Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(1):53-63. doi:10.7150/ijbs.7975 This issue Cite
Research Paper
The First Mitochondrial Genome for Caddisfly (Insecta: Trichoptera) with Phylogenetic Implications
Yuyu Wang, Xingyue Liu ![]() , Ding Yang
, Ding Yang ![]()
Department of Entomology, China Agricultural University, Beijing 100193, China.
Received 2013-10-24; Accepted 2013-11-19; Published 2013-12-13
Abstract
The Trichoptera (caddisflies) is a holometabolous insect order with 14,300 described species forming the second most species-rich monophyletic group of animals in freshwater. Hitherto, there is no mitochondrial genome reported of this order. Herein, we describe the complete mitochondrial (mt) genome of a caddisfly species, Eubasilissa regina (McLachlan, 1871). A phylogenomic analysis was carried out based on the mt genomic sequences of 13 mt protein coding genes (PCGs) and two rRNA genes of 24 species belonging to eight holometabolous orders. Both maximum likelihood and Bayesian inference analyses highly support the sister relationship between Trichoptera and Lepidoptera.
Keywords: Mitochondrial genome, phylogeny, Trichoptera, Holometabola.
Introduction
The Trichoptera (caddisflies) is a holometabolous order of insects, whose egg, larval, and pupal stages usually live in freshwater habitats and adults in terrestrial habitats, except for the period that females of some species reenter the water to lay eggs [1]. Almost 14,300 extant caddisfly species, placed in 49 families and 688 genera, have been described [2] forming the seventh largest order of all insects and second largest extant monophyletic animal group in freshwater [3]. It has been estimated that the world fauna may contain as many as 50,000 species [4] and is distributed in all zoogeographical regions and sub-regions with the exception of the Antarctic [5]. Although caddisflies are not generally considered to be of great economic importance as pests, they are beneficially important in the trophic dynamics and energy flow in aquatic ecosystems. The larvae are also useful as biological indicators for assessing water quality. Since larvae of different species vary in sensitivity to various types of pollution, extensive use of them has been made for this purpose [6-9].
Trichoptera is closely related to the order Lepidoptera and together the two orders constitute the superorder Amphiesmenoptera, and monophyly of these two orders is strongly supported in both morphological and molecular analyses [10-13]. Three major groups Annulipalpia, Spicipalpia, and Integripalpia have been recognized based largely on the differences in the way silk is used [14]. The Annulipalpia (net-making caddisflies) include all of the families whose larvae make retreats and capture nets to help feeding or catch small animals; the Integripalpia (case-making caddisflies) commonly construct portable, predominantly tubular cases; and the Spicipalpia (free-living caddisflies), include the rest families whose larvae are either predators or grazers. The monophyly of the order Trichoptera is very well established [15, 16]. However, there has been considerable disagreement concerning the basal relationships of Trichoptera. There is broad agreement about the monophyly of two of these major taxonomic groups, the Annulipalpia and Integripalpia, and considerable disagreement about the monophyly and relative placement of taxa within the Spicipalpia [17].
Mitochondria are important functional organelles in eukaryotic cells [18], and the mt genome is being widely used for studies on evolutionary biology, because the mt genome sequences can be more phylogenetically informative than shorter sequences of individual genes, and provide multiple genome-level characteristics [19-22]. Until 10 October 2013, 291 complete holometabolous insect mt genomes have been sequenced and lodged in GenBank (http://www.ncbi.nlm.nih.gov), representing 9 orders. However, no trichopteran mt genome has been determined.
In this paper, we present the complete mt genome of a caddisfly species, Eubasilissa regina (McLachlan, 1871) of the family Phryganeidae, representing the first species from the order Trichoptera with entire mt genome sequenced. In order to notarize the phylogenetic position of Trichoptera, a nearly complete mt genome of Apatania sp. (Trichoptera: Apataniidae) was also sequenced. Based on present mt phylogenomic analysis, the sister relationship between Trichoptera and Lepidoptera is confirmed.
Materials and methods
Samples and DNA extraction
The Eubasilissa regina specimen used to determine the mt DNA was collected from Motuo, Xizang Autonomous Region, China, in August 2011. The Apatania sp. specimen was collected from Jiaocheng County, Shanxi Province, China, in July 2011. After collection, they were initially preserved in 95% ethanol in the field, and transferred to -20℃ for the long-term storage upon the arrival at the China Agricultural University (CAU). Total DNA was purified from muscle tissues of the thorax using TIANamp Genomic DNA Kit (TIANGEN). The quality of DNA was assessed through electrophoresis in a 1% agarose gel and staining with Gold View (nucleic acid stain replacing EB). Voucher specimens are deposited in the Entomological Museum of CAU with voucher numbers TRI001 for E. regina and TRI002 for Apatania sp.
PCR amplification and sequencing
The mt genomes of E. regina and Apatania sp. were generated by amplification of overlapping PCR fragments (Supplementary Material: Table S1). Firstly, fourteen fragments were amplified using the universal primers [23]. Then, six specifically designed primers (Supplementary Material: Table S1) based on the known sequences were used for the secondary PCRs.
All PCRs used NEB Long Taq DNA polymerase (New England BioLabs, Ipswich, MA) under the following amplification conditions: 30s at 95℃, 40 cycles of 10 s at 95℃, 50s at 48-55℃, 1kb/min at 65℃ depending on the size of amplicons, and the final elongation step at 65℃ for 10 min. The quality of PCR products were evaluated by agarose gel electrophoresis.
