Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The global ES cell chromatin...
Bivalent modification and gene...
The role of histone acetylation
ATP-dependent chromatin...
Histone methylation and...
Histone variants
DNA methylation
Conclusions and Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(10):1134-1144. doi:10.7150/ijbs.7998 This issue Cite
Review
The Epigenomics of Embryonic Stem Cell Differentiation
Daniel C. Kraushaar ![]() , Keji Zhao
, Keji Zhao ![]()
Systems Biology Center, National Heart, Lung, and Blood Institute, NIH, Bethesda, MD, 20892, USA
Received 2013-10-28; Accepted 2013-11-25; Published 2013-12-9
Abstract
Embryonic stem cells (ESCs) possess an open and highly dynamic chromatin landscape, which underlies their plasticity and ultimately maintains ESC pluripotency. The ESC epigenome must not only maintain the transcription of pluripotency-associated genes but must also, through gene priming, facilitate rapid and cell type-specific activation of developmental genes upon lineage commitment. Trans-generational inheritance ensures that the ESC chromatin state is stably transmitted from one generation to the next; yet at the same time, epigenetic marks are highly dynamic, reversible and responsive to extracellular cues. Once committed to differentiation, the ESC epigenome is remodeled and resolves into a more compact chromatin state. A thorough understanding of the role of chromatin modifiers in ESC fate and differentiation will be important if they are to be used for therapeutic purposes.
Recent technical advances, particularly in next-generation sequencing technologies, have provided a genome-scale view of epigenetic marks and chromatin modifiers. More affordable and faster sequencing platforms have led to a comprehensive characterization of the ESC epigenome and epigenomes of differentiated cell types. In this review, we summarize and discuss the recent progress that has highlighted the central role of histone modifications, histone variants, DNA methylation and chromatin modifiers in ESC pluripotency and ESC fate. We provide a detailed and comprehensive discussion of genome-wide studies that are pertinent to our understanding of mammalian development.
Keywords: Epigenomics, chromatin, embryonic stem cells, differentiation
Introduction
Embryonic stem cells (ESCs) are derived from the inner cell mass of the d4.5 blastocyst. They are defined by their unique properties of self-renewal and pluripotency, which make them an invaluable tool for studies of development and therapeutic medicine. Continuous self-renewal can be readily achieved in culture and maintains the pluripotent plasticity of ESCs. When transplanted into blastocyst-stage embryos, ESCs can contribute to somatic tissues from all three germ layers as well as the germ line of the developing mouse demonstrating the retention of their pluripotent potential during culture [1]. As ESCs exit self-renewal and commit to differentiation into cell types representative of the three germ layers ectoderm, mesoderm and endoderm they recapitulate embryonic differentiation programs that take place during in vivo development. For this reason, ESCs provide a valid model for the study of early development. Furthermore, defined culture protocols have been developed that allow for the differentiation of ESCs into cell types that may be used for cell therapeutic purposes.
The transition of a pluripotent stem cell to a committed and more developmentally restricted cell type is accompanied by rapid and global changes in transcription that result in stable silencing of pluripotency genes and activation of lineage-specific genes. At the same time, stage specific genes of multipotent progenitors have to remain competent for activation during further differentiation whereas tissue-specific genes that are considered outside the progenitor's differentiation potential have to be terminally turned off. A number of cytokines, growth factors, morphogens and their co-factors regulate early lineage fate decisions and ultimately the transcription programs of ESCs and their roles are becoming increasingly defined and often coincide with their functions in vivo [2-6]. Extracellular signals are complemented by the action of intracellular transcription factors that drive gene expression patterns required for cell differentiation and lineage specification. Regulators of chromatin states and epigenetic modifiers have emerged as additional players of cell fate. Numerous genetic studies that have disrupted genes, which encode for chromatin modifiers, have resulted in embryonic lethality [7-9]. These studies have demonstrated the importance of ATP-dependent chromatin remodelers, histone modifying enzymes and DNA methyltransferases in embryonic development.
Despite significant advances in the technologies used to map the genome-wide distribution of histone marks and a continuous decrease in required cell numbers the study of dynamic epigenetic chromatin modification during in vivo development remains challenging. At the same time, ESCs can be cultured in abundance and provide a powerful tool for the study of epigenetic mechanisms and cell fate. Genome-wide maps of epigenetic factors have revealed a unique epigenetic signature in pluripotent ESCs and have contributed models to explain their plasticity. Also, comparisons of ESCs with cells differentiated from ESCs have proven powerful in understanding the dynamics of epigenetic marks during development. Thus, we have learned that each specific cell type has not only its own specific expression profile or 'transcriptome' but is also featured by its own epigenetic signature or 'epigenome' that is transmitted as heritable information through cell divisions [10-12].
In 2006, Yamanaka et al. and subsequently Thompson et al. achieved the reprogramming of mouse and human fibroblasts respectively into ESC-like pluripotent cells by ectopic expression of Oct4, Sox2, c-myc and Klf4 [13, 14]. The successful reprogramming into induced pluripotent stem cells (iPSCs) ignited immense excitement since ethical concerns that arise from the derivation of ESCs may be circumvented using this alternative approach. In addition, a pool of patient-specific stem cells could now be generated with a possibility for autologous regenerative transplants. Since then, we have come to understand that the epigenome of somatic cells must be reset into an ESC-like state and further highlights the importance of epigenetic regulation in maintaining pluripotency [15-18].
Here, we review our current understanding of the global epigenetic landscape of pluripotent ESCs, how it is set up by chromatin remodeling enzymes, and how it resolves during the course of differentiation. To this end, we have focused our discussion primarily on studies that have utilized genome-wide sequencing technologies such as ChIP-Chip or ChIP-Seq to map histone modifications and epigenetic modifiers.
