Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(2):212-224. doi:10.7150/ijbs.8056 This issue Cite
Research Paper
Bufalin Induces the Interplay between Apoptosis and Autophagy in Glioma Cells through Endoplasmic Reticulum Stress
Shuying Shen1, Yi Zhang1, Zhen Wang1, Rui Zhang2, Xingguo Gong1 ![]()
1. Institute of Biochemistry, Zhejiang University, Hangzhou, 310058, PR China;
2. Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, No. 300 Guangzhou Road, Nanjing 210029, China.
Received 2013-11-5; Accepted 2014-1-16; Published 2014-2-6
Abstract
Malignant gliomas are common primary tumors of the central nervous system. The prognosis of patients with malignant glioma is poor in spite of current intensive therapy and thus novel therapeutic modalities are necessary. Bufalin is the major component of Chan-Su (a traditional Chinese medicine) extracts from the venom of Bufo gargarizan. In this study, we evaluated the growth inhibitory effect of bufalin on glioma cells and explored the underlying molecular mechanisms. Our results showed that bufalin inhibited the growth of glioma cells significantly. Mechanistic studies demonstrated that bufalin induced apoptosis through mitochondrial apoptotic pathway. In addition, bufalin was also found to induce ER stress-mediated apoptosis, which was supported by the up- regulation of ER stress markers, CHOP and GRP78, and augmented phosphorylation of PERK and eIF2α as well as cleavage of caspase-4. Downregulation of CHOP using siCHOP RNA attenuated bufalin-induced apoptosis, further confirming the role of ER stress response in mediating bufalin-induced apoptosis. Evidence of bufalin-induced autophagy included formation of the acidic vesicular organelles, increase of autophagolysosomes and LC3-II accumulation. Further experiments showed that the mechanism of bufalin-induced autophagy associated with ATP deleption involved an increase in the active form of AMPK, decreased phosphorylation levels of mTOR and its downstream targets 4EBP1 and p70S6K1. Furthermore, TUDC and silencing of eIF2α or CHOP partially blocked bufalin-induced accumulation of LC3-II, which indicated that ER stress preceded bufalin-induced autophagy and PERK/eIF2α/CHOP signaling pathway played a major part in the process. Blockage of autophagy increased expression of ER stress associated proteins and the ratio of apoptosis, indicating that autophagy played a cytoprotective role in bufalin induced ER stress and cell death. In conclusion, bufalin inhibits glioma cell growth and induces interplay between apoptosis and autophagy through endoplasmic reticulum stress. It will provide molecular bases for developing bufalin into a drug candidate for the treatment of maglinant glioma.
Keywords: ER stress, autophagy, apoptosis, bufalin, glioma cancer.
Introduction
Autophagy is an important cellular catabolic process which functions to maintain homeostasis by degrading excessive or unnecessary proteins and dysfunctional cellular organelles in living cells [1]. In eukaryotic cells, autophagy is a key mechanism for long-lived protein degradation and organelle turnover, which serves as a critical adaptive response that recycles energy and nutrients during starvation or stress [2]. In addition to its important homeostatic role, this degradation pathway is involved in various human disorders, including metabolic conditions, infectious diseases and cancers [3]. Recently, the frontier of autophagy and cancer has experienced an explosion of research. Cancer cells may utilize autophagy to enhance fitness to survive with altered metabolism in the hostile tumor microenvironment, suggesting deployment of therapeutic strategies to block autophagy for cancer therapy. On the other hand, high levels of autophagy might directly lead to autophagic cell death (type II programmed cell death) in cancers [4]. Indeed, the final life-or-death destiny of cancer cells is influenced by the interaction of autophagy and apoptosis, which depends on the type, concentration and duration of chemotherapeutic drugs as well as the type of cancer cells.
Several independent studies support that autophagy could be induced by unfolded protein response (UPR), the major endoplasmic reticulum (ER) stress pathway [5]. In essence, the UPR functions to relieve the ER burden by inhibiting protein translation, reducing the amount of proteins entering the ER and increasing degradation of misfolded proteins [6]. UPR is primarily regulated by three ER-located sensors such as activating transcription factor 6 (ATF6), inositol requiring enzyme 1 (IRE1), and PKR-like ER kinase (PERK). In response to ER stress, ATF6 is cleaved by site 1 and site 2 proteases (S1P and S2P) and then migrates to the nucleus to activate the transcription of GRP78. IRE1 initiates gene expression by processing the mRNA of the transcription factor X box-binding protein 1 (XBP1) via its endonuclease activity. PERK phosphorylates a subunit of the eukaryotic translation initiation factor 2α (eIF2α), leading to a general reduction in protein synthesis as a way to counteract ER stress [7]. It also mediates transcriptional activation of Atg5 and LC3 proteins via the C/EBP homologous protein (CHOP) and activating transcription factor 4 (ATF4), respectively. Signaling mechanisms linking ER stress to autophagy vary with specific stress conditions and organisms [8].
ER stress can trigger the apoptotic machinery, and ultimately lead to cell death under severe or chronic stress conditions, for instance, when adaptive responses are exceeded or a dysfunctional UPR is unable to correct the balance of ER stress [9]. CHOP is the best characterized mediator in the transition of ER stress to apoptosis, which is a key proapoptotic transcription factor induced during ER stress [10]. In addition to CHOP, caspase-4, an ER-resident caspase, is another major pro-apoptotic factor that is involved in ER stress-induced cell death pathway [11].
Bufalin, a soluble digoxin-like immunoreactive component of Chansu (a traditional Chinese medicine), which is an extract of dried toad venom from the skin glands of Bufogargarizans or Bufomelanostictus, can exert growth inhibition, cell cycle arrest, autophagy and apoptosis in hepatocellular carcinoma [12] and colorectal cancer [13]. Therefore, bufalin may act as a promising new candidate for adjuvant therapy against some malignant tumors by activation of cell death. However, the precise mechanisms remain unknown and no detailed studies have so far been reported on its action on human glioma cells.
Given the diverse effects described for bufalin in different model systems, we initiated a study to measure the changes in cellular protein levels and activities in response to bufalin treatment to gain further insight into the metabolic and regulatory pathways. The results revealed that bufalin induced mitochondrial-mediated apoptosis, autophagy via the AMPK-mTOR signaling and PERK/eIF2α/CHOP pathway, endoplasmic reticulum (ER) stress and unfolded protein response (UPR). This study also unveiled the concomitant interplay between these cellular events and their impact on human glioma cells in response to bufalin. Analysis of the expression of ER chaperones and markers of UPR showed that ER signaling pathway may play a pivotal role in modulating the crosstalk between autophagy and apoptosis in our cell lines.