All fragments were sequenced in both directions using the BigDye Terminator Sequencing Kit (Applied Bio Systems) and the ABI 3730XL Genetic Analyzer (PE Applied Biosystems, San Francisco, CA, USA) with two vector-specific primers and internal primers for primer walking.
Bioinformatic analysis
The complete mt genome of E. regina and the nearly complete mt genome of Apatania sp. has been deposited in GenBank under accession number KF756943 and KF756944 respectively. Mt DNA sequences were proof-read and aligned into contigs in BioEdit version 7.0.5.3 [24]. Sequence analysis was performed as follows. Firstly, the tRNA genes were identified by tRNAscan-SE Search Server v.1.21 [25] using invertebrate mitochondrial predictors with a COVE cutoff score of 1, or by sequence similarity to tRNAs of other Lepidoptera insects [26, 27]. PCGs were identified as open reading frames corresponding to the 13 PCGs in metazoan mt genomes. The rRNA gene boundaries were interpreted as the end of a bounding tRNA gene and by alignment with other lepidopteran gene sequences [26-29]. The base composition, codon usage, and nucleotide substitution were analyzed with MEGA 4.0 [30]. The GC and AT asymmetry was measured in terms of GC and AT skews using the following formulae: AT-skew = (A-T)/(A+T) and GC-skew = (G-C)/(G+C) [31]. Secondary structures of the small and large subunits of rRNA were inferred using models predicted for Drosophila yakuba [32], Apis mellifera [33], and Manduca sexta [34]. Stem-loops were named according to M. sexta [34].
Phylogenetic analysis
Phylogenetic analysis was carried out based on 26 complete or nearly complete mt genomes from GenBank. The ingroup taxa include 24 species from 24 families, which represent eight orders of Holometabola with available mt genomes (Supplementary Material: Table S2). Two Paraneoptera taxa, namely Hydrometra sp. (Hemiptera), and Thrips imaginis (Thysanoptera) were selected as outgroups for their relatively close relationships with Holometabola [35]. The saturation of different codon positions was assessed by DAMBE [36].
DNA alignment was inferred from the amino acid alignment of 13 PCGs using Clustal X [37]. RNA alignment was conducted by G-blocks Server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) by more stringent selection. Alignments of individual genes were then concatenated excluding the stop codons. MrBayes Version 3.1.2 [38] and a PHYML online web server [39, 40] were employed to reconstruct the phylogenetic trees. Model selection was based on Modeltest 3.7 [41] for nucleotide sequences. According to the Akaike information criterion, the GTR+I+G model was optimal for analysis with nucleotide alignments and MtREV model for amino acid sequence. In Bayesian inference, two simultaneous runs of 2,000,000 generations were conducted. Each set was sampled every 200 generations with a burnin of 25%. Trees inferred prior to stationarity were discarded as burnin, and the remaining trees were used to construct a 50% majority-rule consensus tree. In the maximum likelihood (ML) analysis, the parameters were estimated during analysis and the nodal support values were assessed by bootstrap re-sampling (BP) [42] calculated using 100 replicates.
Results and discussion
Genome organization
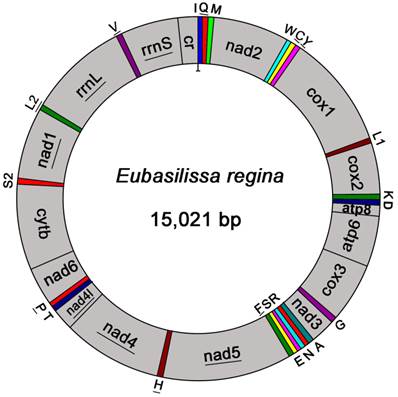
The complete mt genome of E. regina is a typical circular DNA molecule of 15,021 bp in length (GenBank accession number KF756943; Figure 1) and contains the ancestral 37 genes, with 9 PCGs and 14 tRNAs encoded in the major strand, while 4 PCGs, 8 tRNAs and 2 rRNAs encoded in the minor strand (Figure 1, Supplementary Material: Table S3). Notably, the genome of this species is the smallest one compared with the holometabolous mt genomes presently studied in the phylogenomic analysis. Such a short mt genome is mainly attributed to the small size of the A+T-rich region (270 bp) and large non-coding regions absence. Within the holometabolous mt genomes studied in this work, the length variation is minimal in PCGs, tRNAs, rrnL and rrnS, but very different in the putative control region (Figure 2; Supplementary Material: Table S4). In addition to the control region, there were 42 nucleotides dispersed in 10 intergenic spacers, ranging from 1 to 13. The longest spacer sequence (13 nucleotides) was located between tRNALys and tRNAAsp. Gene overlaps were also found at 7 gene junctions involving 22 nucleotides with the longest overlap (8 nucleotides) between tRNATrp and tRNACys (Supplementary Material: Table S3). The partial complete mt genome of Apatania sp. is 14,218 bp in length and contains 34 genes, with 12 complete PCGs and partial sequences of nad2, 19 tRNAs and 2 rRNAs.
Mitochondrial map of Eubasilissa regina. The tRNAs are denoted by the color blocks and are labelled according to the IUPACIUB single-letter amino acid codes. Gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left.
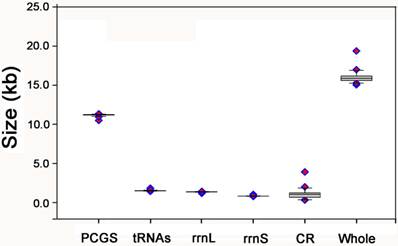
The size of PCGs, rrnL, rrnS, and CR, respectively, among the Holometabolous mt genomes used in this study.
