Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(10):1032-1042. doi:10.7150/ijbs.7273 This issue Cite
Research Paper
Asiatic Acid Isolated From Centella Asiatica Inhibits TGF-β1-induced Collagen Expression in Human Keloid Fibroblasts via PPAR-γ Activation
Difei Bian1, Jizhou Zhang1, Xin Wu1, Yannong Dou1, Yan Yang1, Qian Tan2, Yufeng Xia1, Zhunan Gong3, Yue Dai1 ![]()
1. State Key Laboratory of Natural Medicines, Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, Nanjing 210009, China;
2. Department of Burns and Plastic Surgery, Nanjing Drum Tower Hospital, Medical School of Nanjing University, Nanjing 210008, China;
3. Center for New Drug Research & Development, College of Life Science, Nanjing Normal University, Nanjing 210024, China.
Received 2013-7-27; Accepted 2013-9-15; Published 2013-10-25
Abstract
Keloids are fibroproliferative disorders characterized by exuberant extracellular matrix deposition and transforming growth factor (TGF)-β/Smad pathway plays a pivotal role in keloid pathogenesis. Centella asiatica extract has been applied in scar management for ages. As one of its major components, asiatic acid (AA) has been recently reported to inhibit liver fibrosis by blocking TGF-β/Smad pathway. However, its effect on keloid remains unknown. In order to investigate the effects of AA on cell proliferation, invasion and collagen synthesis, normal and keloid fibroblasts were exposed to TGF-β1 with or without AA. Relevant experiments including 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, 5-ethynyl-2-deoxyuridine (EdU) incorporation assay, Transwell invasion assay, enzyme-linked immunosorbent assay, Western blot, quantitative polymerase chain reaction and RNA interference assay were conducted. As a result, keloid fibroblasts showed higher responsiveness to TGF-β1 stimulation than normal fibroblasts in terms of invasion and collagen synthesis. AA could suppress TGF-β1-induced expression of collagen type I, inhibit Smad 2/3 phosphorylation and plasminogen activator inhibitor-1 (PAI-1) expression, while elevate Smad 7 protein level. Noteworthy, the effects of AA on keloid fibroblasts could be abrogated by PPAR-γ antagonist GW9662 and by silencing of PPAR-γ. The present study demonstrated that AA inhibited TGF-β1-induced collagen and PAI-1 expression in keloid fibroblasts through PPAR-γ activation, which suggested that AA was one of the active constituents of C. asiatica responsible for keloid management, and could be included in the arsenal for combating against keloid.
Keywords: keloid, TGF-β1, asiatic acid, PPAR-γ, collagen
Introduction
Wound healing is a complicated process, there are roughly three overlapping phases, inflammation, granulation and remodeling[1]. Any derailment in the process would lead to a variety of ailments, including chronic wound healing[2], hypertrophic scar[3] and keloid[4].
As excessive healing, keloid and hypertrophic scar are both aberrant responses to insults such as burn, trauma and surgery, resulting in exuberant collagen deposition. However, keloid differs from hypertrophic scar in several aspects including invasive growth beyond the original wound margins; hardly regress with time; and high recurrence rate after excision[5]. Furthermore, keloids have specific clinical features as locating predominately on ear lobe and shoulder, occurring readily during puberty, and having a predilection for dark skin color[6]. The major concerns of keloids are pain and pruritus, together with aesthetic and psychological problems. Several therapeutic modalities are available to date, however, none of them are universally effective[7]. Hence, there is unmet need for developing better therapeutic alternatives in keloid treatment.
The pathogenesis of keloid still remains elusive. However, transforming growth factor (TGF)-β, a potent profibrotic factor that stimulates collagen synthesis, is the focus in keloid researches. Among the three isoforms, TGF-β1 has been of most well investigated. Previous studies showed higher expression of TGF-β1 in keloid tissue as well as in keloid-derived fibroblasts[8, 9]. As in a milieu of elevated TGF-β1 level, keloid fibroblasts exhibited highly phosphorylated Smad 2/3, compared with normal fibroblasts[10]. On the contrary, Smad 7, which plays an inhibitory role by antagonizing Smad 2/3 signaling, was downregulated in keloids[11]. Therefore, it is a promising approach for the development of keloid therapeutics by targeting TGF-β/Smad pathway.
Centella Asiatica is an annual herbs growing in tropical swampy areas, which is frequently blended as a daily drink and eaten as vegetable in Southeast Asia. Moreover, C. Asiatica extract has been applied for scar management for ages[12]. Previously, we screened the anti-scar effects of asiaticoside, madecassoside, asiatic acid and madecassic acid, four major triterpenoid ingredients in C. Asiatica extract, and identified that asiatic acid (AA) was the most effective constituent. AA also exhibited apoptotic effects in several tumor cell lines[13-15]. Recent study reported that AA can inhibit liver fibrosis by abrogating TGF-β/Smad pathway[16]. Moreover, as the corresponding glycoside of AA, asiaticoside suppresses collagen expression in keloid fibroblasts[17]. As keloid is a kind of cutaneous fibrosis and considered as benign tumor sometimes, the effect of AA on keloid has yet not been explored. The current study was to investigate the effects of AA on cell proliferation, invasion and collagen synthesis and the underlying mechanisms.
Materials and methods
Chemicals and Reagents
Asiatic acid (C30H48O5, MW: 488.70) was gifted by Dr. Zhunan Gong, and the purity (≥98%) was determined by HPLC-ELSD. A voucher specimen (Gong 0703) was maintained in the Center for New Drug Research & Development, College of Life Science, Nanjing Normal University. 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), GW9662 and Type I Collagenase were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Rosiglitazone (Ros) was the product of Cayman Chemical Co. (Ann Arbor, MI, USA).
