International Journal of Biological Sciences 10
Impact Factor
ISSN: 1449-2288
Impact Factor
ISSN: 1449-2288
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(3):285-295. doi:10.7150/ijbs.7730 This issue Cite
Research Paper
Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53
Junzhe Jin, Guofu Lin, Hong Huang, Dong Xu, Hao Yu, Xu Ma, Lisi Zhu, Dongyan Ma, Honglei Jiang ![]()
The Fourth Affiliated Hospital of China Medical University, Shenyang, China.
Received 2013-9-23; Accepted 2014-1-25; Published 2014-2-21
Citation:
Jin J, Lin G, Huang H, Xu D, Yu H, Ma X, Zhu L, Ma D, Jiang H. Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53. Int J Biol Sci 2014; 10(3):285-295. doi:10.7150/ijbs.7730. https://www.ijbs.com/v10p0285.htm
Other stylesAbstract
Capsaicin is the major pungent ingredient in red peppers which is world widely consumed. Except its potent pain relieving efficacy as reported, capsaicin also exerted its antitumor activity in several tumor models. Here, we reported that capsaicin had a profound anti-proliferative effect on human colon cancer cells via inducing cell cycle G0/G1 phase arrest and apoptosis, which was associated with an increase of p21, Bax and cleaved PARP. The underlying mechanism of capsaicin's antitumor potency was mainly attributed to the stabilization and activation of p53. Capsaicin substantially prolonged the half-life of p53 and significantly elevated the transcriptional activity of p53. Through suppressing the interaction between p53 and MDM2, MDM2-mediated p53 ubiquitination was remarkably decreased after capsaicin treatment, which resulted in the stabilization and accumulation of p53. The results of p53-shRNA experiment further demonstrated that p53 knockdown severely impaired the sensitivity of tested cells to capsaicin, G0/G1 phase arrest and the apoptosis induced by capsaicin in p53-knockdown cells was also dramatically decreased, implicating the important role of p53 played in capsaicin's antitumor activity. In summary, our data suggested that capsaicin, or a related analogue, may have a role in the management of human colon cancer.
Keywords: colon cancer, capsaicin, p53, MDM2, ubiquitination.
Introduction
Capsaicin is the principal pungent component in hot red peppers and widely consumed in many South Asian and Latin American countries [1]. Through mediating transient potential vanilloid receptor (TRPV-1), which was predominantly present in sensory neurons [2], Capsaicin had potent efficacy on pain relieving and originally been used to treat a variety of neuropathic pain conditions including rheumatoid arthritis, diabetic neuropathy, cluster headaches, herpes zoster [3-5]. Except aforementioned therapeutic value, several studies also have demonstrated that capsaicin could be functioned as a chemopreventive and chemotherapeutic agent in different human cancer models. As reported, capsaicin decreased the growth of human leukemic cells [6], gastric [7], nasopharyngeal [8], prostate [9], and hepatic carcinoma cells [10] in vitro because of its ability to mediate cell cycle arrest and induce cell apoptosis. However, the molecular mechanism underlying capsaicin-induced growth inhibition and apoptosis was not exhaustively elucidated. So far, several mechanisms have been suggested to involve in capsaicin-induced apoptosis, including inhibition of NF-κB nuclear translocation [9], activation of AMPK signaling pathway and the c-Jun NH2-terminal kinases [11], dysfunction of ubiquitin-proteasome systems [12] as well as the up-regulation of several pro-apoptotic proteins and activation of intrinsic pathway to drive caspase activation [13].
p53 plays an important role in protecting the integrity of genome and is described as the “guardian of genome”. Mutation of TP53 gene is found in approximately 50% tumors, which exemplifies the importance of p53 in maintaining genome stability [14]. In response to various cellular stress signals, p53 is activated and functions as a transcription factor to transcribe a program of genes to accomplish a number of different functions, such as activating DNA repair, inducing cell cycle arrest, and initiating cell apoptosis. Owing to the multiplicity of its function, activation of p53 is tightly controlled. p53 is subjected to a diverse and complex array of covalent post-translational modifications. The most commonly reported modifications include phophorylation of serine and/or threonines and acetylation, ubiquitylation, and sumoylation of lysine [15]. Encountered with stressed signals, e.g. DNA damage caused by ionizing radiation or UV irradiation, Ser15 of p53 is phosphorylated rapidly and appears to represent a “priming event” for the subsequent series of modifications, for instance, acetylation [16]. Phosphorylation and acetylation of p53 often drives p53 transcriptional activation via inducing p53 stabilization, accumulation and activation in the nucleus [17]. Under unstressed conditions, through the interaction with MDM2, a RING-finger ubiquitin E3 ligase, p53 is normally maintained at low level via continuous ubiquitination and subsequent degradation by proteasome system [18]. Several studies indicated MDM2 not only mediated p53 degradation but also regulated its localization. Monoubiquitylation might act as a signal of nuclear export for p53, and the different levels of MDM2 could induce both mono- or poly-ubiquitination in a dose-dependent manner and determine the fate of p53 [19]. In many tumor types, overexpression of MDM2 was observed, which led to the aberrant deactivation of p53 and poor prognosis of patients [20-23]. Other modifications of p53 including glycosylation and ribosylation also were reported, but the significance of these modifications in tumor development was unclear and little information was available [24].
In present study, we investigated the antitumor efficacy of capsaicin in human colon cancer cells and the role of p53 played in capsaicin's antitumor activity, as well as the mechanism by which capsaicin induced p53 stabilization and activation. The results showed that capsaicin had profound inhibitory effect on the growth of human colon cancer cells in vitro. After capsaicin treatment, p53 was significantly stabilized and activated via dissociating from the interaction with MDM2 and decreasing MDM2-mediated ubiquitination. p53 shRNA experiments further demonstrated that the activity of capsaicin in colon cancer cell was highly correlated with p53 accumulation and activation.