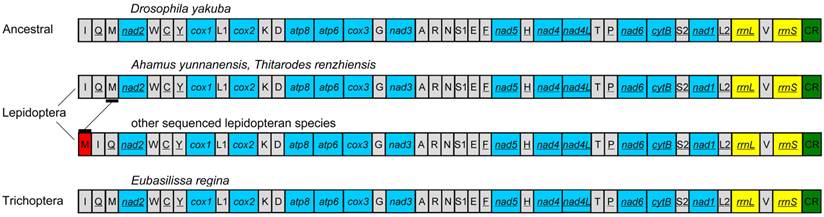
The gene order of the E. regina mt genome is the same as the ancestral gene order of Drosophila yakuba Burla, 1954, which is considered to exhibit the ground pattern of insect mt genomes [32], and all gene boundaries in D. yakuba are conserved in the mt genome of E. regina. The gene order of the partial Apatania sp. mt genome is also the same as D. yakuba. There are two types of gene orders in the lepidopteran mt genomes sequenced to date (89 species until 13 November 2013): one is the same as the ancestral gene order (i.e. Ahamus yunnanensis (Hepialidae) and Thitarodes renzhiensis (Hepialidae)) [27], the other contains the typical rearrangement of tRNAMet for the rest of sequenced lepidopteran mt genomes (Figure 3).
The mt genome gene orders of Eubasilissa regina, Drosophila yakuba and sequenced lepidopteran insects.
Protein-coding genes
The total length of all 13 PCGs of E. regina was 11,148 bp, accounting for 74.22% of the entire length of E. regina mt genome (Supplementary Material: Table S5). The overall A+T content of PCGs was 75.93%, ranging from 66.84% (cox1) to 80.98% (nad6) (Supplementary Material: Table S6). Start and stop codons were determined based on alignments with the corresponding genes of other Lepidoptera species. All but one of the PCGs initiated with ATN as the start codon, in which five genes (atp6, cox3, cytb, nad4, nad4l) use the standard ATG start codon, three genes (atp8, cox2, nad3) initiate with ATC, three genes (nad1, nad2, nad6) initiate with ATT, and nad5 initiates with ATA. The only exception was the cox1, which used CGA (coding for R) as a start codon. Six genes employ a complete translation termination codon TAA (atp6, atp8, cox3, nad4, nad4l, nad6), whereas the remaining three have incomplete stop codons, either T (cox1, cytb, nad1, nad2, nad3, nad5) or TA (cox2) (Supplementary Material: Table S3). The presence of incomplete stop codons is common in metazoan mt genomes [43] and these truncated stop codons were presumed to be completed via post-transcriptional polyadenylation [44]. The common stop codons TAA or TAG could always overlap several nucleotides within the down-stream tRNA, which was supposed to act as “backup” to prevent translation read through if the transcripts were not properly cleaved [45]. The absence of some G/C-rich codons was found in E. regina: the codon UAG and CCG were not used (Supplementary Material: Table S7). This result suggests that the A/T codon bias of the mt genomes affects the amino acid frequency of the encoded proteins [46].
All 11 of the 12 complete PCGs of Apatania sp. initiated with ATN as the start codon, in which six genes (atp6, atp8, cox2, nad1, nad3, nad6) use the standard ATT start codon, four genes (cox3, cytb, nad4, nad4l) initiate with ATG and nad5 initiates with ATC. The only exception was the cox1, which uses CGA (coding for R) as a start codon. Ten genes employ a complete translation termination codon TAA (atp6, atp8, cox2, cox3, cytb, nad2, nad3, nad4, nad4l, nad6), whereas the remaining three have incomplete stop codons T (cox1, nad1, nad5).
In many insect mt genomes, the atp8/atp6 and the nad4l/nad4 gene pairs overlap seven nucleotides (ATGNTAA) which are thought to be translated as a bicstron [47]. In the E. regina mt genome, the overlap nucleotides were conserved for atp8/atp6 (ATGATAA) but not conserved for nad4l/nad4.
Transfer RNAs
The entire complement of 22 typical tRNAs in the arthropod mt genomes was found in E. regina and schematic drawings of their respective secondary structures are shown in Figure 4. Most of the tRNAs could be folded as classic clover-leaf structures, with the exception of tRNASer(AGN), in which its DHU arm simply forms a four-nucleotide loop. This phenomenon was a typical feature of metazoan mt genomes [43]. Within the 22 tRNA genes, 14 genes were encoded by the J-strand, while the remains were coded by the N-strand.
Inferred secondary structure of 22 tRNAs of Eubasilissa regina. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Inferred Watson-Crick bonds are illustrated by lines, whereas GU bonds are illustrated by dots.