Primary cell origin and culture
Keloid specimens were obtained from eight female Chinese patients after surgical excision (ranging from 18 to 31 years with mean age of 25.1 years). All included patients had not been received any treatment nor with systemic disease. Three foreskin samples were collected from patients with circumcision surgery (ranging from 17 to 27 years with mean age of 22.3 years). All patients were informed with written consent. The study was conducted in accordance with the Declaration of Helsinki and its subsequent amendments, and further approved by the Ethics Committee of Drum Tower Hospital (Nanjing, China). Primary keloid and normal fibroblasts were isolated from tissue samples by enzymatic digestion, as previously described[18]. Cells were cultured in Dulbecco's modified Eagle medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal calf serum (Gibco, Grand Island, NY, USA), 100 U/mL streptomycin and penicillin. Only cells from passage 2 to 6 were used in the study.
Cell viability assay
The viability of fibroblasts was detected by MTT assay. Normal and keloid fibroblasts (5 × 103 cells/well) seeded in 96-well plates were incubated with AA (1, 3, 10, 30 and 100 μM) and TGF-β1 (5 ng/mL, PeproTech, Rocky Hill, NJ, USA) for 24 h. Four hours before the end of the incubation, aliquots (20 μL) of MTT were added into each well. The medium were aspirated and 150 μL DMSO were added to dissolve the formazan crystals. The absorbance at 570 nm presents cell viability.
Cell proliferation assay
The proliferation of fibroblasts was determined by 5-ethynyl-2-deoxyuridine (EdU) incorporation kit (RiboBio, Guangzhou, China). Fibroblasts (3 × 103 cells/well) seeded in 96-well plates were incubated with AA (3, 10 and 30 μM) and TGF-β1 (5 ng/mL) in addition to EdU for 24 h. Cells were fixed and stained after incubation according to the manufacturer's instructions. The proliferation rate was calculated by normalizing the number of EdU-positive cells to Hoechst 33342-stained cells in five random fields.
Collagen expression assay
Cells (3 × 104 cells/well) were seeded into 48-well plates and further incubated with AA (3, 10 and 30 μM) and TGF-β1 (5 ng/mL) for 24 h. The content of collagen type I in supernatants was detected by enzyme-linked immunosorbent assay (ELISA) using a commercially available kit (R&D Systems, Minneapolis, MN, USA).
Cell invasion assay
Normal and keloid fibroblasts (1 × 105 cells/well) were inoculated into the upper chamber of 8 μm pore size Transwell insert coated with Matrigel (BD Bioscience, Bedford, MA, USA). After 24 h incubation with AA (3, 10 and 30 μM) and TGF-β1 (5 ng/mL), migrated cells on the lower side of insert were fixed with 4 % formalin. Crystal violet were stained and further extracted with 10% acetic acid. The absorbance at 540 nm represents the number of cells invaded across the Matrigel.
Western blot analysis
Western blot was conducted as previously described[18] with specific primary antibodies for Smad 2/3, phosphorylated-Smad 2, phosphorylated-Smad 3 (Cell Signaling Technology, Beverly, MA, USA), plasminogen activator inhibitor-1 (PAI-1), PPAR-γ (Bioworld Technology, Louis Park, MN, USA) and Smad 7 (Epitomics, Burlingame, CA, USA). Protein bands were developed through Luminata Crescendo Western HRP Substrate (Millipore, Billerica, MA, USA), and the optical density of each band was measured with Image J (US NIH, Bethesda, MD, USA).
Quantitative polymerase chain reaction (qPCR) assay
qPCR with SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA, USA) was performed using Bio-Rad MyiQ2 Detection System (Bio-Rad, Hercules, CA, USA). Primers for PPAR-γ (forward: 5'-CAT GGC AAT TGA ATG TCG TGT C-3'; reverse: 5'-CCG GAA GAA ACC CTT GCA T-3') and Smad 7 (forward: 5'-GAA TCT TAC GGG AAG ATC AAC CC-3'; reverse: 5'-CGC AGA GTC GGC TAA GGT G-3') were analyzed together with the reference gene GAPDH (forward: 5'-GGT GAA GGT CGG TGT GAA CG-3'; reverse: 5'-CTC GCT CCT GGA AGA TGG TG-3'). The transcription of the target gene was analyzed by ΔΔ cycle threshold (2-ΔΔCt) method to determine the fold change.
Gene-silencing by small interfering RNA (siRNA)
Three pairs of siRNA targeting human PPAR-γ together with one pair of scrambled RNA were designed and synthesized by RiboBio Co. (RiboBio, Guangzhou, China). The transfection of siRNA was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at a final concentration of 50 nM according to manufacturer's instruction. Forty-eight hours after transfection, cells were incubated with TGF-β1 (5 ng/mL) and AA (30 μM) or Ros (2 μM) for another 24 h. The supernatants and protein extracts were harvested for further analysis.
Statistical Analysis
All data were expressed as mean ± standard deviation (SD). Differences between control and experimental groups were analyzed by one-way analysis of variance followed by Dunnett's test, and p < 0.05 was considered as statistically significant. All calculations were performed using SPSS statistical software (SPSS, Chicago, IL, USA).