The global ES cell chromatin landscape
ESCs are singular in exhibiting higher global transcription levels than somatic cells. High global transcription levels can be attributed to elevated transcription of sequences that are silent in differentiated cells and even higher transcription levels of genes that are constitutively expressed in both ESCs and differentiated cells [19, 20]. Given such great differences in transcriptional activity one would anticipate substantial differences in chromatin organization between pluripotent and differentiated cell types. In fact, differences in global chromatin configuration can be readily detected by ultrastructural examination of ESC chromatin as well as chromatin from terminally differentiated cells. Chromatin of undifferentiated ESCs appears decondensed and plastic in structure whereas differentiated cells display distinct foci of heterochromatin [21]. Overall, ESC differentiation is accompanied by an increase in heterochromatic foci and a decrease in mobility and turnover of chromatin-associated proteins [21-23]. Genome-wide analyses of histone marks show that modifications, generally associated with transcriptional activity such as H3K4me3, H3K9ac, H3K14ac, H3K36me2 and H3K36me3, are present at high levels in ESCs and become reduced upon differentiation. An opposite trend holds true for repressive marks such as H4K20me3, H3K9me2 and H3K9me3that are more abundant in differentiated cells [24, 25].
Bivalent modification and gene poising by PRC2 and MLL complexes
An open chromatin configuration may merely be a reflection of the hyper-transcriptional activity found in ESCs. However, the identification of both active H3K4me3 and repressive H3K27me3 at silent genes lead to the idea, that developmental genes are bivalently marked and thus primed for activation prior to ESC differentiation [26]. Therefore an open chromatin configuration and abundance of active histone marks is not only a reflection of high transcriptional activity but also of cell plasticity. Initial ChIP-Chip and later ChIP-Seq studies revealed that H3K4me3 co-localizes with H3K27me3 to promoters of a subset of developmental genes [24, 26, 27]. Unlike H3K4me3, which is present at almost three quarters of all gene promoters, H3K27me3 is only present at a small subset of developmental genes but is almost always found together with H3K4me3. The majority of bivalent promoters are transcriptionally initiated but not enriched with the elongating form of Pol II or H3K36me3 [20]. Besides gene promoters, bivalent histone modifications, in the form of H3K4me1 and H3K27me3, also decorate enhancer regions, which silence proximal gene clusters [28, 29].
As ESCs differentiate into more restricted germ layer-type precursor cells, 85% of bivalent genes resolve to monovalent H3K27me3, H3K4me3, or carry neither mark [24, 30]. Lineage-related genes will retain bivalency with the idea that they remain poised for subsequent expression at later developmental stages, and unrelated genes become terminally silenced. Gene priming and gene bivalency though, are not restricted to ESCs and are found in other cell types including hematopoietic stem cells as well as T-cells where bivalent histone modifications facilitate rapid gene activation upon T-cell differentiation [31-33].
The progressive restriction of differentiation potential alongside loss of gene bivalency at lineage-unrelated loci does not always hold true: hematopoietic stem cells derived from ESCs, retain bivalency on non-related neural genes [34]. Similarly, the Th1-specific gene Tbx21 retains both H3K4me3 and H3K27me3 in nTreg cells. In this case, Tbx21 bivalency translates into additional plasticity as illustrated by the capacity of nTreg cells to express Tbx21 in Th1-inducing conditions [35]. Some promoters may also gain bivalency during the course of differentiation, illustrating that gene priming is by no means a unidirectional process [33, 36].
Bivalent chromatin marks are established by the functionally opposing Polycomb (PcG) and Trithorax (Trx)/MLL group (hereafter, MLL complexes) proteins. PcG proteins are comprised of two major complexes PRC1 and PRC2, which mediate H3K27me3 and gene silencing, whereas MLL complexes mediate H3K4 methylation and gene activation. Apart from a few specialized subunits, MLL complexes contain either SET1, MLL1, MLL2, MLL3, or MLL4 as a catalytic subunit and WDR5, RbBP5, and Ash2L as integral core subunits that are necessary for the methylation activity of the complexes [37].
Based on insights into the mechanisms of gene poising, a model that involves H2A ubiquitination has evolved (Fig. 1): Bivalently marked genes assemble DNA Polymerase II (Pol II), yet elongation does not take place consistent with detection of the paused form of Pol II phosphorylated at Ser5 [20]. Ezh2, a subunit of PRC2, catalyzes H3K27 trimethylation. PRC1 binds to the product of PRC2, namely H3K27me3, and subsequently monoubiquitinylates H2A K119 via its E3 ubiquitin ligases, Ring1A and Ring1B. Loss of Ring1A or Ring1B that results in reduction of H2AK119 ubiqitination leads to de-repression of bivalent genes and apparent release of poised Pol II from these sites and replacement with hyperphosphorylated and transcriptionally active Pol II [20, 38, 39]. H3K4me3 mediates gene activation by various mechanisms that include interactions with ATP-dependent remodeling factors as well as recruitment of histone acetyltransferases and Pol II for transcription. Hence the net result of gene priming equals the recruitment of Pol II, mediated by H3K4me3, and simultaneously prevention of transcriptional elongation by H3K27me3, which in effect results in low-level transcription of bivalent genes.
Resolution of gene bivalency upon cell fate commitment. MLL and PcG complexes deposit H3K4me3 and H3K27me3 respectively at bivalent promoters. PRC1 binds to H3K27me3 marks, and via Ring1 ubiquitinates H2A. In ESCs, bivalent domains are found on numerous CpG-rich promoters. Poised RNA Pol II facilitates rapid activation upon presentation of developmental cues. Primed promoters may remain bivalent during differentiation or bivalency may resolve to monovalency.
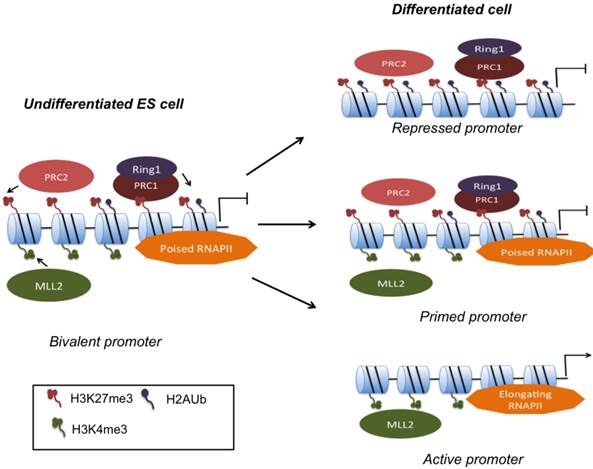
Genome-wide mapping of core subunits of both PRC1 and PRC2 complexes reveal occupation of bivalent genes close to the TSS [40]. Disruption of PcG functional subunits such as Eed, Ezh2, or Suz12 results in de-repression of bivalent differentiation genes, but typically does not affect ESC self-renewal probably because the expression of pluripotency genes remains largely unaffected [26, 27, 41, 42]. Upon differentiation, H3K27me3 marks will have to be removed for timely activation of developmental genes, yet the enzyme that removes H3K27me3 is currently unknown. UTX, a JmjC domain-containing H3K27me3 demethylase is expressed in ESCs, but does not regulate global H3K27me3 levels or H3K27me3 on HOX genes [43].