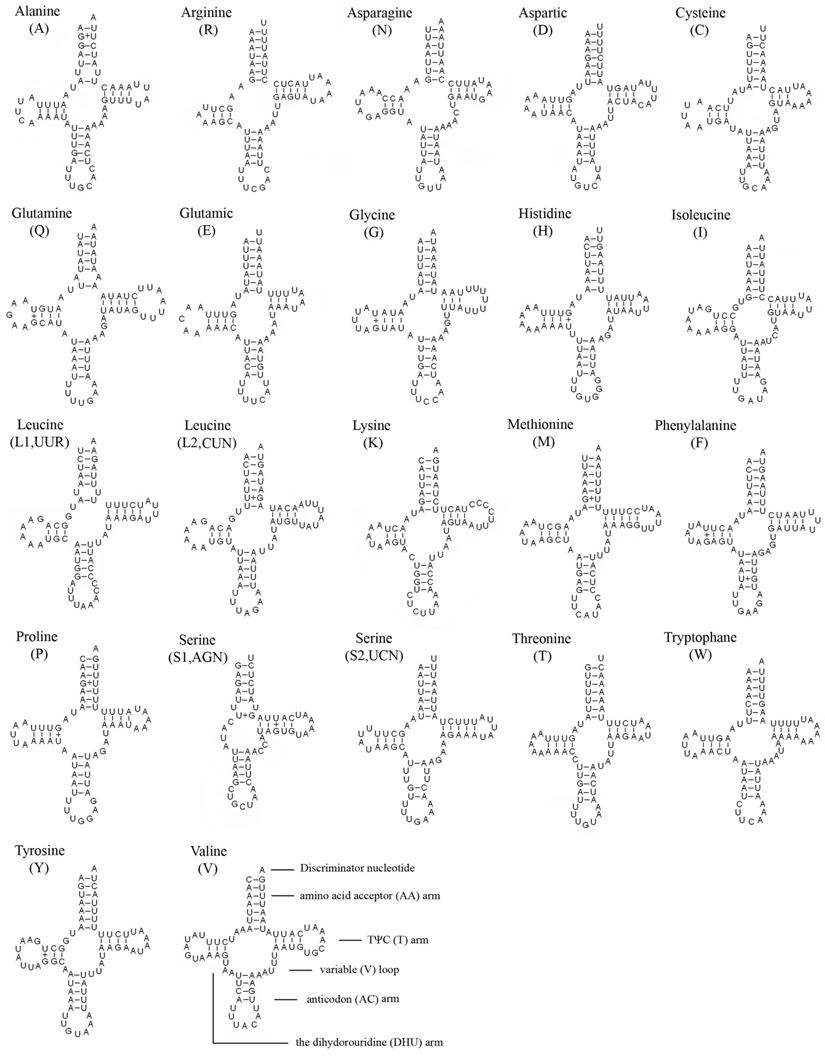
The length of tRNAs ranged from 56 to 70 bp. The aminoacyl (AA) stem (7 bp) and the AC loop (7 nucleotides) were invariable except tRNALeu(UUR) whose AC loop was 9 nucleotides. The DHU and TΨC (T) stems are variable while the loop size (3-8 nucleotides) was more variable than the stem size (0-5 bp). The size of the anticodon (AC) stems was constantly 5 bp except tRNALys whose anticodon stem was 4 bp.
19 mismatched base pairs were found in E. regina tRNAs based on the secondary structure. Thirteen of them were G-U pairs located in the AA stem (5 bp), the DHU stem (6 bp), the AC stem (1 bp) and the T stem (1 bp). The remaining were U-U (5 bp) and A-A (1 bp) mismatches (Figure 4).
Ribosomal RNAs
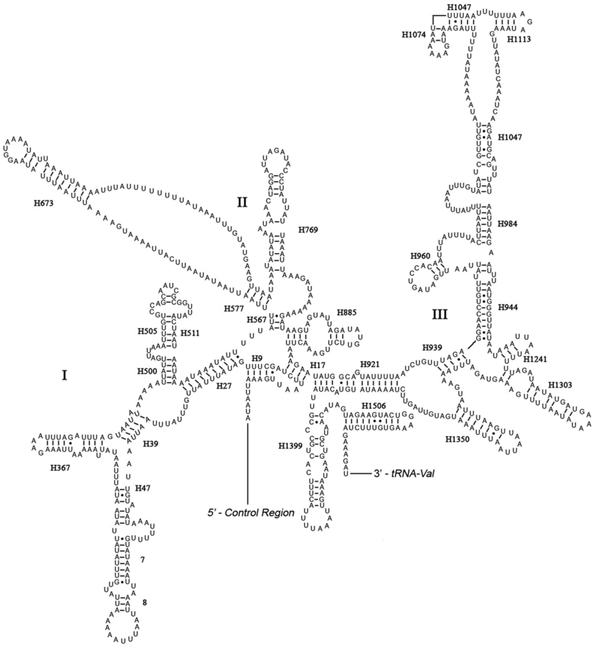
The rrnL was assumed to fill up the blanks between tRNALeu(CUN) and tRNAVal. For the boundary between the rrnS and the non-coding putative control region, alignments with homologous sequences in other Lepidoptera mt genomes were applied to determine the 3'-end of the gene [26-29]. The length of rrnL and rrnS of E. regina was determined to be 1,344 bp and 779 bp, respectively. Both rrnL and rrnS are generally congruent with the secondary structure models proposed for other insects [33, 34, 48-51]. The structure of rrnL of E. regina largely resembles previously published structures for Manduca sexta (Linnaeus, 1763) (Insecta: Lepidoptera: Sphingidae) [34], and the inferred secondary structure presents five canonical domains (I-II, IV-VI) with domain III absent, which is a typical trait in arthropods [48] (Figure 5), and includes 44 helices. The rrnS of E. regina is largely in agreement with those proposed for other Holometabola orders, including three domains and 28 helices (Figure 6).
Predicted secondary structure of the rrnL gene in Eubasilissa regina. Inferred Watson-Crick bonds are illustrated by lines, GU bonds by dots.