Results
Effects of AA on cell viability in normal and keloid fibroblasts
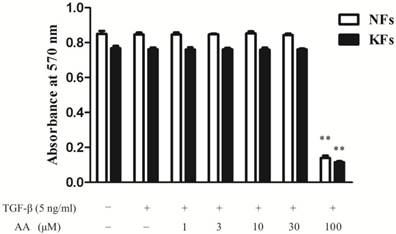
As shown in Fig. 1, AA (1, 3, 10 and 30 μM) did not influence the cell viability in either normal or keloid fibroblasts, while AA (100 μM) inhibited both normal and keloid fibroblasts viability significantly. Therefore, AA (3, 10 and 30 μM) were applied in the following experiments.
Effects of asiatic acid (AA) on cell viability in normal and keloid fibroblasts (NFs and KFs). Cells were incubated with different concentrations of AA and TGF-β1 (5 ng/mL) for 24 h. Cell viability was determined by the absorbance at 570 nm. Each column represents the mean ± SD from three independent experiments. **p < 0.01 vs. model.
Effects of AA on TGF-β1-induced cell proliferation in normal and keloid fibroblasts
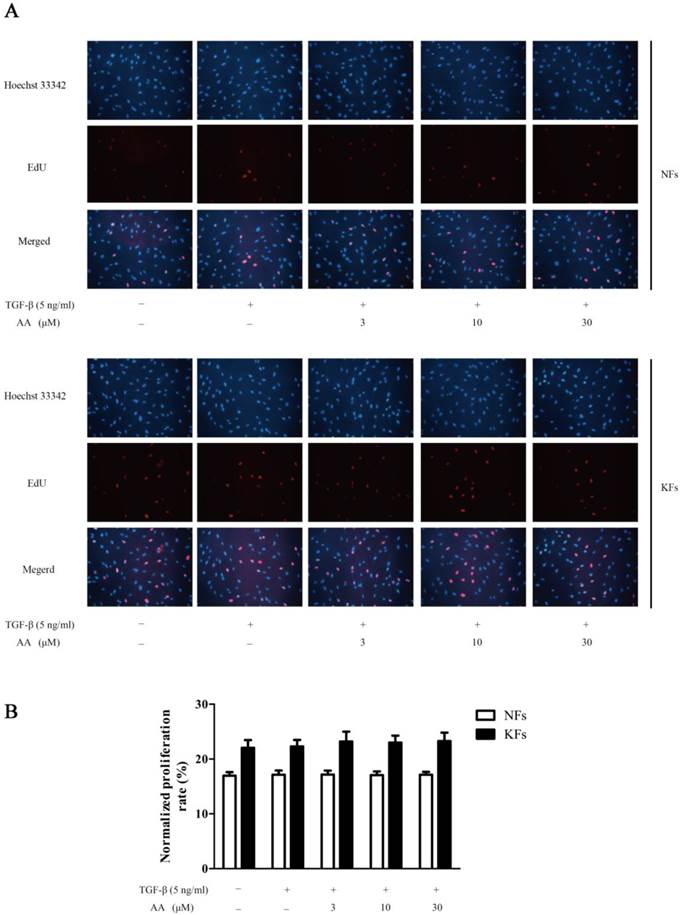
As shown in Fig. 2, keloid fibroblasts were more proliferative than normal fibroblasts. TGF-β1 (5 ng/mL) stimulation, however, did not show apparent enhancement on cell proliferation. Moreover, different concentrations of AA showed no effect on TGF-β1-stimulated cell proliferation in either normal or keloid fibroblasts.
Effects of asiatic acid (AA) on TGF-β1-induced cell proliferation in normal and keloid fibroblasts (NFs and KFs). A: Cells were immunofluorescence stained for 5-ethynyl-2-deoxyuridine (EdU) and Hoechst 33342 after TGF-β1 (5 ng/mL) and different concentrations of AA incubation for 24 h; B: Proliferation rates were calculated by normalizing the numbers of EdU-positive cells to Hoechst-stained cells in five random fields at 250× magnification. Each column represents the mean ± SD from three independent experiments.
Effects of AA on TGF-β1-induced collagen type I expression in normal and keloid fibroblasts
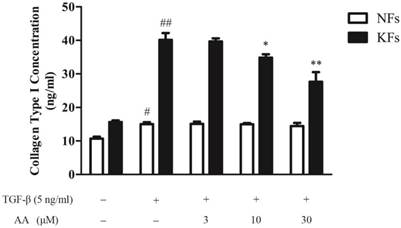
As shown in Fig. 3, keloid fibroblasts had a higher baseline level and responded more briskly to TGF-β1 stimulation in collagen type I expression, compared to normal fibroblasts. AA (10 and 30 μM) significantly decreased TGF-β1-induced collagen type I expression in keloid fibroblasts, while had little effect on normal fibroblasts.
Effects of asiatic acid (AA) on TGF-β1-induced collagen type I expression in normal and keloid fibroblasts (NFs and KFs). Cells were treated with AA (3, 10 and 30 μM) together with TGF-β1 (5 ng/mL) for 24 h. Collagen type I concentrations in supernatants were determined by ELISA. Each column represents the mean ± SD from three independent experiments. #p < 0.05, ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. model.
Effects of AA on TGF-β1-induced invasion in normal and keloid fibroblasts
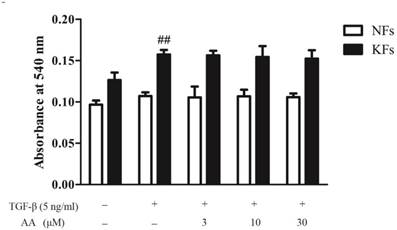
As shown in Fig. 4, invasion capability of keloid fibroblasts was higher than that of normal fibroblasts, and TGF-β1 (5 ng/mL) treatment significantly elevated cell invasion in keloid fibroblasts. However, AA incubation could not affect TGF-β1-induced invasion in either normal or keloid fibroblasts.