Depletions of MLL complex core proteins such as Dpy-30, RbBP5 and Wdr5 in ESCs typically result in global reductions in H3K4 methylation and have made it difficult to decipher their role specifically in gene bivalency [44, 45]. A delay in differentiation is observed upon Dpy-30 knockdown that has been proposed to be a result of ineffective H3K4 methylation needed for upregulation of differentiation markers [45]. Among, MLL proteins, MLL2, has been identified as the methyltransferase that specifically methylates bivalent promoters including those of HOX genes. MLL2 null ESCs also display a delay in differentiation, although no strong link with H3K4me3 was established [46]. Interestingly, knockdown of MLL2 and reduction of H3K4me3 had no effect on gene induction upon differentiation, challenging the idea of gene bivalency as a catalyst for rapid gene activation [47].
An important, and easily overlooked factor in gene priming are the relative levels of active and inactive marks that need to be finely balanced in order to maintain gene poising without causing aberrant gene expression. For instance, LSD1 (lysine specific demethylase 1), which mediates H3K4me1/2 demethylation, is bound to developmental genes, and is specifically overrepresented at bivalently marked genes. Knockdown of LSD1 results in loss of pluripotency and aberrant expression of LSD1 target genes as a result of increased H3K4me2 that indirectly leads to increases in H3K4me3 [48]. Kdm5b, another H3K4 demethylase is primarily required for removal of H3K4me3 from pluripotency regulators such as Oct4 and Nanog, in order to initiate timely differentiation commitment [49].
Despite a growing understanding of PcG function, how PcG and Trx proteins become recruited to their target genes and how the rearrangement of bivalent genes is orchestrated during differentiation is still very much central to current investigation. Mechanistically, gene bivalency provides an unstable steady state of gene repression that may allow for rapid transition to gene activation upon the presentation of developmental stimuli. Thus, a fine-tuned regulation of bivalent domains is necessary for proper ESC differentiation.
The role of histone acetylation
Histone acetylation neutralizes the positive charge of histones and thereby decreases the affinity between histones and DNA [50]. At the same time, it generates recognition signals for chromatin proteins containing bromo domains [51] The steady state of chromatin acetylation levels is modulated by both histone acetyltransferases (HATs) and histone deacetylases (HDACs). Histone deacetylases (HDACs), negatively control histone acetylation by removing acetyl groups from histone tails. HDACs repress transcription by preventing rapid acetylation and Pol II binding to promoters [52]. HDACs act as co-repressors in complexes such as the nucleosome remodeling and histone deacetylation (NuRD), SIN3 complexes and CoREST [53, 54]. Unexpectedly, HDACs are predominantly detected at active genes of T- as well as ESCs [52, 55]. HDAC activity at active genes prevents excessive acetylation that would otherwise lead to chromatin instability and cryptic transcription [56]. In ESCs, HDAC1 is bound predominantly to active genes that include Oct4, Sox2 and Nanog although some lineage-specific genes are also occupied, suggesting both positive and negative regulation of gene expression [55]. It may be for that reason why studies with chemical HDAC inhibitors have not provided consistent results. HDAC inhibition with TSA treatment has been reported to result in spontaneous differentiation and downregulation of pluripotency genes but also inhibition of differentiation in another case [55, 57].
HDAC1 knockout ESCs display normal self-renewal and upregulation of pluripotency-associated genes, [55, 58]. Treatment of fibroblasts with HDAC inhibitors such as trichostatin A, suberoylanilide hydroxamic acid and valproic acid (VPA) substantially improves their reprogramming efficiency [59-62]. Both HDAC1 knockout and the improvement of reprogramming by means of HDAC inhibition, suggest that histone deacetylation principally antagonizes the undifferentiated state of ESCs. Essentially, HDACs may hold a dual role in ESCs: firstly they repress the expression of lineage-specific genes that need to be inactive and entirely free of acetylation. Secondly, deacetylation at active genes will prevent excessive acetylation, which could lead to indiscriminate transcription.
Among, HATs, Tip60/p400 has been identified as a critical requirement for ESC self-renewal and pluripotency [63]. Tip60/p400 contains both H2A.Z deposition and HAT activities combined. The mammalian p400 contains SWI/SNF-like ATPase, and is part of a complex that contains the Tip60 acetyltransferase. Loss of pluripotency is observed upon knockdown of both p400 and Tip60 subunits and is caused by upregulation of developmental genes [63]. Surprisingly, knockdown of p400 and Tip60 results in diminished H4 acetylation levels even when p400/Tip60 target genes become de-repressed. Hence, gene repression may be conferred by deposition of H2A.Z rather than histone acetylation. An examination of H2A.Z deposition in the absence of Tip60/p400 will be important, especially in the light of H2A.Z's important function in facilitating accessibility of PcG complexes to developmental genes in ESCs (see histone variant section). Until then, the exact mechanism by which p400/Tip60 regulates pluripotency remains somewhat obscure.
Two additional HATs, namely p300 and Mof have been characterized on a genome-wide level in ESCs. The acetyltransferase p300 is commonly found at enhancers and genome-wide binding data shows that p300 shares a substantial number of enhancers with the Oct4/Sox2 and Nanog cluster in ESCs and that its recruitment is directly dependent on these pluripotency-associated transcription factors [64].