Material and Methods
Cell Line and reagents HCT116 and LoVo cells were obtained from American Type Culture Collection (ATCC) and cultured in a 37°C incubator with 5% CO2 according to ATCC protocols. Capsaicin, Cycloheximide (CHX) and MG132 were purchased from Sigma (St. Louis, MO, USA). Anti-p53, anti-MDM2, anti-β-actin, anti-rabbit IgG-HRP, anti-mouse IgG-HRP, and normal mouse/rabbit IgG were products of Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p21, anti-Bax, anti-cleaved-PARP and anti-caspase3 antibodies were products of Cell Signaling Technology (Beverly, MA, USA). Lipofectamine™ 2000 was product of Invitrogen. pGL3-p53 firefly luciferase reporter plasmid and pRL-SV40 (renilla luciferase) plasmid were purchased from Promega (Fitchburg, WI, USA). Lentivirus plasmids (pLKO.1-shp53 #1, TRCN0000003753; pLKO.1-shp53 #2, TRCN0000003756) were obtained from Thermo Scientific (Huntsville, AL, USA).
Cell proliferation assay Appropriate tumor cells (2×103/well) were seeded in 96-well plate, 24 hrs later, cells were treated with capsaicin of indicated concentrations and triplicates for per concentration. After incubation for different time points (12, 24, 48, or 72 hrs), 100 μl/well CellTiter-Glo reagent (Promega, Fitchburg, WI, USA) was added and the mixture was incubated at room temperature for 15 mins, then the luminescence was measured with infinite M1000 (TECAN, Männedorf, Schweiz). For anchorage-independent growth assay, colon cancer cells (8×103/ml) were suspended in 1 ml of 0.3% agar (BactoTM, BD) with basal medium Eagle's medium, 10% FBS, 1% antibiotics and different concentrations of capsaicin overlaid into six-well plate containing a 0.6% agar base. The plate was cultured in a 37°C, 5% CO2 incubator for 2 weeks and the number of colonies was counted under a microscope using the Imaga-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA ).
Flow cytometry For cell cycle analysis, tumor cells (2 × 105/well) were seeded into six-well plate and cultured in completed culture medium for 24 hrs, then exposed to different concentrations of capsaicin for 24 hrs. Cells were harvested and washed with PBS twice and fixed with cold 70% ethanol overnight at 4°C. Cells were then stained with 50 μg/ml Propidium Iodide (Biolegend, San Diego, CA, USA) and 100 μg ribonuclease A (QIAGEN, Venlo, Netherland) in 400 μl PBS at 25°C for 30 mins. Stained cells were detected and quantified with FACSort Flow Cytometer (BD, San Jose, CA, USA). For apoptosis analysis, cells (2 × 105/well) were seeded into six-well plate and incubated with different concentrations of capsaicin for 24 h. Cells were trypsinized and washed twice with cold PBS and then re-suspended with Binding Buffer. Annexin V-FITC (Biolegend, San Diego, CA, USA) and Propidium Iodide were added and incubated at room temperature for 15 mins avoiding light. The stained cells were subjected to FACS analysis. All FACS results were analyzed with FlowJo software (Version 7.6).
Western blotting Cells were harvested by trypsinization and pelleted by centrifugation at 500g for 5 mins. The pellets were lysed in NP40 lysis buffer (50 mmol/L Tris-HCl, pH 8.0; 150 mmol/L NaCl; 0.5% NP40) supplemented with protease cocktail (Roche, Germany). Protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA, USA). Proteins were separated by SDS-PAGE and electrically transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). After blocking in 5% non-fat dry milk in TBS, the membranes were probed with specific primary antibodies overnight at 4°C, washed three times with TBS-Tween 20, and then incubated with HRP-conjugated secondary antibodies at room temperature for 1h. Then the membranes were washed with TBS-Tween 20 and the protein bands were visualized using ECL chemiluminescence reagents (Pierce Chemical Co., Rockford, lllinois, USA). In ubiquitination assay, Endogenous p53 was immunoprecipitated from 1 mg whole cell lysate by using p53 antibody and immunoblotted with anti-p53 antibody to capture poly-ubiquitinated p53.
Immunoprecipitation Tumor cells (2×106) were seeded in 10 cm dish and treated with various concentrations of capsaicin for 24 hours, then the cells were harvested and washed twice with ice-cold PBS and lysed in NP40 lysis buffer with protease cocktail (Roche, Germany). All immunoprecipitation procedures were carried out at 4°C. First, 1 mg cell extractions were pre-cleared with 30 μl (50% slurry) agarose A/G beads for 2 hrs rocking at 4 °C. The beads were removed, 30 μl (50% slurry) fresh agarose A/G beads and appropriate antibodies (2 μg) were added to the precleared lysate overnight at 4 °C. The beads were washed, mixed with 6×SDS sample buffer, boiled, and then subjected to Western blotting.
P53 half life measurement For p53 protein stability experiments, HCT116 cells were treated with or without 100 μM capsaicin for 24 hrs, and then 30 μg/ml CHX was added to the culture medium to inhibit protein synthesis. At different time points as indicated (0, 15, 30, 60, or 90 mins), tumor cells were processed as described in Western blotting, and the amount of p53 was quantified by densitometric measurement.
luciferase reporter assay HCT116 cells (5×104/well) were seeded in 24-well plate, and then co-transfected with 800 ng firefly luciferase reporter plasmid and 80 ng pRL-SV40 plasmid by Lipofectamine™ 2000 following manufacturer's instructions. 24 hrs later, cells were treated with different concentrations of capsaicin and incubated for 24 hrs. Cell lysates were analyzed for firefly and renilla luciferase activity according to the protocol of the Dual Luciferase Reporter Assay Kit (Promega, Fitchburg, WI, USA). The firefly luciferase activity was normalized to the value of pRL-SV40 activity for p53 transcription efficiency.
Lentiviral infection HCT116 cells (2×106) were seeded in 10 cm dish and pLKO.1-shp53 was co-transfected into 293T cells together with PSPAX2 and PMD2-G. 48 hrs after transfection, viral supernatant fractions were collected and then infected into HCT116 cells along with 10 μg/ml polybrene. 24 hrs after infection, the medium was replaced with fresh medium containing the appropriate concentration of puromycin. The appropriate experiments were performed with these cells until the control cells (without infection) completely died (usually 2-3 days) in the puromycin medium.