Effects of asiatic acid (AA) on TGF-β1-induced cell invasion in normal and keloid fibroblasts (NFs and KFs). Cells were inoculated in Transwell upper chamber and were incubated with various concentrations of AA and TGF-β1 (5 ng/mL) for 24 h. Migrated cells were stained with Crystal violet and the numbers of migrated cells were determined by absorbance at 540 nm. Each column represents the mean ± SD from three independent experiments. ##p < 0.01 vs. control.
PPAR-γ antagonist abrogated the inhibition of AA and Ros on collagen type I expression and the Smad 2/3 phosphorylation induced by TGF-β1 in keloid fibroblasts
Peroxisome proliferator-activated receptors (PPARs) are ubiquitously expressed in cutaneous tissues, while PPAR-γ was closely related to fibrosis. PPAR-γ agonists have been reported to block TGF-β1 signaling pathway in organ fibrosis, and troglitazone could suppress TGF-β1-induced collagen type I expression in keloid fibroblasts. Therefore, we investigated whether PPAR-γ antagonist GW9662 could abrogate the inhibition of AA on TGF-β1-induced collagen type I expression in keloid fibroblasts. As shown in Fig. 5A, AA (30 μM) and Ros (2 μM) significantly suppressed collagen type I expression induced by TGF-β1, and this inhibition was abrogated by GW9662 (1 μM). GW9662 also increased the Smad 2/3 phosphorylation in keloid fibroblasts (Fig. 5B & 5C), which was consistent with that of collagen type I expression.
PPAR-γ antagonist (GW9662) abrogated the inhibition of asiatic acid (AA) and rosiglitazone (Ros) on collagen type I expression and Smad 2/3 phosphorylation induced by TGF-β1 in keloid fibroblasts. A: Cells were treated as indicated and collagen type I concentrations in the supernatant were determined by ELISA; B: Phosphorylation of Smad 2/3 was measured by Western blot assay; C: Optical density of each band normalized to corresponding GAPDH band. Each column represents the mean ± SD from three independent experiments. ##p < 0.01 vs. control; **p < 0.01 vs. indicated group.
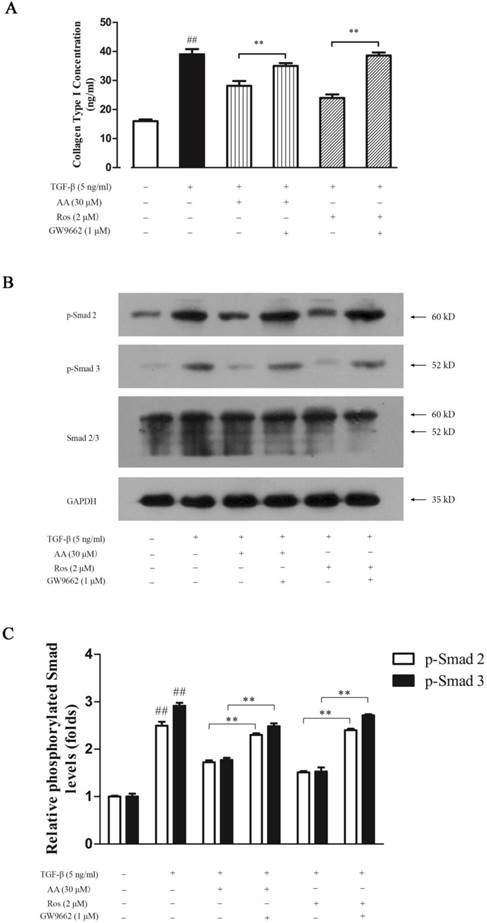
PPAR-γ antagonist abrogated the promotion of AA and Ros on Smad 7 expression induced by TGF-β1 in keloid fibroblasts
As an inhibitory factor in TGF-β/Smad signaling pathway, Smad 7 expressions in both mRNA and protein levels were suppressed under TGF-β1 treatment. As shown in Fig. 6, Smad 7 was significantly inhibited in TGF-β1-treated keloid fibroblasts, while AA (30 μM) and Ros (2 μM) nearly completely reversed this suppression, which was abrogated by GW9662 (1 μM).
PPAR-γ antagonist (GW9662) abrogated the promotion of asiatic acid (AA) and rosiglitazone (Ros) on Smad 7 expression suppressed by TGF-β1 in keloid fibroblasts. A: Cells were treated as indicated and the mRNA levels of Smad 7 were determined by qPCR and were calculated by using 2-ΔΔCt method; B: Cells were treated as indicated and cell lysate were collected for Western blot detection; C: Optical density of each band was normalized to corresponding GAPDH band. Each column represents the mean ± SD from three independent experiments. ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. indicated group.