Mof mediates H4K16ac and is found in two distinct complexes in mammals: Mof-Msl and Mof-Msl1v1. Unlike p400/Tip60 and p300, which primarily regulate the expression of differentiation genes, Mof directly regulates the expression of pluripotency genes [63, 65, 66]. Mof occupies both Oct4 and Nanog and also overlaps substantially with Nanog targets that carry H3K4me3 on their promoters. In fact, Mof activity is required for recruitment of Wdr5 and H3K4me3 at shared Mof/Wdr5 loci, revealing a direct link between Mof and the MLL complex [65].
Overall, the close association of HATs with pluripotency factors and their target sites is striking, and suggests a functional role for HATs in regulating pluripotency-associated transcription factor targets each within different “compartments” of the self-renewal circuitry. Mof facilitates the expression of pluripotency factors and represents an upstream regulator of MLL-associated gene priming. The primary function of Tip60/p400 lies in the repression of developmental genes. p300 has a mostly enhancer-specific function that is carried out in close association with the Oct4/Sox2/Nanog cluster.
ATP-dependent chromatin remodeling
ATP-dependent chromatin modifiers are important regulators of lineage fate and embryonic development. Among them, the Baf complex also called Swi/Snf, and NuRD complexes are examples that are crucial for ESC pluripotency. The Swi/Snf complex has a unique ESC- specific subunit composition and has been termed esBAF [67]. Genetic studies showed that esBAF is required for self-renewal and pluripotency. Brg1, a subunit of the esBAF complex, interacts with Oct4, Sox2 and Nanog, and co-occupies many sites with the same transcription factors [68-70]. One of the primary functions of Brg1 is to potentiate LIF signaling by facilitating STAT3 accessibility to its target genes (Fig. 2A). When Brg1 is lost, Brg1-activated STAT3 target genes that include important pluripotency genes such as Oct4, Nanog and c-myc, become occupied by PcG and silenced by H3K27me3 modification [70]. Yet, at classical PcG- occupied targets including HOX genes, Brg1 co-operates with PcG in the repression of bivalent genes (Fig. 2B). Hence, Brg1 function is highly context-dependent and facilitates both gene activation and repression by means of modulating nucleosome stability to maintain 'stemness' and pluripotency-associated chromatin competence.
Context-dependent roles of esBAF/Brg1 in ESC gene regulation. A: esBAF antagonizes PcG binding in order to potentiate LIF/STAT3 signaling. B: esBAF and PcG act synergistically in the repression of HOX genes. C: esBAF/Brg1 antagonizes Mbd3/NuRD and thereby fine-tunes gene expression levels.
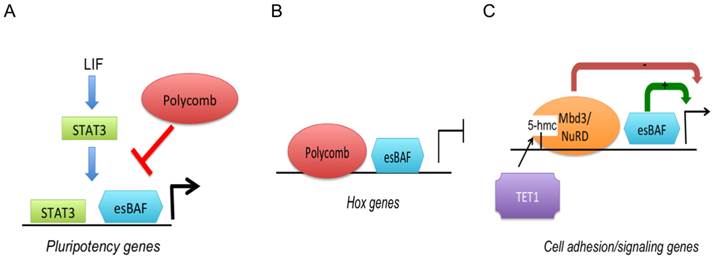
The PcG complex is not the only chromatin modifier that antagonizes Brg1. Mbd3, a subunit of the Nucleosome Remodeling Deacetylase (NuRD) complex co-occupies with Brg1 several hundred promoters associated with cell signaling and adhesion [71]. The NuRD complex contains both a Swi2/Snf2-related ATPase (Mi-2) and HDAC1 and HDAC2. Expression of Brg1/Mbd3 co-occupied genes is fine-tuned through positive regulation by Brg1 and negative regulation through NuRD (Fig. 2C). The role of Mbd3 has become clearer recently after its enrichment was mapped and found to co-localize on bivalent genes with the methylcytosine modification 5'hydroxyl methylcytosine (5'hmC), whose function remained elusive for some time [71, 72]. 5'hmC is created by the TET family of Fe(II) + 2 oxoglutarate-dependent enzymes. Both TET1 and TET2 are required for ESC self-renewal [73, 74]. TET-mediated 5'hmC facilitates the recruitment of Mbd3, which via its HDAC activity decreases H4 acetylation. Reduced histone acetylation in turn results in nucleosome occupation and gene silencing. TET1 further mediates CpG-containing gene repression by facilitating PRC2 recruitment [75]. Bivalent H3K4me3/H3K27me3 promoters are typically decorated with 5'hmC and carry TET1 [72, 76]. Hence, TET1/5'hmC have dual roles in gene repression by recruiting both NuRD and PcG complexes to developmental genes.
ATPase subunits of the chromo domain helicase DNA binding (CHD) proteins contain 2 chromo domains with affinity for methylated histones. They carry out diverse functions in transcriptional regulation [77]. Altogether, 9 CHD proteins in 3 families exist and most of them are important for embryonic development. CHD1 is a transcriptional activator that associates with promoters of active genes in ESCs. CHD1-deficient ESCs accumulate foci of H3K9me3 suggesting that CHD1 antagonizes heterochromatin formation. CHD1-/- ESCs loose pluripotency, which is manifested by their failure to form primitive endoderm [78]. A solid connection between H3K9me3 and block of primitive endoderm formation has not been established however. The mode of function for inhibition of heterochromatin formation is not known but may involve deposition of the H3 variant H3.3 that generally does not carry H3K9me3 [79].
CHD7, another member of the CHD group of ATPase chromatin remodelers is primarily bound to enhancers of active genes and appears to both promote negative and positive regulation of transcription [80, 81]. Further studies are required to decipher the exact roles and mechanisms of action of these interesting chromatin remodelers.
The studies of ATP-dependent chromatin remodeling factors have revealed Brg1 as a central player of pluripotency. Brg1 antagonizes other chromatin enzymes such as PcG and the NuRD complex.