Statistical analysis All the statistical analysis was performed by SPSS software (version 13.0). The experiments were performed in triplicate. All the quantitative data are expressed as mean values ± standard deviation, the significant differences between two groups was assessed by a two-tailed Student's t test. A probability value of P < 0.05 was considered to represent a statistically significant difference.
Results
Capsaicin inhibited colon cancer cell proliferation and anchorage-independent growth
First, we investigated the inhibitory effect of capsaicin against cell proliferation in HCT116 and LoVo. At low concentration (0-40 μM), capsaicin had shown little effect on the growth inhibition, but at high concentration (80-160 μM), long-term (48-72h) treatment with capsaicin substantially inhibited cell proliferation (Figure 1A and B). Anchorage-independent growth is one of the hallmarks of cell transformation and is considered the most accurate and stringent in vitro assay for detecting malignant transformation of cells. Therefore, next we have studied the effects of capsaicin on the anchorage-independent growth. As the result shown in Figure 1C and D, capsaicin could potently inhibit the anchorage-independent growth at 40 μM and the number of colonies formed in soft agar was remarkably decreased, at high concentrations, there was nearly no colony was observed. All these results showed that capsaicin had a profound antitumor efficacy in human colon cancer cells in vitro.
Figure 1
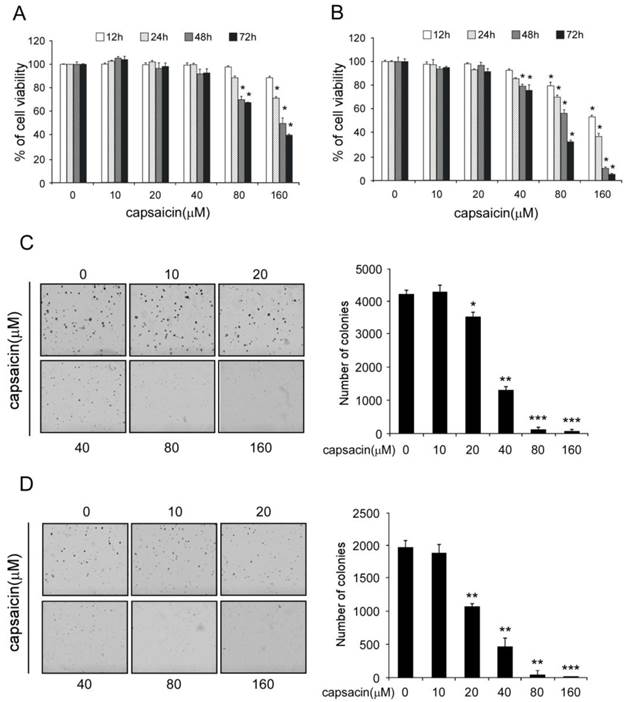
Capsaicin inhibited the proliferation of colon cancer cells and anchorage independent growth. A and B, capsaicin inhibited the proliferation of colon cancers. Human colon cancer cells HCT116 (A) and LoVo (B) were treated with indicated concentrations of capsaicin for 12, 24, 48, or 72 hrs. Cell proliferation was analyzed by the CTG assay. The asterisk (*, p<0.05) indicated a significant decrease of colon cell proliferation after treated with capsaicin. C and D, capsaicin suppressed the anchorage independent growth. Colon cancer cells HCT116 (C) and LoVo (D) were incubated with various concentrations of capsaicin as described and subjected to anchorage independent growth assay. Representative photographs (left panels) were shown, and the graph (right panel) showed the data of at least three independent experiments expressed as means ± SD, the asterisks (*, p<0.05, **, p<0.01, ***, p<0.001, Student's t test) indicated a significant decrease of colony formation after capsaicin treatment in contrast with the control.
Capsaicin induced cell cycle arrest and apoptosis in colon cancer cells
Next, we studied capsaicin's activity on cell cycle and apoptosis. As the result shown in Figure 2A and 2B (Supplementary Figure 1A and 1B), when cell was treated with capsaicin at low concentration (10-20 μM) for 24 hrs, no obvious cell cycle arrest was observed. With the increase of concentration, G0/G1 phase arrest was observed and the percentage of G0/G1 phase cell was elevated. At the high concentration (300 μM), substantial cells had undergone apoptosis with the dramatic increase of sub-G0/G1 peak. Further analysis of the cell cycle distribution of live cells had demonstrated that a large propotion of live cells were at G0/G1 phase(70-80%) after 300 μM capasaicin treatment, which demonstrated the significant G0/G1 arrest induced by capsaicin (Data shown in Supplementary Figure 1A and 1B). In addition, we also adopted Annexin V-FITC/PI double staining to confirm that capsaicin significantly induced cell apoptosis in a dose-dependent manner (Figure 2C and D, Supplementary Figure 1C and 1D), application of 300 μM capsaicin resulted in 20-30% cancer cells to undergo apoptosis. In order to validate the activity of capsaicin, we also detected the expression of p21, Bax and cleaved-caspase3 after capsaicin treatment (Figure 3B). Consistent with the result of flow cytometry analysis, the expression of p21, which is a key regulator of cell cycle progression at G1phase, was significantly increased, suggesting that capsaicin-induced G0/G1 arrest was closely correlted with p21 elevation. Bax is a pro-apoptotic protein and involved in induction of cell apoptosis, moreover, the cleavage of PARP is considered to be an important marker for detection of apoptosis. Western blotting result demonstrated that the expression of Bax and cleaved PARP were remarkbly increased in a dose-depedent manner after capsaicin treatment. All these results confirmed that capsaicin treatment potently induced cell cycle G0/G1 arrest and apoptosis in human colon cancer cells.
