Y., Zhou, X., Feng, J.Q. (2013). BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate. International Journal of Biological Sciences, 9(9), 895-906. https://doi.org/10.7150/ijbs.7508.
ACS
Jing, J.; Ren, Y.; Zong, Z.; Liu, C.; Kamiya, N.; Mishina, Y.; Liu, Y.; Zhou, X.; Feng, J.Q. BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate. Int. J. Biol. Sci. 2013, 9 (9), 895-906. DOI: 10.7150/ijbs.7508.
NLM
Jing J, Ren Y, Zong Z, Liu C, Kamiya N, Mishina Y, Liu Y, Zhou X, Feng JQ. BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate. Int J Biol Sci 2013; 9(9):895-906. doi:10.7150/ijbs.7508. https://www.ijbs.com/v09p0895.htm
CSE
Jing J, Ren Y, Zong Z, Liu C, Kamiya N, Mishina Y, Liu Y, Zhou X, Feng JQ. 2013. BMP Receptor 1A Determines the Cell Fate of the Postnatal Growth Plate. Int J Biol Sci. 9(9):895-906.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.