Histone methylation and heterochromatin
Histone methylation marks including H4K20me3 and H3K9me2/3 are marks associated with heterochromatin and are important for preventing transcription at centromeres, transposons and tandem-repeats. As mentioned previously, global levels of heterochromatic marks and heterochromatin-associated proteins increase upon lineage commitment of ESCs as their chromatin transitions into a less dynamic and less plastic state [21]. H3K9me2 displays a non-overlapping distribution when compared to H3K9me3, and is enriched in large blocks of conserved non- coding stretches of DNA. During differentiation, these H3K9me2 islands increase substantially, and contribute to the silencing of developmental genes in lineage-specific manner. However overall, the gene density that falls into H3K9me2 islands is generally low [25].
Genome-wide mapping of the heterochromatic marks H3K9me3 and H4K20me3 revealed that they are highly co-localized primarily at telomeres, satellites and long-tandem repeats [24, 82]. Several enzymes are involved in the catalysis of H3K9 methylation and include G9a, ESET/Set1b and Suv39h. Genome-wide binding data is currently only available for Set1b. Knockdown of Set1b results in upregulation of differentiation markers due to failure to silence developmental genes in the ESC state. Set1b localizes to a number of bivalent genes that are also enriched in H3K9me3 [82]. Therefore, H3K9me3 adds additional stability to the repression of bivalent genes.
H3K9 demethylases including Jmjd1a and Jmjd2c are expressed in ESCs and are important for ESC self-renewal and pluripotency by keeping ESC- specific genes expressed [83]. Genome-wide binding data would facilitate the delineation between Jmjd1a/Jmjd2c binding, H3K9me3 levels and the expression of their target genes. In summary, H3K9 methylation transferases may act relatively indiscriminately on developmental genes, and gene activation has to be accomplished through recruitment of H3K9 demethylases to a subset of genes that must be expressed. Somewhat surprisingly, these studies have implicated H3K9 methylation not only in terminal silencing of lineage specific genes upon differentiation but also prevalently in gene repression of lineage markers in the undifferentiated state of ESCs.
Histone variants
Nucleosomes can be modified not only by post-translational marks but also by the dynamic incorporation of histone variants. Histone variants of histones H3, H2A, H2B with the exception of H4 have been described. Variants of any one histone type may differ in only a few amino acids (e.g. H3 variants) or may contain significant dissimilarities (e.g. H2 variants) [84, 85]. Canonical histones are defined by being primarily expressed during S-phase at which point a major supply of histones for newly replicated DNA must be ensured. Non-canonical or 'replacement' histones are expressed throughout the cell cycle. The incorporation of histone variants is tightly regulated by histone chaperones, and how their activities in nucleosome assembly are controlled has been subject to intense research over the past few years. In mammalian cells, histone H3 is represented by two cell-cycle dependent histone, H3.1 and H3.2 and the replication-independent H3 variant H3.3. Three chaperones have been identified for incorporation of H3.3, HIRA, ATRX and DAXX. DAXX associates with ATRX and deposits H3.3 primarily at telomeres whereas HIRA deposits H3.3 at non-heterochromatic regions such as active and inactive genes [84].It has been postulated that non-canonical histones may merely function as replacement histones when canonical histones are in short supply such as in non-replicating cells. However, despite small differences in amino acid sequence, each variant is distributed differentially across the genome and carries its own characteristic histone modification signature, strongly suggesting distinct functional roles for each H3 variant [86]. Histone marks that are associated with gene activation such as acetylation marks and H3K4me3 are typically found on H3.3 whereas H3K27me2 and H3K9me3 are found on H3.2. Marks associated with gene silencing are predominantly found on H3.1 [87, 88]. Hence, histone variants may act as determinants of selective histone modifications. Nonetheless, specific histone modifications are not completely fixed to any one variant. Basal transcription still occurs in the absence of H3.3 suggesting that canonical histones can take over transcriptional activation [89]. Vice versa H3.3 can be deposited at DNA replication sites where normally only canonical histones are incorporated, showing that variants may be redundant to some extent [90].
Among H3 variants H3.3 and H3.2 have been mapped in heterozygous epitope tagged H3 variant ESC lines [86, 90]. In non-pluripotent cells, H3.3 is generally enriched at active genes [91, 92]. The unique ESC epigenetic landscape sets itself apart from this pattern and H3.3 can be detected at promoters of both active as well as inactive genes, that include but are not confined to bivalent genes. However, unlike inactive genes, active genes also contain H3.3 in their gene body where its density directly correlates with transcriptional activity. Deposition of H3.3 in gene bodies indicates a direct connection to Pol II-driven elongation of transcription. In fact, a significant correlation between enrichment of HIRA-dependent H3.3 and the elongating form of Pol II has been observed. Furthermore a physical interaction between HIRA and Pol II has been demonstrated [90]. Transcription-linked deposition of H3.3 may help to reset nucleosome occupation and chromatin integrity after each round of transcription.
Considering the strong correlation between H3.3 enrichment and gene activity one would anticipate a role for H3.3 in ESC pluripotency and specifically gene activation. Yet, ESCs deficient in the H3.3-specific chaperone, HIRA, or H3.3- deficient ESCs display no obvious compromise in self-renewal and gene expression appears unaffected at large accompanied by unaltered levels of H3K4me3 and H3K36me3 [86, 93]. However, H3K27me3 and PRC2 recruitment become significantly reduced at bivalent promoters upon loss of H3.3 and/or HIRA, showing that H3.3 is involved in the establishment of PcG-mediated gene priming [93]. It remains somewhat unclear as to why knockdown of H3.3 and the resulting reduction in H3K27me3 do not result in greater de-repression of developmental genes as has been observed from PRC2-deficient ESCs. H3.3 also plays an important role in chromatin integrity as suggested by enrichment of H3.3 at heterochromatic repeat regions such as telomeres. Knockdown of H3.3 or ATRX and Daxx result in telomere dysfunction phenotypes [94, 95].