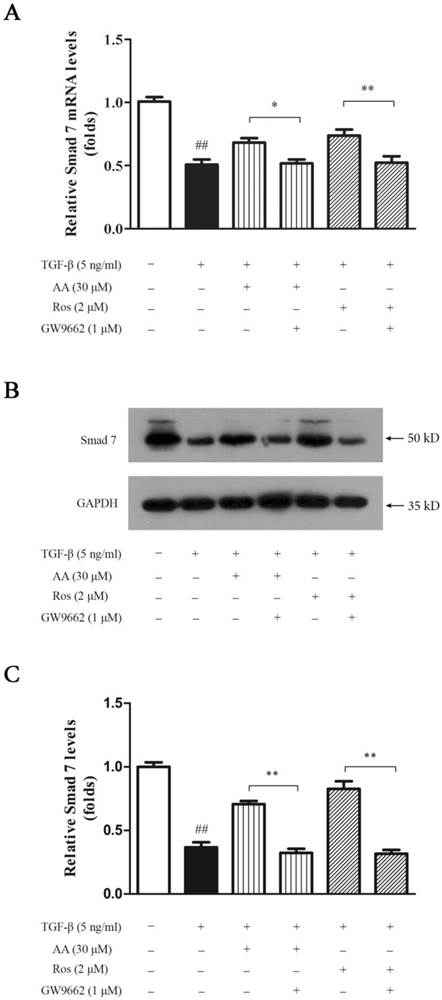
PPAR-γ antagonist abrogated the inhibition of AA and Ros on PAI-1 expression induced by TGF-β1 in keloid fibroblasts
As a direct target of TGF-β/Smad signaling pathway, plasminogen activator inhibitor (PAI)-1 is highly expressed in keloid which inhibits urokinase-type and tissue-type plasminogen activators, and thus prevents collagen degradation. As shown in Fig. 7, TGF-β1 significantly elevated PAI-1 expression in keloid fibroblasts, while both AA (30 μM) and Ros (2 μM) suppressed the PAI-1 upregulation induced by TGF-β1 in keloid fibroblasts, which was abrogated by GW9662 (1 μM).
PPAR-γ antagonist (GW9662) abrogated the inhibition of asiatic acid (AA) and rosiglitazone (Ros) on plasminogen activator inhibitor (PAI)-1 expression induced by TGF-β1 in keloid fibroblasts. A: Cells were treated as indicated and cell lysate were harvested for Western blot detection; B: Optical density of each band was normalized to corresponding GAPDH band. Each column represents the mean ± SD from three independent experiments. ##p < 0.01 vs. control; **p < 0.01 vs. indicated group.
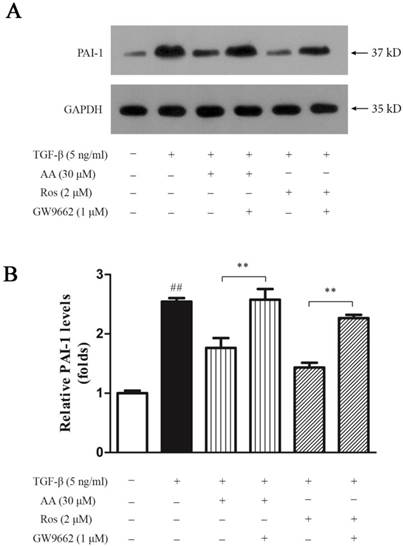
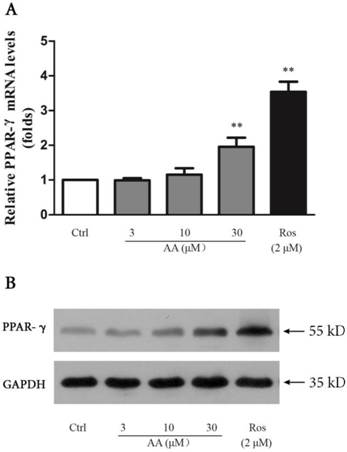
Effects of AA and Ros on PPAR-γ expression in keloid fibroblasts
AA suppressed TGF-β1-induced collagen type I expression in keloid fibroblasts, and this suppression was abrogated by PPAR-γ antagonist GW9662. Thus, we investigated whether AA could activate PPAR-γ pathway. As shown in Fig. 8, AA (10 and 30 μM) concentration-dependently enhanced PPAR-γ expression while Ros (2 μM) as a full PPAR-γ agonist, strongly elevated PPAR-γ mRNA level in keloid fibroblasts. The activation of PPAR-γ pathway was further evidenced by Western blot assay, as PPAR-γ protein expression was also increased after AA and Ros treatment.
Effects of asiatic acid (AA) and rosiglitazone (Ros) on PPAR-γ mRNA and protein expression in keloid fibroblasts. Cells were treated with AA (3, 10 and 30 μM) or Ros (2 μM) for 24 h. A: The mRNA levels of PPAR-γ were determined by qPCR and were calculated by using 2-ΔΔCt method. B: The protein levels of PPAR-γ were determined by Western blot. Each column represents the mean ± SD from three independent experiments. **p < 0.01 vs. control.