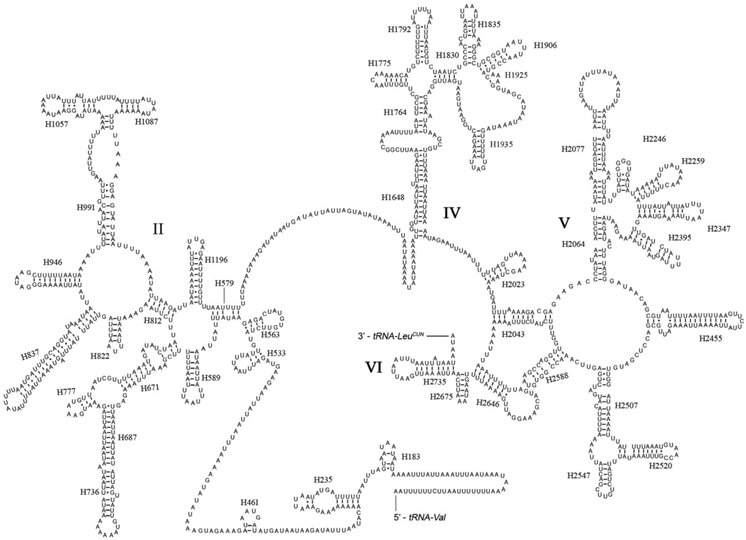
Predicted secondary structure of the rrnS gene in Eubasilissa regina. Roman numerals denote the conserved domain structure. Inferred Watson-Crick bonds are illustrated by lines, GU bonds by dots.
The control region
There are eleven non-coding regions of the whole mt genome of E. regina ranging from 1 to 270 nucletides. The largest non-coding region between rrnS and tRNAIle corresponds to the control region identified in other insects which includes the origin sites for transcription and replication [52]. The A+T content of this region (90.00%) is much higher than that of the coding regions. Several conserved structures of other lepidopteran mitogenomes were also observed in the A+T-rich region of E. regina mt genomes (Figure 7). The ON (origin of minority or light strand replication) in Bombyx mori (Linnaeus, 1758) (Lepidoptera: Bombycidae) has the motif ATAGA preceded by an 18 bp poly-T stretch, is located 21 bp upstream from rrnS [53]. This motif is conserved across Lepidoptera although the length of the poly-T stretch varies between species and is also conserved in E. regina. A poly-A commonly observed in other lepidopteran mt genomes was also found immediately upstream of the tRNAMet gene [26]. The majority of the mt control region of E. regina is made up of non-repetitive sequence but includes a microsatellite-like dinucleotide repeat region, (TA)5, located 21 bp from rrnS. The most abundant AT motif occurs 54 times, ATT occurs 19 times, AAT occurs 17 times, AATT occurs 5 times, ATATAA occurs 4 times. Some short tandem repeat sequences were detected in the control region of E. regina, such as (AT)3, (AT)4, (TA)5, (ATT)3. These tandem repeat elements can be considered as microsatellites and are useful in the study of geographical structure and phylogenetic relationship of species [54].
Features present in the control region of Eubasilissa regina.

Nucleotide composition and codon usage
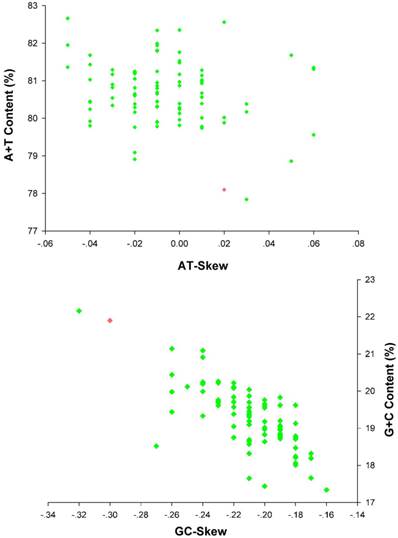
The nucleotide composition of E. regina mt genome is significantly biased toward A/T nucleotides (A = 39.86%, T = 38.25%, G = 7.66%, C = 14.23%; Supplementary Material: Table S5). The overall AT content (78.10%) of E. regina is lower than nearly all of AT content of the lepidopteran mt genomes except Ochrogaster lunifer (Thaumetopoeidae) (Supplementary Material: Table S8, Figure 8). A+T content of isolated PCGs, tRNAs, rRNAs and the control region (CR) is 75.93%, 83.19%, 84.27 and 90.00%, respectively. The metazoan mt genomes usually present a clear strand bias in nucleotide composition [55, 56], and the strand bias can be measured as AT- and GC-skews [31]. AT- and GC-skews of E. regina mt genomes are consistent to most of the strand biases of metazoan mtDNA (positive AT-skew and negative GC-skew for the J-strand), while species of three distantly related families of insects, i.e. Philopteridae (Phthiraptera), Aleyrodidae (Hemiptera) and Braconidae (Hymenoptera), have positive GC-skew and negative AT-skew for the J-strand [57]. A comparative analysis of A+T% vs AT-skew and G+C% vs GC-skew across all available mt genomes of Amphiesmenoptera (Lepidoptera + Trichoptera) is shown in Figure 8. The average AT-skew of the lepidopteran mt genomes is -0.02, ranging from 0.06 to -0.05, whereas the average GC-skew of the lepidopteran mt genomes is -0.20, ranging from -0.16 to -0.32. The codon-based nucleotide composition of E. regina indicates that the A/T content at the third codon position (86.01%) is higher than that of the first and second codon positions (71.56% and 70.24%, respectively).