The histone H2A variant, H2A.Z, is ubiquitously expressed and is more abundant in ESCs than in differentiated cells, suggesting that this variant may play an important role in preserving 'stemness' [88, 96]. In metazoans, enrichment of H2A.Z shows a direct correlation with gene activity and is generally detected in small regions flanking the TSS and enhancers, and usually only at larger regions at telomeric sequences or centric heterochromatin [97]. In ESCs, H2A.Z displays a rather untypical pattern of distribution in that it occupies a large set of silent developmental genes and almost always co-localizes with H3K4me3. Interestingly, H2A.Z co-occupies most developmental genes with PcG, although H2A.Z deposition is independent of PcG [98, 99]. Knockdown of H2A.Z leads to compromised self-renewal and aberrant differentiation with a roughly equal number of up and downregulated genes suggesting context-dependent transcriptional regulation by H2A.Z [99]. Benzonase digestion as well as ChIP-seq with antibodies against Oct4, RbBP5 and Suz12 shows that H2A.Z is a universal facilitator of accessibility enabling binding of pluripotency factors, MLL complex proteins to active genes, and PcG proteins to repressed genes (Fig. 3). [99]. H2A.Z histone exchange renders nucleosomes extremely unstable and ultimately leads to high nucleosome turnover and nucleosome depletion that allows for increased accessibility [91, 100, 101].
Differential roles of H2A.Z in gene activation and repression, modulating self-renewal and differentiation of ESCs. H2A.Z facilitates chromatin accessibility by modulating nucleosome stability. In the undifferentiated state, H2A.Z allows pluripotency transcription factors such as Oct4 to bind to its target sites and thereby facilitate gene activation. At the same time, allowing PcG access to developmental genes modulates gene repression. Upon cell fate commitment, other master transcription factors require H2A.Z for accessibility to their targets.
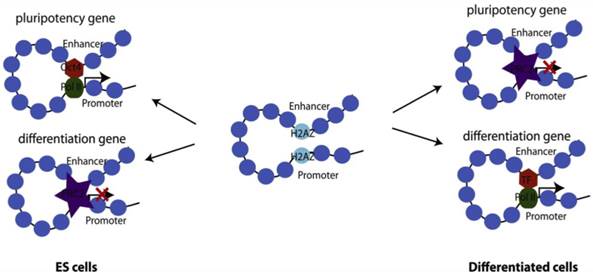
In summary, the histone variant H2A.Z is an important regulator of accessibility at developmental genes and affects gene expression in context-dependent manner. Despite being present at many active loci, H3.3 has surprisingly not been identified as an absolute requirement for gene activation. Paradoxically it appears to be required at silent genes for the establishment of PcG-mediated H3K27 trimethylation.
DNA methylation
A family of DNA methyltransferases that include Dnmt1, -3a and -3b catalyzes DNA methylation. DNA methylation is important for many developmental processes including gene silencing, genomic imprinting and X-chromosome inactivation [102]. It confers gene silencing by several different mechanisms: Methylated DNA impedes with the binding of transcription factors to target sites. For example, c-myc binding to its recognition site or E-box is inhibited by DNA methylation [103]. Furthermore, DNA methylation interferes with the establishment of histone modification marks that are associated with gene activation. For instance, MLL proteins only interact efficiently with unmethylated CpG sequences [104]. In addition, members of the methyl binding proteins that include Mbd1-4 and MeCP2 can recruit histone deacetylases (HDACs) and chromatin remodelers that promote a repressive chromatin environment [105-107]. Besides promoter regions, DNA methylation is widely detected in gene bodies of transcribed genes, although its function there is not clear. Recent results suggest that gene body DNA methylation contributes to inclusion of alternatively spliced exons by recruiting HDACs to decrease nucleosome acetylation and by slowing down Pol II movement [108].
ESCs that lack any of the Dnmts maintain self-renewal but exhibit aberrant lineage commitment [109, 110]. Dnmt1-/- knockout embryoid bodies retain a large number of undifferentiated Oct4-positive cells suggesting that de novo methylation is required for cell fate commitment and efficient silencing of pluripotency genes [110].
High resolution bisulfite deep sequencing and gene ontology analyses in undifferentiated ESCs have shown correlations with histone modifications and most striking is the anti-correlation with H3K4me3 [111, 112]. A subset of non-bivalent, late differentiation genes and signal transduction genes carry DNA methylation [113]. Therefore generally speaking two distinct sets of repressed developmental genes exist in undifferentiated ESCs: one set of genes will have to be turned on early upon lineage commitment. These genes are bivalently marked and DNA hypomethylated. Another smaller set of genes that is induced at later stages of differentiation exhibits DNA hypermethylation at their promoters and DNA methylation will have to be removed during the course of differentiation.
Upon differentiation into the three germ layers, promoters of early developmental genes are typically CpG rich and will not engage in DNA methylation in non-expressing lineages but rather become decorated with H3K27me3 for gene silencing. Promoters of late developmental genes are CpG poor and will become DNA methylated in non-related lineages [114]. Loss of H3K4me3 or H3K27me3 results in partial increases of DNA methylation and loss of both marks leads to hypermethylation and H3K9 trimethylation. Hence bivalent marks that resolve during differentiation will form heterochromatin and ultimately become irreversibly silenced [111].
Comparison between fibroblasts and ESCs shows that an estimated 25% of methylation is in a non-CpG context whereas CpG methylation makes up almost 100% in fibroblasts. During ESC differentiation, non-CpG methylation is lost; on the other hand, non-CpG methylation is restored during reprogramming and illustrates that non-CpG methylation is a unique characteristic of pluripotent cells [115]. Unlike CpG-methylation, non-CpG methylation generally shows a positive correlation with gene activity and is usually enriched in gene bodies. The functional role of non-CpG methylation remains unclear in particular within the context of pluripotency. Given that non-CpG methylation is frequently associated with genes related to RNA processing, RNA splicing and RNA metabolic processes, a function associated with RNA splicing seems plausible [115].
As mentioned previously, TET1/5'hmC are involved in gene repression of developmental genes in collaboration with PcG and NuRD complexes. TET-mediated 5'hmC hydroxylation also plays an important role in gene activation by facilitating active demethylation in mammalian cells [75, 116]. In this context, TET1/5'hmc are present on several pluripotency genes e.g. Nanog, Esrrb and Tcl1, in order to prevent 'accidental' methylation of these genes [75].