Materials and methods
Materials
Bufalin was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in dimethylsulfoxide (DMSO) at 0.4 mM and diluted with fresh medium to achieve the desired concentration. Caspase-3 colorimetric assay kit was purchased from BioVision, Inc. (U.S.A). 3-Methyladenine (3-MA), 3-(4,5-dimetrylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), fetal bovine serum (FBS), temozolomide (TM), Dihydroethidium (DHE), wortmannin (WORT), Chloroquine (CQ), monodancylcadaverin (MDC), acridine orange (AO) and tauroursodeoxycholate (TUDC) were purchased from Sigma (St. Louis, MO, USA). Rabbit antibodies specific for Cleaved caspase-3, Cleaved caspase-4, Cleaved PARP, Bcl-2, Bax, LC3B, AMPK, p-AMPKα(Thr172), mTOR, p-mTOR, p-4EBP, p70S6K, p-p70S6K, Atg5, Beclin1, ACC, p-ACC, CHOP, GRP78, GRP94, ATF6, PERK, p-PERK, IRE1α, p-IRE1α, eIF2α, p-eIF2α, cytosolic cyto c and GAPDH were purchased from Cell Signaling Technology (MA, USA). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was obtained from Santa Cruz Biotechnology.
Cell culture
U87MG and LN229 glioma cells, obtained from American Type Culture Collection (Manassas, VA, USA), were cultured in DMEM medium (Sigma), supplemented with 10% FBS, 100 units/ml penicillin, and 100 units/ml streptomycin. The cells were incubated at 37℃ in a humidified atmosphere containing 5% CO2 inside a CO2 incubator.
MTT assay
MTT assay was employed to examine the effects of bufalin on the proliferation of glioma cells. Briefly, the cells were seeded in 96-well plates at 5×103 cells/well in 200μl medium. Then the cells in the wells were treated with various concentrations of bufalin and cultured for 24 h or 48 h. At the end of culture, MTT solution (0.5 mg/mL in 20μL PBS) was added to each well and incubated for 4 h at 37°C. A enzyme-labeled instrument (Thermo) was used to measure the absorbance of each well at 570 nm. Data were calculated from three independent experiments, each performed in sextuplicate.
Apoptosis analysis by flow cytometry
Different stages of apoptosis were distinguished using an Annexin V-FITC/propidium iodide apoptosis kit (MACS Miltenyi Biotec). Briefly, cells were cultured with bufalin at various concentrations for 24 h, and then 1×106 cells were harvested, washed twice with ice-cold PBS. Apoptotic cells were evaluated by double staining with annexin V-FITC and PI in binding buffer using by flow cytometry. All experiments were carried out in triplicate.
Investigation of cellular redox state
DHE was used to determine the cellular ROS level. Single-cell suspensions of cells treated with different concentrations of bufalin (or DMSO as a blank control, 10μM H2O2 for 2h as a positive control) for 24 h were washed and incubated with 1μM DHE at 37 ℃ for 30 min. Cells were then washed three times with probe-free PBS, and the fluorescence intensities of cells were measured by flow cytometry.
Detection of acidic vesicular organelles
Formation of acidic vesicular organelles (AVOs), a morphological characteristic of autophagy, was detected by acridine orange (AO) staining. Cells were stained with 1 μg/ml acridine orange for 20 min and the samples were observed under a laser scanning confocal microscopy (excitation, 546 nm; emission, 575/640 nm).
Visualization of autophagic vacuoles
The autofluorescent agent MDC was used as a specific autophagolysosome marker to analyze the autophagic process. Glioma cells were treated with 80 nM bufalin or 100 μM temozolomide for 24 h. Autophagic vacuoles were labeled with MDC by incubating cells with 50 μM MDC in PBS at 37℃ for 20 min. After incubation, cells were washed three times with PBS and immediately analyzed by a laser scanning confocal microscopy (excitation, 390 nm; emission, 460 nm).
Transfection with small interfering RNA
Cells were seeded at 2 × 105 cells in 6-well plates 24 h before transfection. Then, the cells were transfected with 60 nM of specific or nontargeting siRNA using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. After 6 h, the opti-MEM was replaced by complete medium, and cells were incubated for another 48 h. After the incubation, cells were treated with various concentrations of bufalin for 24 h and used for subsequent experiments. The sequences of siRNAs are as follows: si-CHOP (5'-AAGAACCAGCAGAGGUCACAA-3'); si-Atg5(5'-GGATGAGATAACTGAAAGG-3'); si-AMPK(5'-AUGAUGUCAGAUGGUGAAU-3'); si-Beclin1(5'-CAGTTTGGCACAATCAATA-3'); si-eIF2α(5'-CACAAATTTCCTGAGGTGGAAGA-3'); si-control(5'-GAGCGCUAGACAAUGAAG-3'). All above siRNAs were purchased from GenePharma (Shanghai, China).
Measurement of ATP content
ATP level was measured as described previously [14]. Briefly, control and bufalin-treated cells were trypsinized and collected, counted in triplicate using a Coulter Z2 Particle Count and Size Analyzer (Coulter Corporation, Miami, FL), after that cells (2,500-104cells/50 ml) were placed in a 96-well plate, six wells per sample. ATP level was measured by the luciferinluciferase method using an ATP Bioluminescence Assay Kit (CellTiter-Glo Luminescent Cell viability assay, Promega, USA). A standard curve was generated from indicated concentrations of ATP for each experiment to reassure the reproducibility of each experiment. Each experiment was performed in triplicate.
Western blot analysis
Whole cellular protein was extracted from glioma cells prepared with lysis buffer for western blotting. Briefly, the cells were lysed in RIPA buffer for 30 min on the ice. Protein levels were quantified using Lowry method. Equivalent amounts of protein (30 μg/lane) were separated by 7-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose blotting membranes. The membranes were blocked in TBST containing 5% non-fat dry milk (w/v) for 2h, and then incubated with primary antibodies at 1/1000 dilution overnight at 4°C and corresponding HRP-conjugated secondary antibodies at room temperature (25℃) for 1 h. Protein bands were visualized by chemiluminescence detection.
Statistical analysis
The data were presented as the mean ± standard deviation (SD) from experiments performed in triplicate. The difference between two mean values was evaluated using the Student's t-test and p < 0.05 was considered to be statistically significant. The statistical analyses were performed using the SPSS software 13.0 (SPSS Inc.,Chicago, IL, USA).