PPAR-γ silencing abrogated the inhibition of AA and Ros on collagen type I expression induced by TGF-β1 in keloid fibroblasts
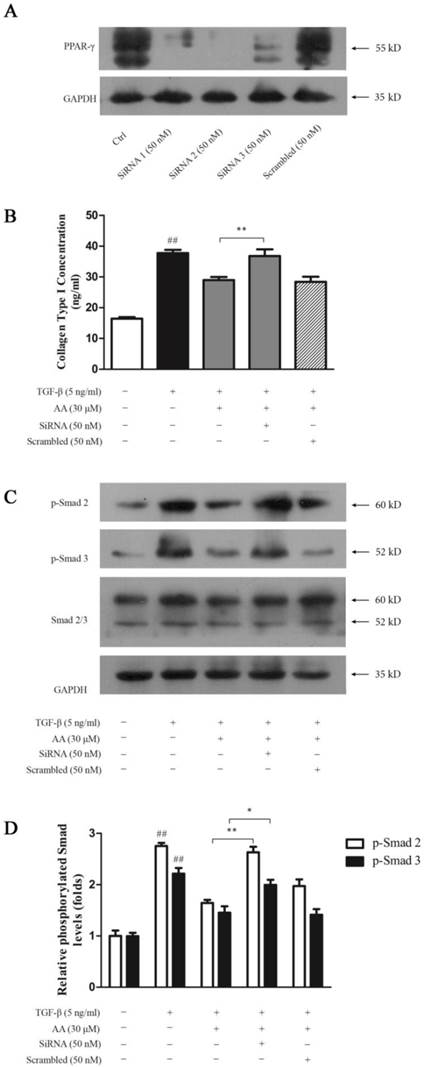
To investigate whether PPAR-γ is essential in inhibition of AA on TGF-β1-induced collagen type I expression in keloid fibroblasts, we silenced PPAR-γ gene with siRNA. As shown in Fig. 9A, all three pairs of siRNA inhibited PPAR-γ expression to various degrees while Pair 2 was of the best efficiency. PPAR-γ silencing abrogated suppression of AA on TGF-β1-induced collagen type I expression (Fig. 9B) as well as Smad 2/3 phosphorylation in keloid fibroblasts, while scrambled RNA exhibited no effect (Fig. 9C and 9D).
PPAR-γ silencing abrogated the inhibition of asiatic acid (AA) and rosiglitazone (Ros) on collagen type I expression and Smad 2/3 phosphorylation induced by TGF-β1 in keloid fibroblasts. A: Cell PPAR-γ protein levels were determined by Western blot after incubation with three pairs of small interfering RNA (SiRNA) or scrambled RNA for 6 h; B: Cells were treated as indicated and collagen type I concentrations in the supernatant were determined by ELISA; C: Phosphorylation of Smad 2/3 was measured by Western blot assay; D: Optical density of each band normalized to corresponding GAPDH band. Each column represents the mean ± SD from three independent experiments. ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. indicated group.
Discussion
Keloid, a result of abnormal wound healing, is characterized as exuberant collagen deposition and invasive growth beyond original wound margins. The high recurrence rate after surgical excision and lack of effective therapeutic modalities still remain unresolved[19]. In our current study, we demonstrated for the first time that AA suppressed phosphorylation of Smad 2/3 induced by TGF-β1 stimulation, and thus inhibited collagen type I expression in keloid fibroblasts. Moreover, AA elevated Smad 7 protein level and suppressed PAI-1 expression in TGF-β1-treated keloid fibroblasts. Noteworthy, the effects of AA on keloid fibroblasts were mediated by PPAR-γ since these effects were abrogated by PPAR-γ antagonist GW9662 and by PPAR-γ silencing.
As a form of cutaneous fibrosis, keloid shares common features with other kinds of fibrosis, mainly excessive extracellular matrix deposition. TGF-β1, one of the members in the TGF superfamily, is a potent profibrotic factor that strongly induces collagen synthesis and thus was well-studied in fibrogenesis [20]. Phosphorylation of receptor-regulated SMADs (R-Smads), namely Smad 2/3 after the binding of TGF-β1 to TGF-β type I and type II receptor, indicates the activation of TGF-β/Smad signaling pathway. Phosphorylated Smad 2/3 further forms heterodimer with common Smad (co-Smad) and is translocated to nuclei to mediate downstream gene expressions, which results in profibrogenesis. Smad 7, an inhibitory Smad (I-Smad), is the transcriptional target and therefore transactivated by TGF-β stimulation. Smad 7 plays a negative feedback role in TGF-β/Smad signaling pathway by antagonizing Smad 2/3 phosphorylation and degrading TGF-β receptors[21]. However, Yu et al. [22] found a decreased expression of inhibitory Smad 7 in keloid fibroblasts, comparing to normal skin and normal scar. Our results showed that TGF-β1 stimulation induced collagen type I expression, phosphorylated of Smad 2/3 and suppressed Smad 7 expression in keloid fibroblasts, which were in accordance with previous and recent published work[23-25]. However, TGF-β1 stimulation failed to affect cell proliferation in either normal or keloid fibroblasts, which was consistent with previous study[23]. Moreover, keloid fibroblasts responded more briskly to TGF-β1 stimulation than normal fibroblasts in terms of collagen expression and cell invasion[9, 23]. AA (10 and 30 μM) inhibited TGF-β1-induced collagen type I expression in keloid fibroblasts in a concentration-dependent manner. This effect might be achieved by suppression of TGF-β/Smad signaling pathway while without interfering with cell numbers.
On the other hand, PAI-1, as a definite target gene of TGF-β/Smad signaling, plays a major role in various kinds of tissue fibrosis[26]. It inhibits activation of tissue-type and urokinase-type plasminogen activators (tPA and uPA) and consequently suppresses degradation of extracellular matrix, which contributes to keloid formation[27]. Our current study demonstrated that AA significantly downregulated PAI-1 protein expression and blocked TGF-β1 stimulated PAI-1 expression in keloid fibroblasts. Due to its effect on balancing the plasminogen proteolytic system, AA could impede keloid formation partially through enhancing collagen degradation.