Conclusions and Perspectives
Recent advances in genome-wide sequencing techniques have provided us with a holistic view of the epigenetic landscape of ESCs. Comparison between undifferentiated ESCs and advanced- or terminally differentiated cell types have allowed us to decipher how epigenetic modifications resolve upon lineage commitment, but has also aided in making somatic cell reprogramming more efficient. Chromatin conformation capture (3C) techniques are constantly improving in resolution and will help to place cell-type specific epigenomes into the context of chromosome topology and long-range interactions.
For a better understanding of mammalian development as well as for regenerative medicine, a more 'lineage-specific' understanding of epigenetic remodeling is required. With this idea in mind, the NIH Epigenomic Roadmap has made it a priority to characterize the epigenome of stem cells and their derivatives in order to gain insight into epigenomic changes of counterpart tissues relevant to disease. Developing robust differentiation systems that yield homogenous cell populations as well as ChIP protocols for small cell numbers remain indispensable goals in this regard. In order to add resolution to our dynamic image of epigenetic remodeling during cell differentiation, more information must be garnered from intermediate transitions of cell differentiation. For instance, recent contributions to the Epigenomic Roadmap have utilized differentiation protocols for human ESC differentiation into ectoderm, mesoderm and endoderm-like cell types.
Of course, more differentiated cell types are not characterized as well as undifferentiated ESCs and comprehensive reference libraries for master transcription factors and epigenetic modifiers must be established over time.
Epigenetic landscapes are highly dynamic and established in a delicate 'ying-yang' of chromatin remodelers. Growth factors and morphogens provide the cues for cell differentiation during development, yet the connection between extracellular cues, signal transduction and chromatin remodeling is poorly defined. It is therefore necessary to integrate global epigenetic information with signal transduction information to establish which parts of the cistrome are responsive to extracellular cues and which ones are not. Although, extracellular cues are the master directors of cell differentiation, epigenetic information that is passed on in heritable fashion at the replication fork, may itself directly affect cell fate. Whether and to what extent epigenetic inheritance forms the basis of ESC fate decisions is an important piece in the puzzle of fundamental development.
Acknowledgements
The research in the authors' laboratory was supported by the Division of Intramural Research, NHLBI, NIH.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature. 1984;309:255-6
2. Lanner F, Rossant J. The role of FGF/Erk signaling in pluripotent cells. Development. 2010;137:3351-60
3. Li Z, Chen YG. Functions of BMP signaling in embryonic stem cell fate determination. Exp Cell Res. 2013;319:113-9
4. Loebel DA, Watson CM, De Young RA, Tam PP. Lineage choice and differentiation in mouse embryos and embryonic stem cells. Dev Biol. 2003;264:1-14
5. Okita K, Yamanaka S. Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr Stem Cell Res Ther. 2006;1:103-11
6. Kraushaar DC, Dalton S, Wang L. Heparan sulfate: a key regulator of embryonic stem cell fate. Biol Chem. 2013;394:741-51
7. Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915-26
8. Bultman S, Gebuhr T, Yee D. et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287-95
9. O'Carroll D, Erhardt S, Pagani M. et al. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330-6
10. Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127-34
11. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7-18
12. Smallwood A, Ren B. Genome organization and long-range regulation of gene expression by enhancers. Curr Opin Cell Biol. 2013;25:387-94
13. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
14. Yu J, Vodyanik MA, Smuga-Otto K. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-20
15. Chin MH, Mason MJ, Xie W. et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111-23
16. Guenther MG, Frampton GM, Soldner F. et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010;7:249-57
17. Mikkelsen TS, Hanna J, Zhang X. et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49-55
18. Maherali N, Sridharan R, Xie W. et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55-70
19. Efroni S, Duttagupta R, Cheng J. et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437-47
20. Stock JK, Giadrossi S, Casanova M. et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428-35
21. Meshorer E, Yellajoshula D, George E. et al. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105-16
22. Wiblin AE, Cui W, Clark AJ, Bickmore WA. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J Cell Sci. 2005;118:3861-8
23. Pajerowski JD, Dahl KN, Zhong FL. et al. Physical plasticity of the nucleus in stem cell differentiation. Proc Natl Acad Sci U S A. 2007;104:15619-24
24. Mikkelsen TS, Ku M, Jaffe DB. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553-60
25. Wen B, Wu H, Shinkai Y. et al. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246-50
26. Boyer LA, Plath K, Zeitlinger J. et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349-53
27. Azuara V, Perry P, Sauer S. et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532-8
28. Creyghton MP, Cheng AW, Welstead GG. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931-6
29. Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273-83
30. Gifford CA, Ziller MJ, Gu H. et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149-63
31. Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A. 2006;103:15782-7
32. Cui K, Zang C, Roh TY. et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80-93
33. Abraham BJ, Cui K, Tang Q, Zhao K. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics. 2013;14:193
34. Mazzarella L, Jorgensen HF, Soza-Ried J. et al. Embryonic stem cell-derived hemangioblasts remain epigenetically plastic and require PRC1 to prevent neural gene expression. Blood. 2011;117:83-7
35. Wei G, Wei L, Zhu J. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155-67
36. Burney MJ, Johnston C, Wong KY. et al. An epigenetic signature of developmental potential in neural stem cells and early neurons. Stem Cells. 2013;31:1868-80
37. Dou Y, Milne TA, Ruthenburg AJ. et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713-9
38. Zhou W, Zhu P, Wang J. et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29:69-80
39. Nakagawa T, Kajitani T, Togo S. et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008;22:37-49
40. Ku M, Koche RP, Rheinbay E. et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242
41. Lee TI, Jenner RG, Boyer LA. et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301-13
42. Chamberlain SJ, Yee D, Magnuson T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells. 2008;26:1496-505
43. Wang C, Lee JE, Cho YW. et al. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc Natl Acad Sci U S A. 2012;109:15324-9
44. Ang YS, Tsai SY, Lee DF. et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183-97
45. Jiang H, Shukla A, Wang X. et al. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513-25
46. Lubitz S, Glaser S, Schaft J. et al. Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol Biol Cell. 2007;18:2356-66