Capsaicin stabilized p53 and activated p53 signaling pathway
Given p21 and Bax are downstream target genes of p53, we detected the effect of capsaicin on p53 expression. As the result shown in Figure 3A and 3B, capsaicin treatment resulted in an increase of p53 expression in a time-dependent and dose-dependent manner. As a short-life protein, p53 was degraded frequently via ubiquitilation under normal condition. Therefore, improvement of its stability was important for p53 to perform its function. We adopted cycloheximide to block protein synthesis in HCT116 cells and detected the expression of p53 after capsaicin treatment. As shown in Figure 3C, without capsaicin, p53 was quickly degraded and the half-life was about 30 mins. However, in capsaicin treatment group, the half-life of p53 was dramatically prolonged and extended to 90 mins, validating the stability of p53 was obviously enhanced. In order to investigate the transcriptional activity of p53 after capsaicin treatment, pGL3-p53 firefly luciferase reporter plasmid was transfected into HCT116 cells. Owing to the expression of luciferase was under the control of p53 transcriptional activity, so we could test p53 activity via measuring luciferase activity. After 40 μM capsaicin treatment, the activity of luciferase was increased nearly 4-fold in comparison with the control group, which suggested that the transcriptional activity of p53 was prominently elevated (Figure 3D).
Figure 2
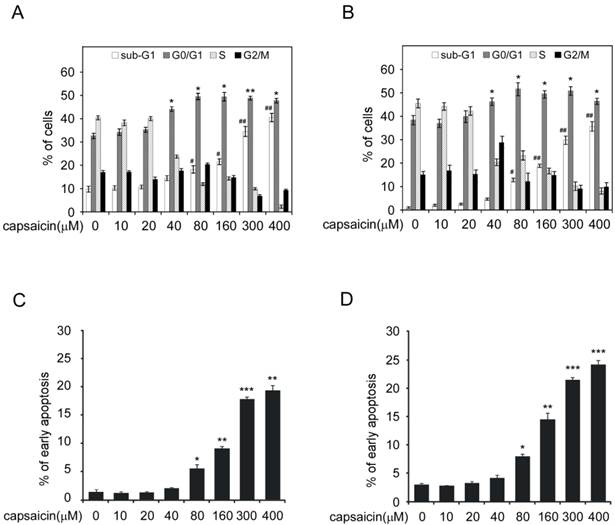
Capsaicin induced G0/G1 cell cycle arrest and apoptosis in colon cancer cells. A and B, capsaicin induced colon cancer cells G0/G1 cell cycle arrest. HCT116 (A) and LoVo (B) cells were treated with various concentrations of capsaicin for 24 hrs. Cellular phase distribution (sub-G1, G0/G1-phase, S-phase, G2/M-phase) was analyzed by staining with Propidium Iodide. C and D, capsaicin induced colon cancer cells apoptosis. HCT116 (C) and LoVo (D) cells were treated with various concentrations of capsaicin for 24 hrs and early-phase apoptosis was detected with Annexin V-FITC/PI double staining. The graph showed the result of three independent experiments expressed as means ± SD, and the symbol (*, p<0.05, **, p<0.01, ***, p<0.001, #, p<0.05, ##, p<0.01, Student's t test) indicated a significant difference.
Figure 3
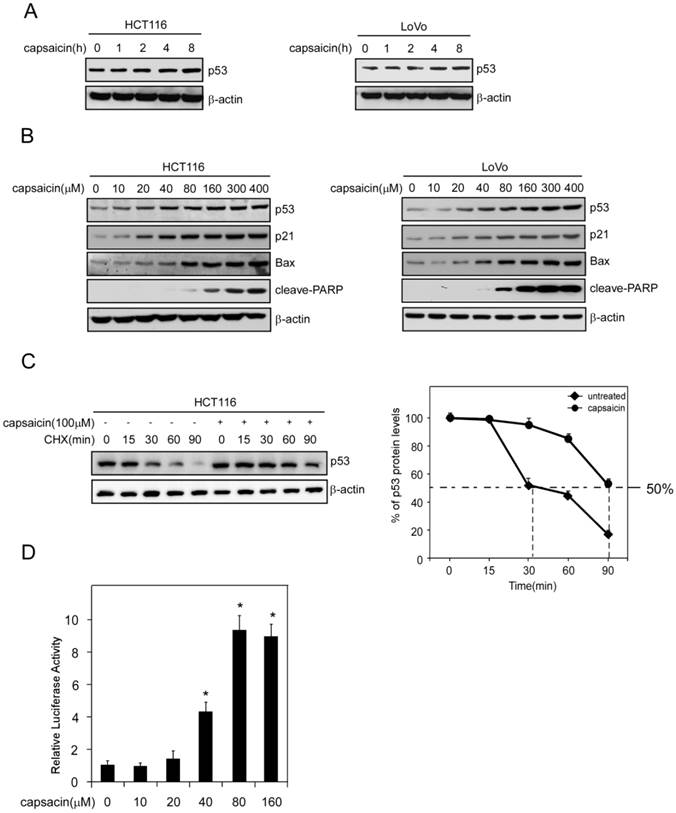
Capsaicin stabilized p53 and activated its transcriptional activity. A, capsaicin increased p53 expression in a time-dependent manner. HCT116 and LoVo cells were exposed to 100 μM capsaicin for indicated times and the cell lysates were subjected to Western blotting analysis. B, capsaicin increased p53 and its target genes p21 and Bax expression in a dose-dependent manner. HCT116 and LoVo cells were treated with different concentrations of capsaicin for 24 hrs and the protein expression were probed with corresponding antibodies. C, capsaicin prolonged p53 half-life time. HCT116 cells were treated with 100 μM capsaicin for 24 hrs, then CHX (20 μg/ml) was added and incubated for indicated time points, then p53 expression level was measured by Western blotting and was quantified by densitometry. D, capsaicin activated p53 transcriptional activity. HCT116 cells were transfected with luciferase reporter plasmids and the activities of luciferase were measured after capsaicin treatment. The relative luciferase activity was normalized to the value of Renilla activity.