Results
Bufalin inhibits the proliferation of glioma cells and induces mitochondria-mediated apoptosis
MTT analysis showed that bufalin treatment caused remarkable growth inhibition in a dose- and time-dependent manner in U87MG cells (Fig. 1A) and LN229 cells (Supplementary Material: Fig. S1A). To identify if the bufalin-induced reduction in viability of glioma cells occurred via apoptosis, Annexin V-FITC/PI double staining analysis was performed. A statistically significant dose-dependent increase of apoptotic cells was observed after exposed to 40nM (early apoptotic:14.4%; late apoptotic and necrotic:12.6%) and 80nM bufalin (early apoptotic: 22.8%; late apoptotic and necrotic:14.8%) for 24 h compared to DMSO-treated controls (4.5%) (Fig. 1B). Similarly, bufalin induced apoptosis in LN229 cells was shown by dose-dependent increase in the number of apoptotic cells (Supplementary Material: Fig. S1B).
Bufalin inhibits U87MG cell growth and induces mitochondria-mediated apoptosis. A: U87MG cells were treated with various concentrations of bufalin for 24 h and 48 h. Cell viability was determined by MTT assay. Data are presented as mean ± SD, n = 3. *p < 0.05 and **p < 0.01 versus control group. B: U87MG cells were treated with bufalin for 24 h, stained with Annexin V and PI, and then analyzed by flow cytometry. Results are representative of three independent experiments. C: Effect of bufalin on cellular ROS production. Results are expressed as a ratio of relative fluorescent intensity. Each bar represents the mean±SD from six experiments. ( *p < 0.05 and**p < 0.01 versus DMSO control ). D: Western blot analysis for the expression of Bcl-2, Bax, cytosolic cytochrome c, cleaved PARP and cleaved caspase-3.
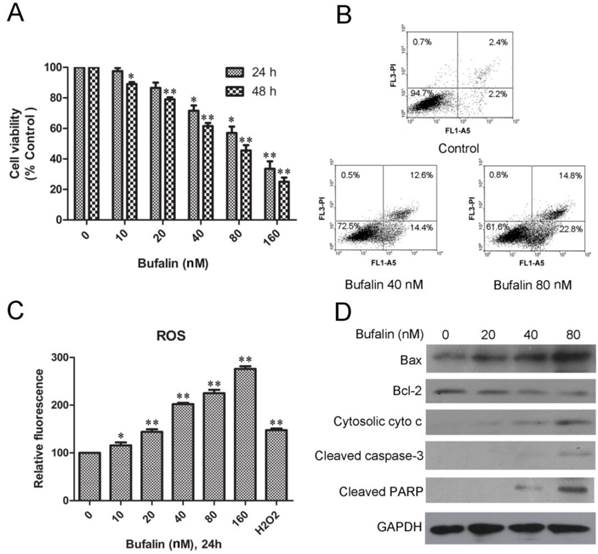
To access if bufalin exposure led to oxidative stress, we quantified ROS in U87MG cell cultures exposed for 24 h up to 160nM bufalin. As shown in Fig. 1C, bufalin treatment induced a significant dose-dependent ROS increase compared to control cells. For the reason that mitochondrial dysfunction is closely related to oxidative stress [15], we next detected mitochondrial apoptosis-related proteins in U87MG cells treated with bufalin. According to our results, bufalin up-regulated the ratio of Bax/Bcl-2 and enhanced the expression of cytoplasmic cytochrome c and PARP as well as cleaved caspase-3 (Fig. 1D). Besides, as shown in Supplementary Material: Fig S1C, the levels of cleaved caspase-3 and PARP were also increased after bufalin exposure for 24 h in LN229 cells. All these results indicate that mitochondrial apoptosis pathway contributes to bufalin-induced glioma cell death.
Bufalin activates autophagy process in glioma cells
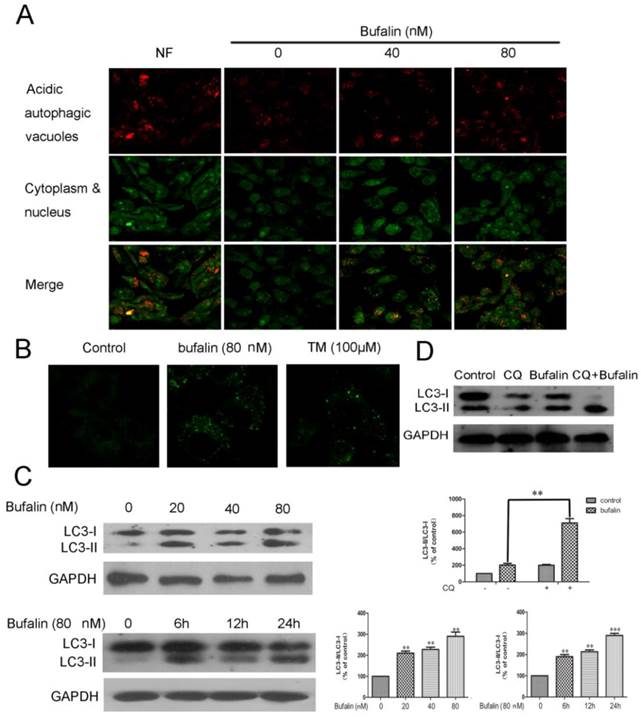
A series of experiments were performed to shed light on the autophagic profiles of bufalin. Acridine orange staining was employed to visualize acidic vesicular organelles in control and bufalin-treated cells. As shown in Fig. 2A, bufalin treatment markedly elevated the quantity of autophagic vacuoles in U87MG cells. Furthermore, we measured the incorporation of MDC, which is a marker for mature autophagic vacuoles (AVs) such as autophagolysosomes [16]. The bufalin treatment significantly increased MDC stained AVs in U87MG and LN229 cells (Fig. 2B, Supplementary Material: Fig S2A), which appeared as distinct dot-like structures distributed within the cytoplasm or localizing in the perinuclear regions. We next analyzed the effect of bufalin on LC3-II protein, which is considered as a determinate marker of autophagy activation [17]. Cellular lysates from cells treated with bufalin were analyzed by LC3 western blotting, showing that LC3-II (16 kDa) migrated faster than LC3-I (18 kDa). An increase in LC3-II protein level in the lysates of bufalin-treated glioma cells was confirmed by the western (Fig. 2C, Supplementary Material: Fig. S2B). In order to monitor autophagy flux, we introduced chloroquine (CQ), a lysosomotropic reagent, in the study. As shown in Figure 2D, the difference in LC3-II levels in the presence and absence of CQ was larger with addition of bufalin, indicating that autophagic flux is increased by bufalin treatment.
Bufalin induces autophagy in U87MG cells. A: Formation of autophagic vacuoles in U87MG cells treated in the nutrient-free (NF) condition or treated with bufalin for 24 h. B: U87MG cells were treated with 80 nM bufalin for 24 h and stained by MDC. MDC staining of cells treated with temozolomide (TM) was presented as a positive control. C: Total cell extracts were assayed by western blotting for expression of LC3-I and LC3-II. D: U87MG cells were treated with bufalin (40 nM) and/or 20 µM Chloroquine (CQ) for 24 h and then subjected to Western Blot. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n =3). **p<0.01 and ***p< 0.001 versus the control or the bufalin treatment alone.