PPARs belong to nuclear receptor superfamily, which include three isoforms, PPAR-α, PPAR-δ and PPAR-γ. They are ubiquitously expressed in tissue and functioning as a classic adipogenic regulator. As a target for insulin sensitization in type II diabetes mellitus (T2DM) treatment, PPAR-γ has been extensively studied. Upon binding its agonist, PPAR-γ, often binds with retinoid X receptor (RXR) α as a heterodimer, undergoes conformational changes and regulates target gene expression by attaching to specific peroxisome proliferation response elements (PPREs) in the promoter region of downstream target gene[28]. In addition to its role in metabolic homeostasis regulation, emerging effects of PPAR-γ have been reported including anti-inflammatory, anti-tumor and anti-fibrotic potentials especially[29-31]. TGF-β/Smad signaling blockage by PPAR-γ activation which leads to decreased collagen deposition and epithelial-mesenchymal transition has been observed in hepatic, pulmonary, and renal fibrosis[32-34]. Furthermore, troglitazone, as a full agonist of PPAR-γ, has been claimed to suppress TGF-β1-induced collagen type I expression in keloid fibroblasts[35]. Ghosh et al. [36] also reported the activation of PPAR-γ could abrogate the profibrotic effect of TGF-β1 in scleroderma, while without interfering with the cellular expression of Smad 7, which is inconsistent with our results. This difference might be partially due to the heterogeneity of keloid fibroblasts and different detection time points when measuring cellular Smad 7 protein level.
Interestingly, PPAR-γ also integrated with TGF-β/Smad signaling pathway in cancer biology. The activation of PPAR-γ could antagonize TGF-β1's effect on cancer growth and metastasis[37]. Taken the apoptotic effect of AA on several tumor cell lines[13-15] together with its recently reported therapeutic effect on liver fibrosis by blocking TGF-β/Smad signaling into consideration[16], we postulated that AA exerted its suppressive effect on TGF-β/Smad signaling through PPAR-γ activation in keloid fibroblasts. Noteworthy, the inhibition of AA on TGF-β1-induced collagen type I expression in keloid fibroblasts could be abrogated by PPAR-γ antagonist GW9662 and by gene silencing. Moreover, elevated PPAR-γ mRNA level after AA incubation in keloid fibroblasts also added credit to our previous hypothesis. Importantly, functioned as a partial PPAR-γ agonist, AA has no such side effects as fluid retention and bone fractures, which occur in full PPAR-γ agonist treatment[38].
Keloid, together with other tissue fibrosis, is intractable disease lacking universally effective therapeutic regimen. Intralesional corticosteroids injection is prevalent in clinical keloid management despite it may cause side effects including tissue atrophy, hypopigmentation or telangiectasia[5]. Pirfenidone is a promising small molecule drug in treating pulmonary fibrosis[39]. However, whether it is effective in keloid treatment remains elusive. Therefore, there is unmet need for alternatives in keloid management, and the study of targeting TGF-β/Smad signaling pathway might lead to clinical application.
In conclusion, this study, for the first time, demonstrated that AA inhibited TGF-β1-induced collagen type I and PAI-1 expression while elevating Smad 7 protein level in keloid fibroblasts. The mechanism was via PPAR-γ activation. Future in vivo experiment is warranted and AA could be exploited as a new approach for keloid treatment.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 30973915), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and Innovative Research Plan for Undergraduate Students of China Pharmaceutical University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314-21 doi:10.1038/nature07039
2. Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010;163:257-68 doi:10.1111/j.1365-2133.2010.09804.x
3. van der Veer WM, Bloemen MC, Ulrich MM, Molema G, van Zuijlen PP, Middelkoop E. et al. Potential cellular and molecular causes of hypertrophic scar formation. Burns. 2009;35:15-29 doi:10.1016/j.burns.2008.06.020
4. Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010;18:139-53 doi:10.1111/j.1524-475X.2009.00553.x
5. Wolfram D, Tzankov A, Pulzl P, Piza-Katzer H. Hypertrophic scars and keloids--a review of their pathophysiology, risk factors, and therapeutic management. Dermatol Surg. 2009;35:171-81 doi:10.1111/j.1524-4725.2008.34406.x
6. Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H. Keloids: current concepts of pathogenesis (review). Int J Mol Med. 2009;24:283-93
7. Viera MH, Caperton CV, Berman B. Advances in the treatment of keloids. J Drugs Dermatol. 2011;10:468-80
8. Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker MT. Expression of transforming growth factor beta 1, 2, and 3 proteins in keloids. Ann Plast Surg. 1999;43:179-84
9. Fujiwara M, Muragaki Y, Ooshima A. Keloid-derived fibroblasts show increased secretion of factors involved in collagen turnover and depend on matrix metalloproteinase for migration. Br J Dermatol. 2005;153:295-300 doi:10.1111/j.1365-2133.2005.06698.x
10. Phan TT, Lim IJ, Chan SY, Tan EK, Lee ST, Longaker MT. Suppression of transforming growth factor beta/smad signaling in keloid-derived fibroblasts by quercetin: implications for the treatment of excessive scars. J Trauma. 2004;57:1032-7
11. Yu H, Bock O, Bayat A, Ferguson MW, Mrowietz U. Decreased expression of inhibitory SMAD6 and SMAD7 in keloid scarring. J Plast Reconstr Aesthet Surg. 2006;59:221-9