Capsaicin suppressed MDM2-mediated p53 ubiquitilation and degradation
MDM2-mediated p53 ubiquitilation plays an important role in the regulation of p53 stability. As the data shown above, p53 stability was substantially enhanced after exposure to capsaicin, so we further investigated whether capsaicin regulated p53 stability in a MDM2-mediated manner. Utilizing p53 antibody to immunoprecipitate p53 and then detecting MDM2-p53 interaction with MDM2 antibody, or inversely, with MDM2 antibody to immunoprecipitate p53 and testing the interaction with p53 antibody, we found that capsaicin strongly suppressed the interaction between MDM2 and p53 in a dose-dependent manner, at the concentration of 40 μM, the binding of MDM2 to p53 was substantially decreased and there was nearly no MDM2 binding was detected at 160 μM (shown in Figure 4A and 4B). Along with the suppression of MDM2-p53 interaction, MDM2-mediated p53 ubiquitylation was also remarkably reduced after capsaicin treatment, which contributed to the stabilization of p53 and the extension of p53 half-life after capsaicin treatment.
Figure 4
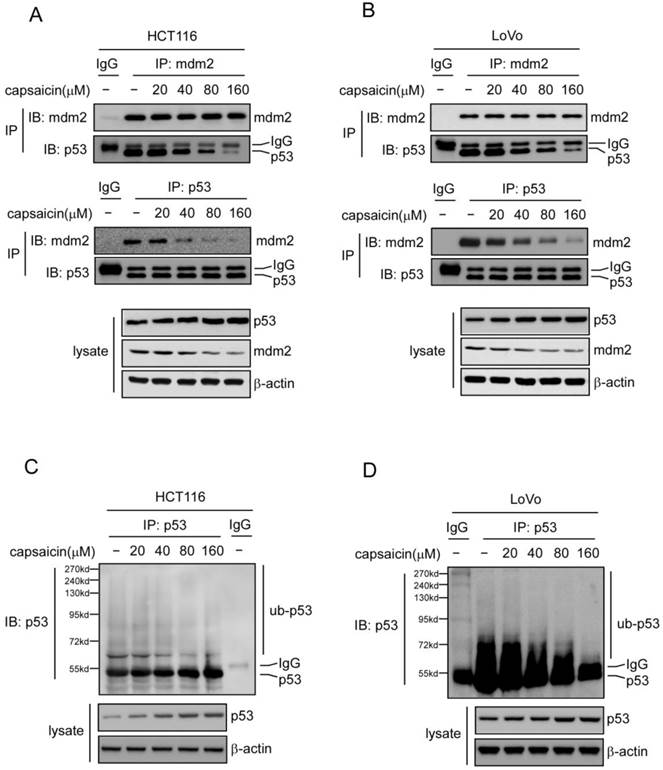
Capsaicin suppressed MDM2-mediated p53 degradation. A and B, capsaicin inhibited the interaction between p53 and MDM2. HCT116 (A) and LoVo (B) cells were treated with various concentrations of capsaicin for 24 hrs and the cell lysates were immunoprecipitated with p53 or MDM2 antibodies, then the binding affinity was analyzed with western blot analysis. C and D, capsaicin inhibited p53 ubiquitination. HCT116 (C) and LoVo (D) cells were treated with various concentrations of capsaicin for 24 hrs, MG132 (20 μM) was added to the culture medium and then incubated for an additional 6 hrs. p53 was immunoprecipitated from 1 mg cell lysate and then detected with anti-p53 antibody to capture poly-ubiquitinated p53.
P53 shRNA decreased the sensitivity of colon cancer cells to capsaicin
In order to confirm the important role of p53 has played in capsaicin antitumor activity, we had developed p53 shRNA to knockdown the expression of p53 in HCT116 cells, and then tested the efficacy of capsaicin in p53-knockdown cells. After HCT116 cells was transfected with p53 shRNA, the expression of p53 was substantially decreased in contrast with mock group, and the number of colonies formed in p53 shRNA group was slightly increased (Figure 5A). As the data shown in Figure 5B, p53 knockdown cells had shown resistance to capsaicin to some extent, after capsaicin treatment, the number of colonies formed in soft agar was significantly increased in comparison with the mock group, in which 40μM capsaicin resulted in remarkable reduction of colony formation. In addition, we also investigated the effect of p53 shRNA on capsaicin-induced cell cycle arrest and apoptosis. As shown in Figure 5C, in p53 shRNA group, capsaicin-induced G0/G1 phase arrest was substantially attenuated, and p53-depedent increase of p21 was also significantly decreased in contrast with the mock group. Similarly, p53 knockdown severely impaired capsaicin-induced apoptosis in HCT116 cells, the percentage of apoptotic cells induced by capsaicin in p53 shRNA group was dramatically decreased. Additionally, p53-depedent increase of Bax after capsaicin treatment was significantly decreased, as well as apoptosis-related protein such as cleaved PARP, cleaved caspase3 (Figure 5D). All these data suggested p53 played an important role in capsaicin-mediated G0/G1 arrest and apoptosis, and the antitumor efficacy of capsaicin was mainly in a p53-dependent manner.