Bufalin targets mTOR signaling via AMPK activation
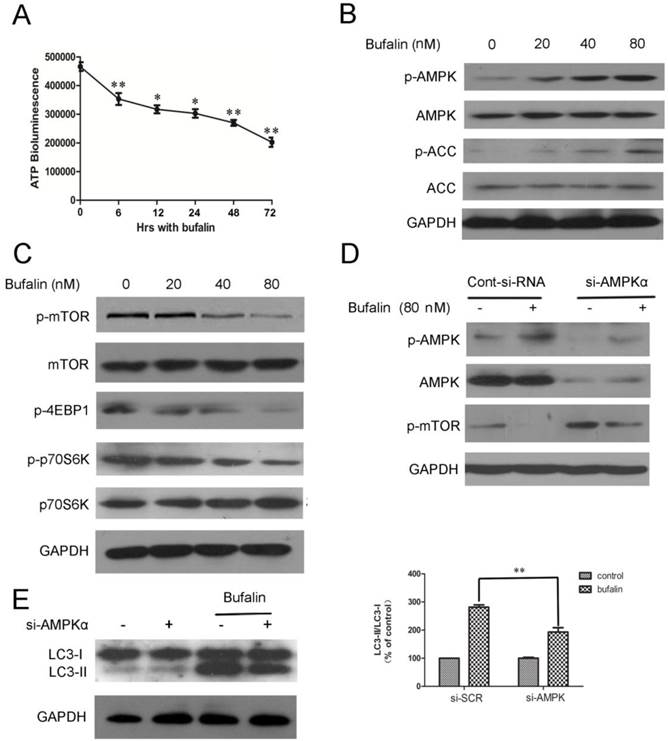
We demonstrated that bufalin damaged the integrity of mitochondrial membrane and resulted in the dysfunction of mitochondria. Mitochondria is the energy source of cell, which is an important determinant for the formation of autophagosomes [18]. Thus, bufalin might induce autophagy by affecting cellular dissipation of energy. To test this possibility, we monitored changes in cellular ATP level in response to bufalin. U87MG cells treated with bufalin showed a significant time-dependent decrease in the level of ATP (Fig. 3A), thereby suggesting that cellular energy depletion could be responsible for bufalin-induced autophagy. The AMP-activated protein kinase (AMPK) is activated directly by elevation in AMP and inhibited by high concentration of ATP [19]. It is well known that one of the major downstream targets regulated by AMPK is the mammalian target of rapamycin (mTOR) [20]. Thus, we investigated whether AMPK and mTOR were involved in bufalin-induced autophagy. Western blot analysis revealed that bufalin increased AMPK phosphorylation without affecting the total AMPK level (Fig. 3B) and subsequently inhibited the activation of its downstream targets, such as mTOR, p70S6K, and 4EBP1 (Fig. 3C). Besides, the upregulation of p-ACC (phospho-acetyl-CoA carboxylase) further confirmed the activation of AMPK (Fig 3B). We also employed a knockdown strategy to further assess the impact of AMPK on bufalin-induced autophagy. As shown in Figure 3E, bufalin-induced LC3-II formation was attenuated by si-AMPK transfection, confirming a direct impact of AMPK knockdown on bufalin-induced autophagic process. Moreover, in parallel with the role of AMPK in down-regulation of mTOR activity, cells transfected with AMPK siRNA displayed an increase of mTOR activation, and bufalin-induced inhibition of mTOR phosphorylation was relatively less pronounced (Fig. 3D). These data indicate that AMPK and its downstream target mTOR are important mediators of bufalin-induced autophagy.
Bufalin targets mTOR signaling via AMPK activation. A: ATP levels of cells treated with 40 nM bufalin for 0-72 h. Data are presented as mean±SD, n =3. *p<0.05, **p<0.01 vs control. B: Cells were incubated with bufalin at various concentrations (0-80nM) for 24 h. The indicated proteins and phosphoproteins were analyzed by western blotting. C: Cell lysates were prepared from U87MG cells subjected to various doses (0-80nM) of bufalin for 24 h and then analyzed by western blotting. D-E: U87MG cells transfected with si-AMPK or si-control were treated with bufalin (80 nM) for 24 h and then harvested for protein analysis. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n=3). **p<0.01 versus the bufalin treatment alone.
Inhibition of autophagy enhances bufalin-induced apoptosis
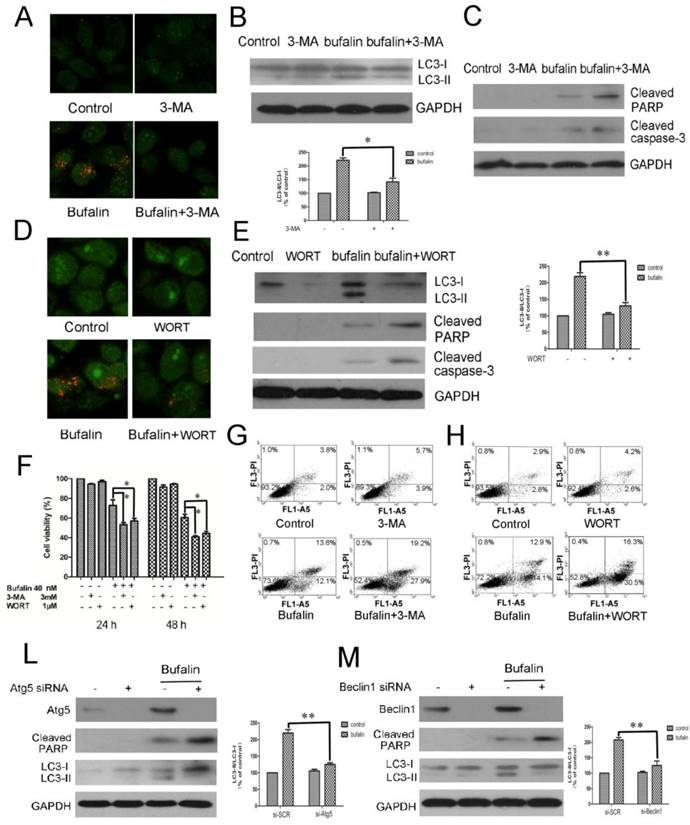
To investigate the role of autophagy in bufalin-induced cell death, autophagy inhibitors, 3-MA and WORT were introduced. Acridine orange staining demonstrated that both 3-MA and WORT suppressed bufalin-induced accumulation of acid vacuoles in glioma cells (Fig. 4A and D, Supplementary Material: Fig S4A and B). Western blot analysis confirmed that co-treatment with 3-MA or WORT inhibited the conversion of LC3-I to LC-II elicited by bufalin (Fig. 4B and E), suggesting that 3-MA and WORT effectively blocked the autophagic effect of bufalin. Next, MTT assay was employed to assess the effects of bufalin together with 3-MA or WORT over cell growth inhibition. In the combination group, the viability of U87MG and LN229 cells decreased faster than in the bufalin group, indicating that autophagy inhibitors enhanced the cytotoxic effect of bufalin in glioma cells (Fig. 4F, Supplementary Material: Fig S4C). To validate the observation that inhibition of autophagy affected cell sensitivity to bufalin, we performed Annexin V-FITC and PI double staining assay. Flow cytometry analysis showed that the percentage of AV+/PI+ and AV+/PI- cells increased in combined treatment group than bufalin treatment group in U87MG cells (Fig. 4G and H). Furthermore, the co-treatment with autophagy inhibitor augmented bufalin-induced apoptosis, which was verified by analysis of the executioners, PARP and caspase 3. In bufalin-treated group, they were all cleaved into their specific active forms, and the activity in the bufalin-treated cells with inhibited autophagy was significantly higher (Fig. 4C and E). Similar changes of PARP were also detected in LN229 cells (Supplementary Material: Fig. S4D and E). To further corroborate the data gained by the pharmacological treatment, we used an RNA interference approach to knock down Atg5 and Beclin1 genes, respectively. Under such conditions, bufalin-induced LC3-I to LC3-II conversion was attenuated with an increase of PARP cleavage (Fig. 4L and M, Supplementary Material: Fig S4G and H). Collectively, these results indicate that bufalin-induced autophagy is a protective mechanism, and blockage of autophagy aggravates the bufalin-induced apoptosis.