AT% vs AT-Skew and GC% vs GC-Skew in Amphiesmenoptera mt genomes. Measured in bp percentage (Y-axis) and level of nucleotide skew (X-axis). Values are calculated on full length mt genomes. Green diamond, Lepidoptera; Red diamond, Trichoptera.
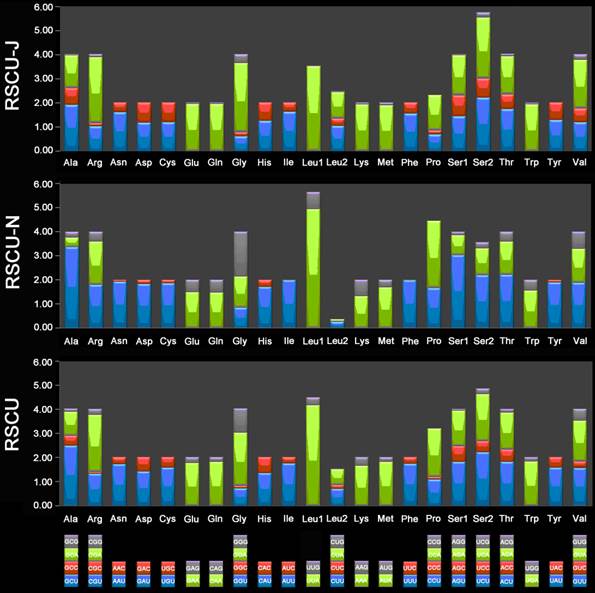
The 13 PCGs of E. regina and 12 complete PCGs of Apatania sp. exhibit the canonical mitochondrial start codons ATN for invertebrate mtDNAs [43] except cox1 genes which use CGA (coding for R) as a start codon. All Lepidoptera species examined to date use R as the initial amino acid for cox1 and the use of non-canonical start codons for this gene is common across insects [58]. Cox1 initiating with codon CGA is probably a synapomorphy of the monophyletic lineage comprising Trichoptera and Lepidoptera. Stop codons for the 13 PCGs were almost invariably complete TAA or incomplete TA/T. The genome-wide bias toward AT was well documented in the codon usage (Supplementary Material: Table S7). At the third codon position, A/T was overwhelmingly represented compared to G or C (Figure 9). There is a strong bias toward AT-rich codons with the six most prevalent codons in E. regina, as in order, TTA-Leu (10.45%), ATT-Ile (10.02%), TTT-Phe (8.89%), ATA-Met (7.55%), AAT-Asn (5.32%), and TAT-Tyr (3.55%) (Supplementary Material: Table S7).
Relative synonymous codon usage (RSCU) in the Eubasilissa regina mt genome. Codon families are provided on the x-axis.
Phylogeny
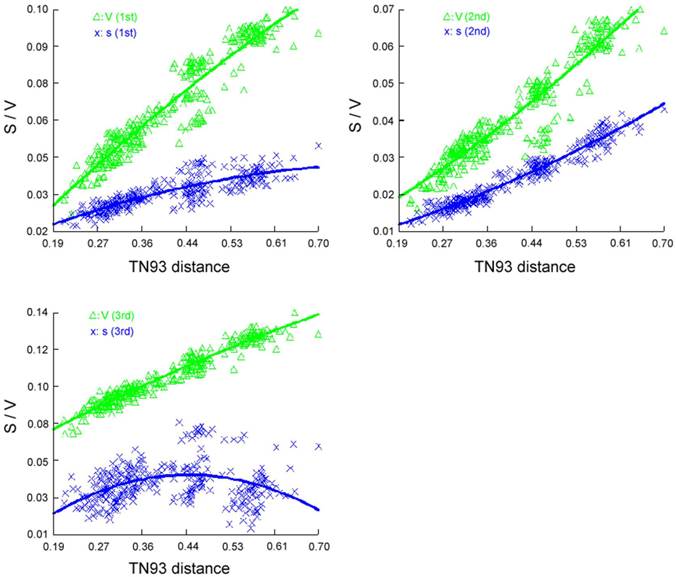
We performed phylogenetic analysis including 24 species, which represent eight orders of Holometabola with available mt genomes as ingroup taxa and two Paraneoptera taxa, namely Hydrometra sp. (Hemiptera) and Thrips imaginis (Thysanoptera) as outgroups (Supplementary Material: Table S2). In order to notarize the phylogenetic position of Trichoptera, nearly complete mt genome of Apatania sp. (Trichoptera: Apataniidae) was also sequenced. Before reconstructing the phylogeny tree, the saturation of different codon positions in PCGs was tested (Figure 10). The results indicate that the third positions of codons have experienced substitution saturation; hence the nucleotides of the third postions are excluded from the datasets to reconstruct the phylogeny tree. Herein, three datasets were used in the present analyses, each representing different types of data partitioning and inclusion/exlusion of particular sites. There were 7553 sites in the PCG12R matrix (containing the first and the second codon positions of PCGs, plus the two rRNA genes), 6620 sites in the PCG12 matrix (containing the first and the second codon positions of PCGs), and 3310 sites in the AA matrix (containing the amino acid sequences).
Saturation curve of different codon position transitions (S) and transversions (V) per TN93 distance.