Figure 5
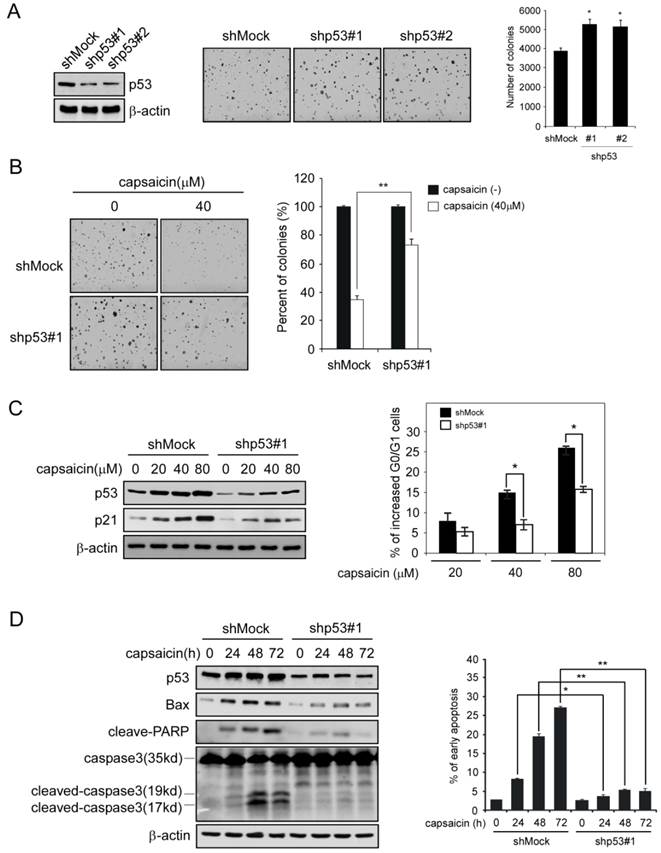
Knockdown of p53 in HCT116 severely impaired its sensitivity to capsaicin. A, knockdown p53 expression in HCT116 with specific p53 shRNA. Validation of p53 expression by Western blotting (left panels), the representative photographs (middle panels), and graph (right panel) of the difference of the anchorage-independent growth between mock and p53 knockdown group. B, sensitivity of HCT116 cells to capsaicin after p53 knockdown. Representative photographs of p53 knockdown HCT116 cells after capsaicin treatment (left panels), the graph (right panel) was the result of three independent experiments expressed as means ± SD. HCT116 cells with Mock shRNA or p53 shRNA were treated with 40 μM capsaicin for anchorage independent assay. C, p21 expression and the G0/G1 arrest in p53-knockdown HCT116 cells after capsaicin treatment. HCT116 cells with Mock shRNA or p53 shRNA were treated with indicated concentrations of capsaicin for 24 hrs, the cells were split into two aliquots, one for cell cycle analysis and the other for immunoblot. D, apoptotic-related protein expression (Bax, cleaved-PARP, caspase 3) and capsaicin-induced apoptosis in p53-knockdown HCT116 cell after capsaicin treatment. HCT116 cells with Mock shRNA or p53 shRNA were treated with 160 μM capsaicin for indicated time points, then the cells were split into two aliquots, one was subjected to Annexin V-PI double staining and the other was subjected to immunoblot. The asterisks (*, p<0.05, **, p<0.01, Student's t test) indicated a significant difference.
Discussion
Colon cancer is an increasingly prevalent health problem in China with over 400,000 new cases and 100,000 deaths occurring as a result of the disease every year [25]. In contrast with the prevalence of this disease, the limited therapeutic options provide a strong stimulus for the development of novel therapeutics. Large numbers of previous experiments have consolidated capsaicin's activity in cancer prevention, and moreover, it was shown that capsaicin -induced apoptosis played an important role in its antitumor activity. However, the underlying mechanism of how capsaicin induce tumor cell to undergo apoptosis were not fully understood. In present study, we have demonstrated that capsaicin had a profound antitumor activity in human colon cancer in vitro. Capsaicin treatment has stabilized and activated p53 by dissociating it from MDM2 and capsaicin exerted its activity in a p53 dependent manner.
In response to a wide variety of stress signals, the p53 pathway is often activated. As reported by previous studies, capsaicin application in tumor cells resulted in stress signals, such as the generation of reactive oxygen species [6], induction of Endoplasmic Reticulum stress [26], dissipation of the mitochondrial membrane potential [27], all these stress signals could lead to activation of p53 [28].As the result of reporter gene experiment shown, the transcriptional activity of p53 was dramatically enhanced after capsaicin treatment. With the activation of p53, the expression of p21 and Bax, which were the two main p53-responsive genes, were also substantially increased. p21 plays an important role in cell cycle regulation while Bax is a key protein involved in the process of cellular apoptosis. Through binding to and inhibiting the activity of cyclin-CDK2 or CDK4 complex, p21 functions as a regulator of cell cycle progression of G1 phase [29]. Bax has a direct effect in induction of mitochondrial depolarization, release of cytochrome c, and activation of a caspase cascade, leading to apoptosis [30]. Furthermore, p53 shRNA experiments clarified that capsaicin exerted its antitumor activity in a p53-dependent manner, knockdown expression of p53 in colon cancer with shRNA severely impaired capsaicin-mediated colony formation inhibition, G0/G1 phase arrest and apoptosis. However, p53 activation mediated capsaicin anti-tumor efficacy was still an open question. In accordance with our result, Ito K et al also suggested that expression of wild-type p53 was necessary for capsaicin-induced cellular growth inhibition and apoptosis in myeloid leukemia cells [6]. In prostate cancer cells, Akio Mori et al reported that capsaicin inhibited of androgen-independent growth in p53 mutant prostate cancer cells via targeting the NF-kappa B signaling pathway [9]. Somatic TP53 gene alterations are frequent in human cancer and it has been reported that p53 mutations are associated with significantly poorer response to intensive chemotherapy and induce drug resistance by interfering with the normal apoptotic pathway in patient [31]. In human colorectal cancer, p53 mutation rate is about 43.2% [32]. Despite the high frequency of TP53 mutation in human cancer, not all mutations will lead to the loss of its transcriptional activity. As reported by Audrey Petitjean et al, in all tested missense mutations of p53 (1070 mutants), 31% (334) were “functional”, 27% (288) were “partially functional”, 4% (46) were “supertrans” and 38% (402) were “nonfunctional” [33]. Therefore, under the condition that the mutant p53 retained its transactivation ability, capsaicin maybe also exerts its antitumor activity via p53 signaling pathway. In another colon cell line HT-29, which harbors 818G>A mutation in TP53, capsaicin's antitumor activity was also observed (data not shown), so the detailed relationship between p53 mutation status and capsaicin's efficacy requires further investigations.