The inhibition of autophagy enhances bufalin-induced apoptosis. A-C: U87MG cells were pretreated or not with 3 mM 3-MA for 1 h and further treated with 40 nM bufalin for 24 h. A: Acridine orange staining was used to detect autophagic vacuoles. B: Western blot showing LC3 in U87MG cells. C: Activation of caspase-3 and its downstream effector PARP was assayed by western blotting. D-E: U87MG cells were pretreated or not with 1 µM Wortmannin (WORT) for 1 h and further treated with 40 nM bufalin for 24 h. D: Acridine orange staining in U87MG cells. E: Western blot showing indicated proteins in U87MG cells. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n =3). *p<0.05 and **p< 0.01 versus the bufalin treatment alone. F: The viability of U87MG cells was measured by MTT assay following 24 h and 48 h of culture with bufalin alone or with 3 mM 3-MA or 1 µM WORT. Data are presented as mean±SD of three separate experiments. *p<0.05 versus bufalin treatment alone. G-H: Apoptotic cell death was detected by Annexin V-FITC/PI double staining. U87MG cells were pretreated or not with 3 mM 3-MA (G) or with 1 µM WORT (H) for 1 h and further treated with 40 nM bufalin for 24 h. L-M: U87MG cells were transfected with control siRNA, Atg5 siRNA (L) or Beclin1 siRNA (M) as described. The cells were exposed to 40 nM bufalin for 24 h and then lysed for immunoblotting with antibodies against Beclin-1, Atg5 and LC3. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n=3). **p<0.01 versus the bufalin treatment alone.
ER sress-mediated apoptosis is involved in bufalin induced cell death
Evidence has accumulated from many studies that ER stress-associated apoptosis could be an intriguing candidate to be responsible for cell death induced by anti-tumor agents [21]. To identify whether ER stress was involved in bufalin induced cell death, we followed the behavior of some ER stress markers. There are three ER stress sensors including PERK, IRE1 and ATF6 that can activate the UPR response under ER dysfunction [22]. We therefore performed western blots to check these proteins and found that p-PERK, p-IRE1α, p-eIF2α and ATF6 fragments increased in a concentration-dependent manner (Fig. 5A). Upon ER stress, GRP78 is released from the UPR sensors, resulting in the activation and transduction of UPR signals [23]. As shown in Figure 5B, protein expression of GRP78 and GRP94 increased in a dose-dependent manner after 24 h of bufalin treatment. Next, we raised the question that whether ER stress was involved in apoptosis induced by bufalin. During ER stress, ATF 6 activates the transcription of CHOP and ultimately leads to caspase cascade activation to complete the execution of ER stress-induced apoptosis [24]. Recent studies have shown that caspase-4, an ER-resident caspase, is activated in response to ER stress. Protein expression analysis showed that bufalin upregulated CHOP and accelerated cleavage of caspase-4 in U87MG cells (Fig. 5C). Accordingly, the increases in GRP78, p-PERK, p-eIF2α and CHOP expression in LN229 cells also suggest that part of the UPR is activated with bufalin (Supplementary Material: Fig. S5). Altogether, these results indicate that ER stress-associated apoptosis is also involved in bufalin-induced glioma cell death.
ER stress-mediated apoptosis is involved in bufalin-induced cell death. A-C: U87MG cells were exposed to various concentrations of bufalin for 24 h. A: The effect of bufalin on ER stress sensors PERK, p-PERK, eIF2α, p-eIF2α, ATF6 f, IRE1 and p-IRE1. B: The expression levels of GRP78 and GRP94 in U87MG cells affected by bufalin. C: Western blot analysis for the expression of ER stress-associated apoptotic proteins CHOP and cleaved caspase-4. D-F: U87MG cells were transfected with CHOP specific siRNA and scrambled siRNA for 48 h, respectively. Then the cells were exposed to bufalin for another 24 h. D: CHOP and cleaved caspase-3 were examined by western blotting. E: The caspase-3 activity was determined by Caspase-3 colorimetric assay kit. The data are presented as the mean±SD, n=3. **p < 0.01 vs. sicontrol-transfected group. F: Cell viability was determined by MTT assay. Data are presented as mean ± SD, n = 3. **p < 0.01 versus control group. G: U87MG cells were transfected with CHOP specific siRNA and scrambled siRNA for 48 h, respectively. Then the cells were exposed to bufalin ( 80 nM ) for 48 h, stained with Annexin V and PI, and analyzed by flow cytometry. Results are representative of three independent experiments. H: Treatment was as described for G. The apoptosis rates are represented as the mean ± SD of three independent experiments. **p<0.01versus si-control group.