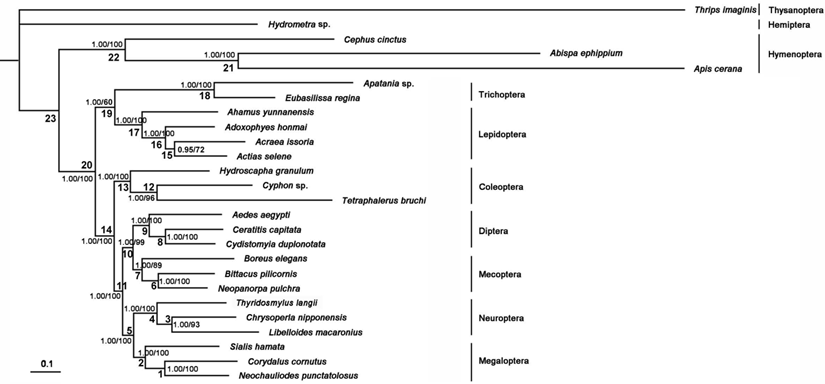
The phylogenetic trees generated from Bayesian and ML inferences have the same topologies (Figure 11) based on the PCG12R and PCG12 matrices, whereas the AA matrix could not result in reasonable phylogenetic trees. The supporting values of the PCG12R matrix are higher than the other matrices shown in Figure 11. Bayesian posterior probabilities and bootstrap values of ML of major nodes recovered by various phylogenetic approaches are listed in Supplementary Material: Table S9. Monophyly of the eight ingroup orders was recovered in all analyses. The Hymenoptera was recovered to be the sister of all other homometabolous lineages, which was consistent with the previous study [59,60]. The sister relationship between Trichoptera and Lepidiptera was recognized in all analyses with high nodal support values. Mecoptera was recovered as a sister-group of Diptera. Neuropterida (Neuroptera + Megaloptera in this study) is supported across all analyses, forming the sister-group to (Diptera + Mecoptera), which is consistent with Wei et al [61] and differs from the presently preferred sister-group relationship of Neuropterida and Coleoptera [11] (Figure 11).
Phylogenetic relationships among the holometabolous insects used in this study. Numbers at the nodes are Bayesian posterior probabilities (left) and ML bootstrap values (right).
Conclusion
This is the first description of the complete mt genome of a caddisfly species E. regina (Trichoptera: Phryganeidae). Firstly, considering the structure of mt genome, the cox1 of the two caddisfly species (E. regina and Apatania sp.) herein studied both start with CGA (coding for R), and all lepidopteran species examined to date also use R as the initial amino acid for cox1 [58], which indicates that cox1 initiating with codon CGA is probably a synapomorphy of the monophyletic lineage comprising Trichoptera and Lepidoptera. Secondly, the mt genomic phylogeny herein reconstructed clearly supports the sister relationship between Trichoptera and Lepidoptera. More robust interfamilial phylogeny within Spicipalpia of Trichoptera should be made in future mt phylogenomic analysis based on a comprehensive sampling.
Abbreviations
atp6 and atp8: genes for the ATPase subunits 6 and 8; cox1-cox3: genes for cytochrome c oxidase subunits I-III; cytb: a gene for apocytochrome b; nad1-nad6 and nad4l: genes for NADH dehydrogenase subunits 1-6 and 4l; rrnL: large (16S) rRNA subunit (gene); rrnS: small (12S) rRNA subunit (gene); tRNAX (where X is replaced by three letter amino acid code of the corresponding amino acid), transfer RNA.
Supplementary Material
Table S1 - Table S9.
Acknowledgements
We express our sincere thanks to Ms. Lihua Wang and Mr. Taiyang Wei (Beijing) for collecting specimens. We are especially thankful to Dr. Changhai Sun (Nanjing) for identifying the specimen of Apatania sp. This research was supported by the National Natural Science Foundation of PR China (Nos. 31320103902 and 31322501) and the Foundation for the Author of National Excellent Doctoral Dissertation of PR China (No. 201178).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Resh VH, Rosenberg DM. The Ecology of Aquatic Insects. New York: Praeger. 1984
2. Zhang ZQ. Animal biodiversity: An outline of higher-level classification and survey. Zootaxa. 2011;3148:1-237
3. Malm T, Johanson KA, Wahlberg N. The evolutionary history of Trichoptera (Insecta): A case of successful adaptation to life in freshwater. Syst Entomol. 2013;38:459-473
4. Schmid F. Essai d'evaluation de la faune mondiale des Trichopteres. In: (ed.) Morse JC. Proceedings of the 4th International Symposium on Trichoptera. The Hague: Dr. W. Junk. 1984:337
5. De Moor F, Ivanov V. Global diversity of caddisflies (Trichoptera: Insecta) in freshwater. Hydrobiologia. 2008;595:393-407
6. Resh VH, Unzicker JD. Water quality monitoring and aquatic organisms: the importance of species identification. Journal (Water Pollution Control Federation). 1975;47:9-19
7. Dohet A. Are caddisflies an ideal group for the biological assessment of water quality in streams. Nova Suppl Ent. 2002;15:507-520
8. Rosenberg DM, Resh VH. Freshwater biomonitoring and benthic macroinvertebrates. New York: Chapman and Hall. 1993
9. Resh V. Recent trends in the use of Trichoptera in water quality monitoring. In: (ed.) Otto C. Proceedings of the 7th International Symposium on Trichoptera. Leiden: Backhuys Publishers. 1993:285-291
10. Kristensen NP. Studies on the morphology and systematics of primitive Lepidoptera (Insecta). Steenstrupia. 1984;10:141-191
11. Whiting MF. Phylogeny of the holometabolous insect orders: molecular evidence. Zool Scr. 2002;31:3-15
12. Beutel RG, Friedrich F, Hörnschemeyer T. et al. Morphological and molecular evidence converge upon a robust phylogeny of the megadiverse Holometabola. Cladistics. 2011;27:341-355
13. Wheeler WC, Whiting M, Wheeler QD, Carpenter JM. The phylogeny of the extant hexapod orders. Cladistics. 2001;17:113-169
14. Ross HH. The caddis flies, or Trichoptera of Illinois. Bull Ill Nat Hist Surv. 1944;23:1-326
15. Weaver JSIII. The evolution and classification of Trichoptera, part I: the groundplan of Trichoptera. In: (ed.) Morse JC. Proceedings of the 4th International Symposium on Trichoptera. The Hague: Dr. W. Junk. 1984:413-419