Except gene mutation, stabilization and activation of p53 is also modulated via multiple post-translational modifications. Without stress stimulation, p53 is maintained at low levels owing to MDM2-mediated ubiquitilation and subsequent degradation, the half-life of p53 is very short [34]. In present study, we found that after capsaicin treatment, the half-life of p53 was substantially extended to more than 90 mins, which resulted in the accumulation of p53. Further investigation demonstrated the extension of p53 half-life was attributed to the dissociation of p53 from MDM2. Previous studies had shown stress signals were often generated after capsaicin treatment in different cell types [6, 13, 35], and these signals would cause a series of post-translational modifications of p53. Ito K et al [6] reported that capsaicin-induced ROS resulted in the phosphorylation of p53 on Ser15 and played essential roles in capsaicin's effect on leukemic cells. It was thought that the post-translational phosphorylation of p53 would free p53 from MDM2 and lead to p53 stabilization. However, some evidence also shown that p53 can be stabilized without these modifications. Blattner et al mutated a series of known stress-induced phosphorylation sites on p53 and demonstrated that these p53 mutant forms also could be stabilized [36, 37]. Moreover, in another study, Vassilev et al reported that Nutilins, a powerful compound which exerted the ability to inhibit the interaction between p53 and MDM2, could induce p53 accumulation in nucleus without upregulating p53 phosphorylation [38]. It was concluded that the single prerequisite for induced stabilization of p53 was its prior destabilization of MDM2 and subsequent disruption of the complex formation [16]. Our result for the first time demonstrated that capsaicin significantly dissociated p53 from MDM2, and reduced MDM2-mediated p53 ubiquitilation. However, whether capsaicin suppressed the interaction between p53 and MDM2 in a direct way (direct binding to the complex), or in an indirect way (stress signals-induced post-translational modifications of p53), or both of them would require further studies. Up to now, significant progresses have been achieved in the design and development of small-molecule inhibitors of the MDM2-p53 interaction as new cancer therapies. A number of these small-molecule inhibitors, such as analogs of MI-219 and Nutlin-3, have progressed to advanced preclinical development or early phase clinical trials [39, 40].
Potential limitation of the use of capsaicin in cancer therapy was the higher concentration required for its activity. An in vivo study showed that the highest concentration of capsaicin in serum after oral administration (30mg/kg) was 1.9ug/ml (6.22uM) [41]. Oral LD50 doses of capsaicin reported in previous animal study were: 118mg/kg for male and 97.4mg/kg for female mice [42]. These data implicated that the concentration of capsaicin required to exert its antitumor activity cannot be reached in vivo. But there were evidences from animal studies demonstrated that also lower doses of capsaicin were sufficient to attenuate tumor growth and induce apoptosis of cancer cells in vivo. Ito et al demonstrated that capsaicin daily injection (50mg/kg) significantly inhibited leukemia cell in vivo growth without any organ damage observed [6]. Zhang et al showed that injection of capsaicin (2.5mg/kg) 5 times a week was effective in reducing AsPC-1 human pancreatic xenograft growth in nude mice [43]. Two investigations also showed that capsaicin at 3mg/kg or 10mg/kg could substantially attenuate the xenograft growth of human colon cancer Colo205 or HT29 respectively [44, 45]. However, a study performed by Yang et al showed that capsaicin may promote colorectal cancer metastasis via modulating Akt/mTOR and STAT-3 pathways in vivo [46]. Therefore, more in vivo experiments will be needed to further confirm capsaicin activity against tumor growth. In summary, we demonstrated a model of capsaicin-induced cell cycle arrest and apoptosis in human colon cancer cells. Via dissociating p53 from the interaction with MDM2, capsaicin substantially inhibited MDM2-mediated p53 ubiquitilation. Meanwhile, our data strongly indicated the role of a functional p53 played in capsaicin's antitumor efficacy. Limited effective treatment for colon cancer highlighted the urgent need for novel therapeutic approaches. As a component of hot peppers, capsaicin is widely consumed as food additive throughout of the world, which indicates it is less toxic to human than current chemotherapeutic agents. Therefore, capsaicin or related analogues maybe have potentials as a novel molecular targeted agent against human colon cancers, especially those patients with wide-type p53 expression.
Supplementary Material
Supplementary Figure 1Acknowledgements
This work was supported by Science and Technology Program of LiaoNing Province (Grant No.2012225019).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacological reviews. 1991;43:143-201
2. Raisinghani M, Pabbidi RM, Premkumar LS. Activation of transient receptor potential vanilloid 1 (TRPV1) by resiniferatoxin. The Journal of physiology. 2005;567:771-86
3. Watson CP, Evans RJ, Watt VR. Post-herpetic neuralgia and topical capsaicin. Pain. 1988;33:333-40
4. Sicuteri F, Fusco BM, Marabini S, Campagnolo V, Maggi CA, Geppetti P. et al. Beneficial effect of capsaicin application to the nasal mucosa in cluster headache. The Clinical journal of pain. 1989;5:49-53
5. Matucci Cerinic M, McCarthy G, Lombardi A, Pignone A, Partsch G. Neurogenic influences in arthritis: potential modification by capsaicin. The Journal of rheumatology. 1995;22:1447-9
6. Ito K, Nakazato T, Yamato K, Miyakawa Y, Yamada T, Hozumi N. et al. Induction of apoptosis in leukemic cells by homovanillic acid derivative, capsaicin, through oxidative stress: implication of phosphorylation of p53 at Ser-15 residue by reactive oxygen species. Cancer research. 2004;64:1071-8
7. Huh HC, Lee SY, Lee SK, Park NH, Han IS. Capsaicin induces apoptosis of cisplatin-resistant stomach cancer cells by causing degradation of cisplatin-inducible Aurora-A protein. Nutrition and cancer. 2011;63:1095-103
8. Ip SW, Lan SH, Lu HF, Huang AC, Yang JS, Lin JP. et al. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Human & experimental toxicology. 2012;31:539-49
9. Mori A, Lehmann S, O'Kelly J, Kumagai T, Desmond JC, Pervan M. et al. Capsaicin, a component of red peppers, inhibits the growth of androgen-independent, p53 mutant prostate cancer cells. Cancer research. 2006;66:3222-9