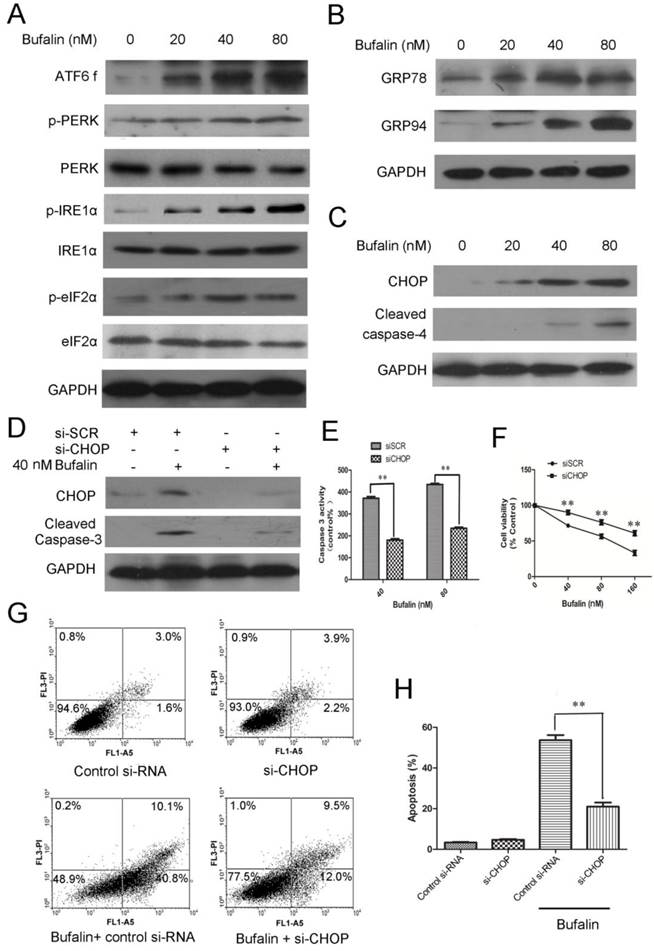
Knockdown of CHOP expression by CHOP-specific siRNA was used to further confirm the role of CHOP in regulation of cell proliferation and apoptotic death. As shown in Figure 5D, CHOP-specific siRNA effectively downregulated CHOP protein levels. Knockdown of CHOP also decreased activity of intracellular caspase 3 (Fig. 5D-E) and reduced cytotoxicity of bufalin to U87MG cells (Fig. 5F). The average apoptotic percentage of U87MG cells exposed to 80 nM bufalin for 48 h, decreased from 53.2% to 21.6% after CHOP knockdown (Fig. 5G-H). These results indicate that CHOP is a pivotal mediator in ER stress-mediated apoptosis induced by bufalin.
Bufalin induces autophagy via activation of ER stress in U87MG cells
The close relationship between ER stress and autophagy is demonstrated by recent reports showing that ER stress is a potent inducer of autophagy [25]. To investigate whether ER stress following bufalin treatment of cells generated an autophagic response, western blotting was performed to probe the expression of GRP78 and LC3-II. As shown in Fig. 6A, tauroursodeoxycholate (TUDC), an ER stress inhibitor, reduced the level of GRP78 and attenuated the expression of LC3-II level induced by bufalin. Consistent with the immunoblotting results, an obvious reduction in autophagy induction was further confirmed by MDC staining. The MDC-positive vacuoles were relatively sparse and weak in fluorescence intensity in cell cultures co-treated with bufalin+TUDC when compared with cultures treated with bufalin (24 h) (Fig. 6B), indicating that incorporation of MDC into vacuoles was inhibited by TUDC. To further investigate the relationship between autophagy and ER stress, the expression levels of p-eIF2α, GRP78 and CHOP were determined by western blotting. As shown in Figure 6C, U87MG cells treated with bufalin in combination with 3-MA upregulated the expression of p-eIF2α, GRP78 and CHOP compared with the group treated with bufalin alone. Our findings with 3-MA indicate that autophagy plays a protective role against bufalin-induced cytotoxicity, probably through relieving ER stress. These results support the conclusion that cells exposed to bufalin experience ER stress that leads to induction of autophagy and suggest that mobilization of the autophagic machinery might generate a feedback signal needed for full induction of ER stress.
Bufalin induces autophagy through stimulation of ER stress in U87MG cells. A-C: U87MG cells were pretreated or not with 500 μM TUDC (A-B) or with 3 mM 3-MA (C) for 1 h and further treated with 40 nM bufalin for 24 h. A: Western blot analysis for the expression of LC3 and GRP78. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n =3). *p<0.05 versus the bufalin treatment alone. B: The number of MDC-labeled vacuoles was observed using a laser scanning confocal microscope (63X). C: Western blot analysis for the expression of p-eIF2α, GRP78 and CHOP. D-E: U87 cells transfected with sieIF2α/ siCHOP or sicontrol were treated with bufalin (40 nM) for 24 h and then harvested for protein analysis. Relative levels of LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean±SD, n =3). *p<0.05 versus the bufalin treatment alone.
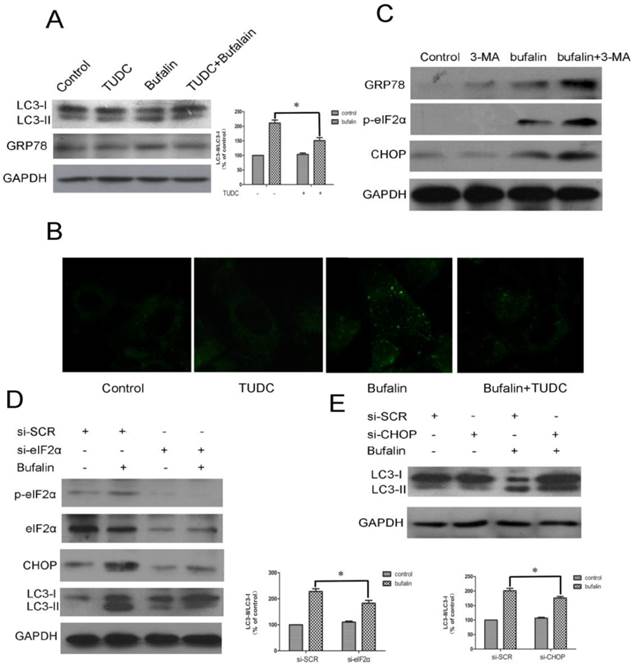
Evidence has accumulated that ER stress could be linked to a persistent activation of the PERK-eIF2α-CHOP axis, which leads to autophagy. To investigate the mechanism underlying ER stress induced autophagy, specific siRNA directed against eIF2α was utilized in U87MG cells. In the sieIF2α-transfected cells, the baseline levels of phospho-eIF2α and eIF2α were largely reduced, and importantly, bufalin-induced upregulation of CHOP was significantly attenuated (Fig. 6D). Similar results were observed in the effect of bufalin on the conversion of LC3-I to LC3-II in sieIF2α-transfected U87MG cells, which was that knockdown of eIF2α effectively attenuated bufalin-induced LC3-II conversion. We next determined whether knockdown of CHOP affected bufalin-induced LC3-II expression. The efficacy of CHOP knockdown was confirmed by western blotting (Fig. 6C). As illustrated in Fig. 6E, bufalin led to a marked increase in the level of LC3-II in sicontrol transfected cells, whereas it failed to promote the conversion of LC3-I to LC3-II in siCHOP-transfected cells. These results indicate that the PERK/eIF2α/CHOP signaling pathway is required for bufalin-induced autophagy in U87MG cells.