16. Kristensen N. Phylogeny of extant hexapods. In: Naumann ID, ed. The insects of Australia, 2nd ed. Melbourne: Melbourne University Press. 1991;1:125-140
17. Kjer KM, Blahnik RJ, Holzenthal RW. Phylogeny of Trichoptera (caddisflies): characterization of signal and noise within multiple datasets. Syst Biol. 2001;50:781-816
18. Koehler CM, Bauer MF. Mitochondrial function and biogenesis. Heidelberg: Springer Verlag. 2004
19. Dowton M, Castro L, Austin AD. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: the examination of genome 'morphology'. Invertebr Syst. 2002;16:345-356
20. Boore JL, Macey JR, Medina M. Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymol. 2005;395:311-348
21. Masta SE, Boore JL. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol Biol Evol. 2008;25:949-959
22. Boore JL. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol Evol. 2006;21:439-446
23. Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Ann Rev Ecol Evol Syst. 2006;37:545-579
24. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95-98
25. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acid Res. 1997;25:955-964
26. Liu QN, Zhu BJ, Dai LS, Wei GQ, Liu CL. The complete mitochondrial genome of the wild silkworm moth, Actias selene. Gene. 2012;505:291-299
27. Cao YQ, Ma C, Chen JY, Yang DR. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: the ancestral gene arrangement in Lepidoptera. BMC genomics. 2012;13:276
28. Hu J, Zhang D, Hao J, Huang D, Cameron S, Zhu C. The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event. Mol Biol Rep. 2010;37:3431-3438
29. Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene. 2006;373:52-57
30. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
31. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
32. Clary DO, Wolstenholme DR. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985;22:252-271
33. Gillespie J, Johnston J, Cannone J, Gutell R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol. 2006;15:657-686
34. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-123
35. Grimaldi DA, Engel MS. Evolution of the Insects. New York: Cambridge University Press. 2005
36. Xia X, Lemey P. Assessing substitution saturation with DAMBE. The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing. 2009:2
37. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acid Res. 1997;25:4876-4882
38. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
39. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
40. Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucl Acid Res. 2005;33:W557-W559
41. Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
42. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791
43. Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173-216
44. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470-474
45. Boore J. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda). BMC genomics. 2006;7:182
46. Haruyama N, Mochizuki A, Sato Y, Naka H, Nomura M. Complete mitochondrial genomes of two green lacewings, Chrysoperla nipponensis (Okamoto, 1914) and Apochrysa matsumurae Okamoto, 1912 (Neuroptera: Chrysopidae). Mol Biol Rep. 2011;38:3367-3373
47. Stewart JB, Beckenbach AT. Insect mitochondrial genomics: the complete mitochondrial genome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome. 2005;48:46-54
48. Cannone J, Subramanian S, Schnare M, Collett J, D'Souza L, Du Y. et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinf. 2002;3:2
49. Buckley T, Simon C, Flook P, Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol Biol. 2000;9:565-580
50. Niehuis O, Naumann CM, Misof B. Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org Divers Evol. 2006;6:17-32
51. Niehuis O, Yen S-H, Naumann CM, Misof B. Higher phylogeny of zygaenid moths (Insecta: Lepidoptera) inferred from nuclear and mitochondrial sequence data and the evolution of larval cuticular cavities for chemical defence. Mol Phylogenet Evol. 2006;39:812-829
52. Taanman J-W. The mitochondrial genome: structure, transcription, translation and replication. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 1999;1410:103-123
53. Saito S, Tamura K, Aotsuka T. Replication origin of mitochondrial DNA in insects. Genetics. 2005;171:1695-1705
54. Li H, Liu H, Shi A, Štys P, Zhou X, Cai W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS One. 2012;7:e29419
55. Hassanin A, Léger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54:277-298
56. Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol. 2006;38:100-116
57. Wei S, Shi M, Chen X. et al. New views on strand asymmetry in insect mitochondrial genomes. PloS One. 2010;5:e12708
58. Fenn J, Cameron S, Whiting M. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol Biol. 2007;16:239-252
59. Savard J, Tautz D, Richards S. et al. Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of Holometabolous insects. Genome Res. 2006;16:1334-1338
60. Wang Y, Liu X, Winterton SL, Yang D. The first mitochondrial genome for the fishfly subfamily Chauliodinae and implications for the higher phylogeny of Megaloptera. PLoS One. 2012;7:e47302
61. Wei S, Shi M, Sharkey MJ, Van Achterberg C, Chen X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics. 2010;11:371
Author contact
![]() Corresponding author: xingyue_liucom, dyangcaucom.
Corresponding author: xingyue_liucom, dyangcaucom.