12. Widgerow AD, Chait LA, Stals R, Stals PJ. New innovations in scar management. Aesthetic Plast Surg. 2000;24:227-34
13. Lee YS, Jin DQ, Kwon EJ, Park SH, Lee ES, Jeong TC. et al. Asiatic acid, a triterpene, induces apoptosis through intracellular Ca2+ release and enhanced expression of p53 in HepG2 human hepatoma cells. Cancer Lett. 2002;186:83-91
14. Park BC, Bosire KO, Lee ES, Lee YS, Kim JA. Asiatic acid induces apoptosis in SK-MEL-2 human melanoma cells. Cancer Lett. 2005;218:81-90 doi:10.1016/j.canlet.2004.06.039
15. Hsu YL, Kuo PL, Lin LT, Lin CC. Asiatic acid, a triterpene, induces apoptosis and cell cycle arrest through activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways in human breast cancer cells. J Pharmacol Exp Ther. 2005;313:333-44 doi:10.1124/jpet.104.078808
16. Tang LX, He RH, Yang G, Tan JJ, Zhou L, Meng XM. et al. Asiatic Acid Inhibits Liver Fibrosis by Blocking TGF-beta/Smad Signaling In Vivo and In Vitro. PLoS One. 2012;7:e31350. doi:10.1371/journal.pone.0031350
17. Tang B, Zhu B, Liang Y, Bi L, Hu Z, Chen B. et al. Asiaticoside suppresses collagen expression and TGF-beta/Smad signaling through inducing Smad7 and inhibiting TGF-betaRI and TGF-betaRII in keloid fibroblasts. Arch Dermatol Res. 2011;303:563-72 doi:10.1007/s00403-010-1114-8
18. Wu F, Bian D, Xia Y, Gong Z, Tan Q, Chen J. et al. Identification of Major Active Ingredients Responsible for Burn Wound Healing of Centella asiatica Herbs. Evid Based Complement Alternat Med. 2012;2012:848093. doi:10.1155/2012/848093
19. Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17:113-25 doi:10.2119/molmed.2009.00153
20. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210 doi:10.1002/path.2277
21. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816-27 doi:10.1096/fj.03-1273rev
22. Yu H, Bock O, Bayat A, Ferguson MW, Mrowietz U. Decreased expression of inhibitory SMAD6 and SMAD7 in keloid scarring. J Plast Reconstr Aesthet Surg. 2006;59:221-9
23. He S, Yang Y, Liu X, Huang W, Zhang X, Yang S. Compound Astragalus and Salvia miltiorrhiza extract inhibits cell proliferation, invasion and collagen synthesis in keloid fibroblasts by mediating transforming growth factor-beta / Smad pathway. Br J Dermatol. 2012;166:564-74 doi:10.1111/j.1365-2133.2011.10674.x
24. Wu CS, Wu PH, Fang AH, Lan CC. FK506 inhibits the enhancing effects of transforming growth factor (TGF)-beta1 on collagen expression and TGF-beta/Smad signalling in keloid fibroblasts: implication for new therapeutic approach. Br J Dermatol. 2012;167:532-41 doi:10.1111/j.1365-2133.2012.11023.x
25. He S, Liu X, Yang Y, Huang W, Xu S, Yang S. et al. Mechanisms of transforming growth factor beta(1)/Smad signalling mediated by mitogen-activated protein kinase pathways in keloid fibroblasts. Br J Dermatol. 2010;162:538-46 doi:10.1111/j.1365-2133.2009.09511.x
26. Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493-507 doi:10.1002/jcp.22783
27. Tuan TL, Hwu P, Ho W, Yiu P, Chang R, Wysocki A. et al. Adenoviral overexpression and small interfering RNA suppression demonstrate that plasminogen activator inhibitor-1 produces elevated collagen accumulation in normal and keloid fibroblasts. Am J Pathol. 2008;173:1311-25 doi:10.2353/ajpath.2008.080272
28. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409-35 doi:10.1146/annurev.med.53.082901.104018
29. Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244-51 doi:10.1016/j.tips.2005.03.003
30. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219-46 doi:10.1146/annurev-physiol-021909-135846
31. Tachibana K, Yamasaki D, Ishimoto K, Doi T. The Role of PPARs in Cancer. PPAR Res. 2008;2008:102737. doi:10.1155/2008/102737
32. Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350:385-91 doi:10.1016/j.bbrc.2006.09.069
33. Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, Huxlin KR. et al. Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 2009;41:722-30 doi:10.1165/rcmb.2009-0006OC
34. Wang W, Liu F, Chen N. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-beta1 in renal interstitial fibroblasts. Mediators Inflamm. 2007;2007:62641. doi:10.1155/2007/62641
35. Zhang GY, Yi CG, Li X, Ma B, Li ZJ, Chen XL. et al. Troglitazone suppresses transforming growth factor-beta1-induced collagen type I expression in keloid fibroblasts. Br J Dermatol. 2009;160:762-70 doi:10.1111/j.1365-2133.2008.08989.x
36. Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305-18 doi:10.1002/art.20104
37. Lee CH, Kim HD, Shin SM, Kim SG. A Novel Mechanism of PPARgamma Regulation of TGFbeta1: Implication in Cancer Biology. PPAR Res. 2008;2008:762398. doi:10.1155/2008/762398
38. Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends Endocrinol Metab. 2012;23:205-15 doi:10.1016/j.tem.2012.03.001
39. Hilberg O, Simonsen U, du Bois R, Bendstrup E. Pirfenidone: significant treatment effects in idiopathic pulmonary fibrosis. Clin Respir J. 2012;6:131-43 doi:10.1111/j.1752-699X.2012.00302.x
Author contact
![]() Corresponding author: Prof. Yue Dai, Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, 24 Tong jia Xiang, Nanjing 210009, People's Republic of China. Tel.: +86-25-8327-1400 Fax: +86-25-8530-1528 Email: yuedaicpucom.
Corresponding author: Prof. Yue Dai, Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, 24 Tong jia Xiang, Nanjing 210009, People's Republic of China. Tel.: +86-25-8327-1400 Fax: +86-25-8530-1528 Email: yuedaicpucom.