Discussion
Malignant gliomas is known as one of the most lethal tumors in humans and currently, the conventional cancer therapies fail to exert positive effects on glioma cells [26]. Patients suffering from glioblastomas usually have extremely poor prognosis which may be partly explained by the low efficiency of targeting their inherent apoptosis-resistant phenotype. The potential exploitation of a novel therapy is highly warranted for the treatment of glioma.
The current studies indicate that during tumorigenesis, ER stress develops in response to harsh environmental cues including nutrient starvation, oxidative stress and other metabolic dysregulations of cells [27]. A group of ER signal transduction pathways, collectively termed the unfolded protein response (UPR), is evolved to facilitate adaptation to the changing environment and re-establish ER function and homeostasis [28]. However, masses of normal cells do not go through an active “stress” response and the UPR remains in a state of quiescence. This difference between tumor and normal cells allows for the drugs that target ER stress to achieve specificity in malignant cancer therapy. A variety of anticancer therapies are associated with the induction of ER stress in cancer cells, suggesting that strategies devised to stimulate its prodeath function or block its prosurvival function, could be envisaged to improve their chemotherapeutic efficiency [29].
Researches on the effect of bufalin on UPR and ER stress has not been reached, in part because the broad range of intracellular effects varies in different cell types. Our results showed that bufalin induced UPR in glioma cells: increased GRP78 and GRP94, cleavage of ATF6, phosphorylation of PERK, IRE1α and eIF2α. Meanwhile, it was accompanied by the enhanced expression of CHOP. The CHOP promoter receives positive input from other components of the UPR, and the upregulation of CHOP protein can provoke caspase activation, thus leading to cell demise [30-31]. Deregulated CHOP activity compromises cell viability, and cells deficient in CHOP are significantly protected from the lethal consequences of ER stress [32]. In this study, knockdown of CHOP using CHOP-specific siRNA suppressed the activation of apoptotic effector caspase 3 and reduced apoptosis rate of U87MG cells exposed to bufalin. Murine caspase 12 and human caspase 4 have been described to be activated by ER stress inducing agents, and to be responsible for ER-induced apoptosis [33]. Besides, cells treated with siRNA targeting caspase 4 are resistant to ER-stress induced apoptosis [34]. In agreement with these reports, western blot analysis revealed that the expression of cleaved caspase-4 increased in bufalin-treated U87MG cells. Overall, our data demonstrate the capacity of bufalin to activate the key proteins of ER stress as well as ER-associated apoptotic proteins, CHOP and caspase-4. These data preliminarily indicate that bufalin induces ER-mediated apoptosis.
Proteasomal degradation and autophagy are the two main mechanisms that are in charge of protein clearance in the cell. Unlike proteasomal degradation that digests soluble ubiquitin-conjugated proteins, autophagy can degrade both soluble and aggregated proteins [35]. Thus, in order to clear the ER from the protein aggregates that cannot be degraded by the proteasome, the UPR may upregulate the autophagy machinery. Besides, a variety of stress signals such as nutrient starvation or treatment with anticancer agents that induce ER stress can also stimulate the autophagy process [36]. Here we noticed the occurrence of autophagy in bufalin-exposed U87MG cells, as proven by the increased number of MDC labeled vesicles, acridine orange stained acidic autophagolysosomes and LC3-II accumulation. Autophagy serves as an alternative energy source in times of nutrient scarcity [37]. As a key energy sensor, decreased ATP production initially activates AMPK, which stimulates autophagy under energy-deprived conditions [38]. Stimulation of AMPK inactivates mTOR through AMPK-mediated phosphorylation of both TSC2 and Raptor [39]. Here we found that bufalin treatment increased phosphorylation of AMPK and inhibited phosphorylation of mTOR and its downstream targets, p70S6K and 4EBP1, which was in accordance with a decreased ATP level. Moreover, knockdown of AMPKα attenuated the bufalin induced autophagy. Considering these results, we suggest that AMPK/mTOR signaling is tightly involved in bufalin-induced autophagy in U87MG cells.
The link between ER stress and autophagy has been investigated only in the last decade, and many questions concerning the signaling pathways associating ER stress with autophagy remain largely unanswered. In this study, TUDC combined with bufalin downregulated the expression of GRP78 and LC3-II proteins in U87MG cells, suggesting that autophagy induced by bufalin might be a consequence of ER stress. Knockdown of eIF2α or CHOP attenuated bufalin-induced LC3-II conversion, confirming the involvement of PERK/eIF2α/CHOP pathway in bufalin-induced autophagy. In line with our results, Tallóczy et al [25] found that cells that carry a nonphosphorylatable mutant of eIF2α fail to induce autophagy in response to starvation. Moreover, we used autophagy inhibitors to better clarify the role of autophagy in ER stress in glioma cells treated with bufalin. The data demonstrated that ER stress was enhanced in bufalin-exposed cells when autophagy was blocked by inhibitors. It is worth noting that several researches have reported that 3-MA and WORT, also known as the phosphatidylinositol 3-kinase (PI3K) inhibitors, inhibit both autophagy and the UPR, indicating that ER stress-induced UPR operates through a pathway in which PI3K plays a role [40]. Our results unveiled that blocking PI3K activity failed to prevent the induction of GRP78, implying that PI3K activity was not neccessary to transduce ER stress signaling in bufalin-treated glioma cells. All together, we conclude that ER stress especially the PERK/eIF2α/CHOP pathway is responsible for the autophagic response and the activation of autophagy alleviates ER stress, which might act as a self-defense mechanism.
However, there is no doubt that the underlying mechanisms of bufalin in glioma therapy still need further investigation. For example, Bcl-2 family members are widely distributed in ER, particularly Bcl-2, which has been shown to be responsible for maintaining Ca2+ homeostasis and regulating protein folding in ER [41]. In addition, the Bcl-2 family proteins are closely associated with the IRE1/JNK pathway, which also participates in ER stress-mediated autophagy [42]. Thus, further exploration to identify a potential molecular player of the cross-talk among apoptosis, autophagy and ER stress may be fascinating.