10. Huang SP, Chen JC, Wu CC, Chen CT, Tang NY, Ho YT. et al. Capsaicin-induced apoptosis in human hepatoma HepG2 cells. Anticancer research. 2009;29:165-74
11. Ying H, Wang Z, Zhang Y, Yang TY, Ding ZH, Liu SY. et al. Capsaicin induces apoptosis in human osteosarcoma cells through AMPK-dependent and AMPK-independent signaling pathways. Molecular and cellular biochemistry. 2013;384:229-37
12. Maity R, Sharma J, Jana NR. Capsaicin induces apoptosis through ubiquitin-proteasome system dysfunction. Journal of cellular biochemistry. 2010;109:933-42 doi:10.1002/jcb.22469
13. Kim SR, Kim SU, Oh U, Jin BK. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. Journal of immunology. 2006;177:4322-9
14. Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochemical and biophysical research communications. 2005;331:834-42
15. Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends in molecular medicine. 2010;16:528-36
16. Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell death and differentiation. 2006;13:941-50
17. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nature reviews Molecular cell biology. 2008;9:402-12
18. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Molecular cell. 2006;21:307-15
19. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972-5
20. Baccouche S, Daoud J, Frikha M, Mokdad-Gargouri R, Gargouri A, Jlidi R. Immunohistochemical status of p53, MDM2, bcl2, bax, and ER in invasive ductal breast carcinoma in Tunisian patients. Annals of the New York Academy of Sciences. 2003;1010:752-63
21. Elkablawy MA, Maxwell P, Williamson K, Anderson N, Hamilton PW. Apoptosis and cell-cycle regulatory proteins in colorectal carcinoma: relationship to tumour stage and patient survival. The Journal of pathology. 2001;194:436-43
22. Gustafsson B, Axelsson B, Gustafsson B, Christensson B, Winiarski J. MDM2 and p53 in childhood acute lymphoblastic leukemia: higher expression in childhood leukemias with poor prognosis compared to long-term survivors. Pediatric hematology and oncology. 2001;18:497-508
23. Mori S, Ito G, Usami N, Yoshioka H, Ueda Y, Kodama Y. et al. p53 apoptotic pathway molecules are frequently and simultaneously altered in nonsmall cell lung carcinoma. Cancer. 2004;100:1673-82
24. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nature reviews Cancer. 2004;4:793-805
25. Guo P, Huang ZL, Yu P, Li K. Trends in cancer mortality in China: an update. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012;23:2755-62
26. Lin S, Zhang J, Chen H, Chen K, Lai F, Luo J. et al. Involvement of endoplasmic reticulum stress in capsaicin-induced apoptosis of human pancreatic cancer cells. Evidence-based complementary and alternative medicine: eCAM. 2013;2013:629750
27. Lin CH, Lu WC, Wang CW, Chan YC, Chen MK. Capsaicin induces cell cycle arrest and apoptosis in human KB cancer cells. BMC complementary and alternative medicine. 2013;13:46
28. Lin WC, Chuang YC, Chang YS, Lai MD, Teng YN, Su IJ. et al. Endoplasmic reticulum stress stimulates p53 expression through NF-kappaB activation. PloS one. 2012;7:e39120
29. Cmielova J, Rezacova M. p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected]. Journal of cellular biochemistry. 2011;112:3502-6
30. Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658-62
31. Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A. et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589-97
32. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology. 2010;2:a001008
33. Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R. et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8424-9
34. Rogel A, Popliker M, Webb CG, Oren M. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Molecular and cellular biology. 1985;5:2851-5
35. Lee MJ, Kee KH, Suh CH, Lim SC, SH O. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology. 2009;264:205-14
36. Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene. 1999;18:1723-32
37. Loughery J, Meek D. Switching on p53: an essential role for protein phosphorylation? Biodiscovery. 2013
38. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8
39. Rew Y, Sun D, Gonzalez-Lopez De Turiso F, Bartberger MD, Beck HP, Canon J. et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. Journal of medicinal chemistry. 2012;55:4936-54
40. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223-41
41. Suresh D, Srinivasan K. Tissue distribution & elimination of capsaicin, piperine & curcumin following oral intake in rats. The Indian journal of medical research. 2010;131:682-91
42. Saito A, Yamamoto M. Acute oral toxicity of capsaicin in mice and rats. The Journal of toxicological sciences. 1996;21:195-200
43. Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13:1465-78
44. Lu HF, Chen YL, Yang JS, Yang YY, Liu JY, Hsu SC. et al. Antitumor activity of capsaicin on human colon cancer cells in vitro and colo 205 tumor xenografts in vivo. Journal of agricultural and food chemistry. 2010;58:12999-3005
45. Zhu LL, Hu WL, Zhang LJ, Yu ZG, Huang CJ, Jiang MZ. et al. Antitumor effect of capsaicin on colorectal carcinoma xenograft in nude mice. Zhonghua zhong liu za zhi. 2013;35:256-61
46. Yang J, Li TZ, Xu GH, Luo BB, Chen YX, Zhang T. Low-concentration capsaicin promotes colorectal cancer metastasis by triggering ROS production and modulating Akt/mTOR and STAT-3 pathways. Neoplasma. 2013;60:364-72
Author contact
![]() Corresponding author: Honglei Jiang Ph.D. M.D. Address: The No.4 Chongshan east road Huanggu district, Shenyang Liaoning, 110032. Tel: 024-62043517 Fax: 024-62255001 E-mail: grkjxzorg.
Corresponding author: Honglei Jiang Ph.D. M.D. Address: The No.4 Chongshan east road Huanggu district, Shenyang Liaoning, 110032. Tel: 024-62043517 Fax: 024-62255001 E-mail: grkjxzorg.
Citation styles
APA
Jin, J., Lin, G., Huang, H., Xu, D., Yu, H., Ma, X., Zhu, L., Ma, D., Jiang, H. (2014). Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53. International Journal of Biological Sciences, 10(3), 285-295. https://doi.org/10.7150/ijbs.7730.
ACS
Jin, J.; Lin, G.; Huang, H.; Xu, D.; Yu, H.; Ma, X.; Zhu, L.; Ma, D.; Jiang, H. Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53. Int. J. Biol. Sci. 2014, 10 (3), 285-295. DOI: 10.7150/ijbs.7730.
NLM
Jin J, Lin G, Huang H, Xu D, Yu H, Ma X, Zhu L, Ma D, Jiang H. Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53. Int J Biol Sci 2014; 10(3):285-295. doi:10.7150/ijbs.7730. https://www.ijbs.com/v10p0285.htm
CSE
Jin J, Lin G, Huang H, Xu D, Yu H, Ma X, Zhu L, Ma D, Jiang H. 2014. Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating p53. Int J Biol Sci. 10(3):285-295.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.