47. Hu D, Garruss AS, Gao X. et al. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol. 2013;20:1093-7
48. Adamo A, Sese B, Boue S. et al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol. 2011;13:652-9
49. Kidder BL, Hu G, Yu ZX. et al. Extended self-renewal and accelerated reprogramming in the absence of Kdm5b. Mol Cell Biol. 2013
50. Shogren-Knaak M, Ishii H, Sun JM. et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844-7
51. Loyola A, Almouzni G. Bromodomains in living cells participate in deciphering the histone code. Trends Cell Biol. 2004;14:279-81
52. Wang Z, Zang C, Cui K. et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019-31
53. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32-42
54. Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143-53
55. Kidder BL, Palmer S. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 2012;40:2925-39
56. Carrozza MJ, Li B, Florens L. et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581-92
57. Lee JH, Hart SR, Skalnik DG. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis. 2004;38:32-8
58. Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242-7
59. Huangfu D, Maehr R, Guo W. et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795-7
60. Huangfu D, Osafune K, Maehr R. et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269-75
61. Liang G, Taranova O, Xia K, Zhang Y. Butyrate promotes induced pluripotent stem cell generation. J Biol Chem. 2010;285:25516-21
62. Mali P, Chou BK, Yen J. et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010;28:713-20
63. Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162-74
64. Chen X, Xu H, Yuan P. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106-17
65. Li X, Li L, Pandey R. et al. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell. 2012;11:163-78
66. Zhong X, Jin Y. Critical roles of coactivator p300 in mouse embryonic stem cell differentiation and Nanog expression. J Biol Chem. 2009;284:9168-75
67. Ho L, Ronan JL, Wu J. et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci U S A. 2009;106:5181-6
68. Ho L, Jothi R, Ronan JL. et al. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A. 2009;106:5187-91
69. Kidder BL, Palmer S, Knott JG. SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells. 2009;27:317-28
70. Ho L, Miller EL, Ronan JL. et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol. 2011;13:903-13
71. Yildirim O, Li R, Hung JH. et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498-510
72. Pastor WA, Pape UJ, Huang Y. et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394-7
73. Koh KP, Yabuuchi A, Rao S. et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200-13
74. Ito S, D'Alessio AC, Taranova OV. et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129-33
75. Wu H, D'Alessio AC, Ito S. et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389-93
76. Wu H, D'Alessio AC, Ito S. et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679-84
77. Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol. 2007;85:463-76
78. Gaspar-Maia A, Alajem A, Polesso F. et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863-8
79. Konev AY, Tribus M, Park SY. et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087-90
80. Schnetz MP, Bartels CF, Shastri K. et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590-601
81. Schnetz MP, Handoko L, Akhtar-Zaidi B. et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010;6:e1001023
82. Bilodeau S, Kagey MH, Frampton GM. et al. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484-9
83. Loh YH, Zhang W, Chen X. et al. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545-57
84. Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421-34
85. Redon C, Pilch D, Rogakou E. et al. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162-9
86. Goldberg AD, Banaszynski LA, Noh KM. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678-91
87. McKittrick E, Gafken PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci U S A. 2004;101:1525-30
88. Hake SB, Garcia BA, Duncan EM. et al. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J Biol Chem. 2006;281:559-68
89. Hodl M, Basler K. Transcription in the absence of histone H3.3. Curr Biol. 2009;19:1221-6
90. Ray-Gallet D, Woolfe A, Vassias I. et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell. 2011;44:928-41
91. Jin C, Zang C, Wei G. et al. H3.3/H2A.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat Genet. 2009;41:941-5
92. Kraushaar DC, Jin W, Maunakea A. et al. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013;14:R121
93. Banaszynski LA, Wen D, Dewell S. et al. Hira-Dependent Histone H3.3 Deposition Facilitates PRC2 Recruitment at Developmental Loci in ES Cells. Cell. 2013;155:107-20
94. Wong LH, McGhie JD, Sim M. et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010;20:351-60
95. Wong LH, Ren H, Williams E. et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2009;19:404-14
96. Kafer GR, Lehnert SA, Pantaleon M. et al. Expression of genes coding for histone variants and histone-associated proteins in pluripotent stem cells and mouse preimplantation embryos. Gene Expr Patterns. 2010;10:299-305
97. Barski A, Cuddapah S, Cui K. et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823-37
98. Illingworth RS, Botting CH, Grimes GR. et al. PRC1 and PRC2 are not required for targeting of H2A.Z to developmental genes in embryonic stem cells. PLoS One. 2012;7:e34848
99. Hu G, Cui K, Northrup D. et al. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013;12:180-92
100. Li Z, Gadue P, Chen K. et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell. 2012;151:1608-16
101. Dion MF, Kaplan T, Kim M. et al. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405-8
102. Senner CE. The role of DNA methylation in mammalian development. Reprod Biomed Online. 2011;22:529-35
103. Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science. 1991;251:186-9
104. Birke M, Schreiner S, Garcia-Cuellar MP. et al. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002;30:958-65
105. Jones PL, Veenstra GJ, Wade PA. et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187-91
106. Nan X, Ng HH, Johnson CA. et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-9
107. Ng HH, Zhang Y, Hendrich B. et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58-61
108. Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013
109. Tsumura A, Hayakawa T, Kumaki Y. et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805-14
110. Jackson M, Krassowska A, Gilbert N. et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862-71
111. Meissner A, Mikkelsen TS, Gu H. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766-70
112. Laurent L, Wong E, Li G. et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320-31
113. Fouse SD, Shen Y, Pellegrini M. et al. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160-9
114. Xie W, Schultz MD, Lister R. et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134-48
115. Lister R, Pelizzola M, Dowen RH. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315-22
116. Guo JU, Su Y, Zhong C. et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423-34
Author contact
![]() Corresponding author: Daniel C. Kraushaar: e-mail: daniel.kraushaargov; Tel: (301) 451-9676 or Keji Zhao: e-mail: zhaoknih.gov; Tel: (301) 496-2098.
Corresponding author: Daniel C. Kraushaar: e-mail: daniel.kraushaargov; Tel: (301) 451-9676 or Keji Zhao: e-mail: zhaoknih.gov; Tel: (301) 496-2098.