In summary, we focuse on the impact of simultaneous initiation of apoptosis, autophagy and ER stress following bufalin treatment, also providing understanding of their interplay in glioma cells. This study unveils that both mitochondrial- and ER mediated apoptosis are involved in glioma cell death induced by bufalin. In addition to apoptosis, bufalin also induces autophagy by activating AMPK/mTOR pathway and PERK/eIF2α/CHOP pathway, which alleviate ER stress and might play a cytoprotective role. Inhibition of autophagy potentiates the proapoptotic effect of bufalin, suggesting appropriate regulation of autophagy is necessary for sensitizing tumor cells to anticancer therapy. Further study could be ongoing to define optimal strategies to modulate ER stress for cancer treatment since ER stress can be involved in both apoptosis and autophagy.
Supplementary Material
Fig.S1 - Fig. S5.
Acknowledgements
This work was supported by Key Laboratory of Microbial Biochemistry and Metabolism Engineering of Zhejiang Province and the State Agricultural S&T Result Transforming Fund, the Ministry of Science and Technology of China (NO.2012C2202001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344-8
2. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255-63
3. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF. et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798-803
4. Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12:1528-34
5. Yien Che Tsai, Allan M. Weissman. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes & Cancer. 2010;7:764-778
6. Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci (Schol Ed). 2012;4:412-31
7. Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling throughphosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3beta in mouse insulinoma cells. Diabetes. 2005;54:968-75
8. Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 2011;140:987-97
9. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013-30
10. Matsumoto H, Miyazaki S, Matsuyama S, Takeda M, Kawano M, Nakagawa H. et al. Selection of autophagy or apoptosis in cells exposed to ER-stress depends on ATF4 expression pattern with or without CHOP expression. Biol Open. 2013;2:1084-90
11. Hiroshi S, Hideaki O, Akihiko Y, Hiroki Y. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci. 2006;119:3958-3966
12. Miao Q, Bi LL, Li X, Miao S, Zhang J, Zhang S. et al. Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis andAutophagy. Int J Mol Sci. 2013;14:1370-82
13. Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med. 2011;51:1365-75
14. Ando T, Imamura H, Suzuki R, Aizaki H, Watanabe T, Wakita T, Suzuki T. Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 2012;8:e1002561
15. Wang YC, Lee CM, Lee LC, Tung LC, Hsieh-Li HM, Lee-Chen GJ. et al. Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12). J Biol Chem. 2011;286:21742-54
16. Li X, Li X, Wang J, Ye Z, Li JC. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci. 2012;8:901-12
17. Mao Y, Da L, Tang H, Yang J, Lei Y, Tiollais P. et al. Hepatitis B virus X protein reduces starvation-induced cell death through activation of autophagy and inhibition of mitochondrial apoptotic pathway. Biochem Biophys Res Commun. 2011;415:68-74
18. Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol. 2010;75:795-800
19. Hamilton SR, O'Donnell JB Jr, Hammet A, Stapleton D, Habinowski SA, Means AR. et al. AMP-activated protein kinase kinase: detection with recombinant AMPK alpha1 subunit. Biochem Biophys Res Commun. 2002;293:892-8
20. Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381-400
21. Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded Protein response in tumorigenesis andanticancer therapies. Oncogene. 2013;32:805-18
22. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900-17
23. Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307-16
24. Muller C, Salvayre R, Nègre-Salvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011;18:817-28
25. Tallóczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ. et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190-5
26. Núria de la Iglesia, Sidharth V. Puram, Azad Bonni. STAT3 Regulation of Glioblastoma Pathogenesis. Curr Mol Med. 2009;9:580-590
27. Lin ML, Chen SS, Lu YC, Liang RY, Ho YT, Yang CY, Chung JG. Rhein induces apoptosis through induction of endoplasmic reticulum stress and Ca2+-dependent mitochondrial death pathway in human nasopharyngeal carcinoma cells. Anticancer Res. 2007;27:3313-22
28. Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician's perspective. J Hematol Oncol. 2011;4:8
29. Ghavami S, Yeganeh B, Stelmack GL, Kashani HH, Sharma P, Cunnington R. et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrialfibroblasts. Cell Death Dis. 2012;3:e330
30. Sok J, Wang X, Batchvarova N, Kuroda M, Harding H, Ron D. CHOP-Dependent Stress-Inducible Expression of a Novel Form of Carbonic Anhydrase VI. Mol Cell Biol. 1999;19:495-504
31. Ma Y, Shimizu Y, Mann M, Jin Y, Hendershot L. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. 2010;15:281-293
32. Ghavami S, Yeganeh B, Stelmack G, Kashani H, Sharma P, Cunnington R. et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012;3:e330
33. Yamamuro A, Kishino T, Ohshima Y, Yoshioka Y, Kimura T, Kasai A. et al. Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J Pharmacol Sci. 2011;115:239-243
34. Thepparit C, Khakpoor A, Khongwichit S, Wikan N, Fongsaran C, Chingsuwanrote P. et al. Dengue 2 infection of HepG2 liver cells results in endoplasmic reticulum stress and induction of multiple pathways of cell death. BMC Res Notes. 2013;6:372
35. Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol. 2010;2010:930509
36. Oh SH, Lim SC. Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J Pharmacol Exp Ther. 2009;329:112-22
37. Singh R, Kaushik S, Wang Y, Xiang YQ, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-1135
38. Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E. et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40-50
39. Liu JL, Mao Z, Gallick GE, Yung WK. AMPK/TSC2/mTOR-signaling intermediates are not necessary for LKB1-mediated nuclear retention of PTEN tumor suppressor. Neuro Oncol. 2011;13:184-94
40. Kang JH, Chang YC, Maurizi MR. 4-O-carboxymethyl ascochlorin causes ER stress and induced autophagy in human hepatocellular carcinoma cells. J Biol Chem. 2012;287:15661-71
41. Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011;3:a004424
42. Meister S, Frey B, Lang VR, Gaipl US, Schett G, Schlötzer-Schrehardt U. et al. Calcium channel blocker verapamil enhances endoplasmic reticulum stress and cell death induced by proteasome inhibition in myeloma cells. Neoplasia. 2010Jul;12(7):550-61
Author contact
![]() Corresponding author: Fax: +86 571 88206549 Tel.: +86 571 88206475. Email: gongxingguocom. Address: Room 345, Institute of Biochemistry, Zhejiang University, Hangzhou, 310058, PR China.
Corresponding author: Fax: +86 571 88206549 Tel.: +86 571 88206475. Email: gongxingguocom. Address: Room 345, Institute of Biochemistry, Zhejiang University, Hangzhou, 310058, PR China.