Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(4):438-447. doi:10.7150/ijbs.8095 This issue Cite
Research Paper
ELK3 Suppresses Angiogenesis by Inhibiting the Transcriptional Activity of ETS-1 on MT1-MMP
Sun-Hee Heo, Je-Yoel Cho ![]()
Department of Veterinary Biochemistry, BK21 Plus, Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul, Republic of Korea.
Received 2013-11-10; Accepted 2014-3-7; Published 2014-3-27
Abstract
Ets transcription factors play important roles in vasculogenesis and angiogenesis. Knockout of the Ets gene family members in mice resulted in disrupted angiogenesis and malformed vascular systems. In this study, the role and mechanism of ELK3, an Ets factor, in angiogenesis was investigated using ELK3-specific siRNA in human vascular endothelial cells (HUVECs) and in vivo implantation assay. The suppression of ELK3 expression resulted in the reinforcement of VEGF-induced tube formation in HUVECs. The in vivo Matrigel plug assay also showed that ELK3 knockdown resulted in increased angiogenesis. Luciferase activity of the MT1-MMP promoter induced by ETS-1 factor was attenuated ELK3 co-transfection. CHIP assay showed the binding of ELK3 on the MT1-MMP promoter. MT1-MMP knockdown in the ELK3 knockdowned cells resulted in the decrease of tube formation suggesting that MT1-MMP transcriptional repression is required for ELK3-mediated anti-angiogenesis effect. Our data also showed that the suppressive effect of ELK3 on the angiogenesis was partly due to the inhibitory effect of ELK3 to the ETS-1 transcriptional activity on the MT1-MMP promoter rather than direct suppression of ELK3 on the target gene, since the expression level of co-repressor Sin3A is low in endothelial cells. Our results suggest that ELK3 plays a negative role of VEGF-induced angiogenesis through indirectly inhibiting ETS-1 function.
Keywords: ELK3, VEGF, MT1-MMP, Sin3A.
Introduction
Angiogenesis is critical to biological processes such as development and reproduction, and diseases such as arthritis, cancer and diabetes [1, 2]. Endothelial and mural cells compose the vessel walls and are embedded in an extracellular matrix (ECM). Angiogenesis comprises 3 steps: the activation of the endothelial cells, the proliferation and migration of the endothelial cells, and the maturation of the newly formed vessels. The conversion of endothelial cells to an angiogenic phenotype is induced by soluble factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). The ECM are dissolved by activated proteases, including matrix metalloproteinases (MMPs), which results in the proliferation and migration of endothelial cells (ECs). Finally, tube formation by the ECs is stabilized by recruiting pericytes [3-5].
The ETS transcription factors are present in a wide range of organisms from sponges to humans and are characterized by a shared DNA binding domain of approximately 85 amino acids that recognizes specific ETS domain binding sites (EBS) containing the purine-rich core sequence 5'-GGAA/T-3' [6-9]. The ETS factors regulate the transcription of over 200 target genes that contribute to diverse biological processes such as apoptosis, development, hematopoiesis, proliferation and migration [6, 10-13]. A subset of ETS factors plays a role in angiogenesis and vasculogenesis. For example, Ets-1 controls the expression of genes associated with cell attachment and the migration of endothelial cells, such as αv integrins, β3 integrins, VE cadherin, intracellular adhesion molecules (ICAMs) and von Willebrand factor [14]. The mutation of Fli-1 in mice resulted in embryonic hemorrhage of the brain and neural tube, which indicate a possible defect in the dorsal aorta [15]. Tel knockout in mice resulted in disrupted yolk sac angiogenesis and disorganized adult bone marrow hematopoiesis [16, 17].
ELK3, also previously known as NET, is a member of the ETS family of transcription factors and is known to act as a transcriptional repressor [18, 19]. ELK3, ELK-1 and SAP1 comprise a subfamily of ternary complex transcription factors and each has a unique chromosomal location [20]. Unlike ELK-1 and SAP1, ELK3 contains the JNK-induced export (JEX) sequence, which is may contribute to the differences between the functions of ELK3 from those of ELK-1 and SAP1. The mRNA expression of ELK3 is detected at the early stages of mouse development, especially at the sites of vasculogenesis, angiogenesis and chondrogenesis [21]. ELK3 expression is highest in human umbilical vein endothelial cells (HUVECs) compared to several tissues and cell lines [22]. Additionally, defects of the lymphatic system and abnormal expression of Egr-1 in the vascular system appeared in ELK3 knockout mice [23].
The MMPs are a family of over 20 zinc-containing endopeptidases that can degrade extracellular matrix (ECM) [24]. MT1-MMP is an MMP-2 activator that regulates various physiological and pathological processes such as angiogenesis, cancer invasion and wound healing [25]. The overexpression of MT1-MMP promotes the migration, invasion and metastasis of cancer cells in vitro and in vivo [26]. In addition, MT1-MMP expression is critical for cancer cell growth in a 3D collagen-based matrix [27].
In this study, we explored the role of ELK3 in vessel endothelial cells. The functions of ELK3 were evaluated during VEGF-induced angiogenesis in vitro and in vivo.
Materials and Methods
Cell culture. Human umbilical vein endothelial cells (HUVECs) were obtained from Modern Cell & Tissue Technologies, Inc. (Seoul, Korea). The HUVECs were cultured in gelatin-coated plates with EC medium (Clonetics catalog CC-3162). Human embryonic kidney 293 cells and human embryonic pulmonary epithelial L-132 cells were cultured in DMEM. The cell lines were incubated in the presence of 5% CO2 at 37°C.
RNA interference of ELK3 and MT1-MMP. Nonspecific control siRNA (D-001810-10), human ELK3 siRNA (L-010320-00-0005, J-010320-05 and J-010320-06) and ON-TARGET plus human MMP14 (MT1-MMP) siRNA (J-004145-13 and J-004145-14) were purchased from Dharmacon (Chicago, IL), Inc. Twenty nM ELK3 siRNA or nonspecific control siRNA were transfected into the HUVECs using Effectene (Qiagen). The efficacy of knockdown was assessed by quantitative RT-PCR using ELK3-specific primers and control GAPDH primers.
Extraction of RNA. Total cellular RNA was isolated from HUVECs using the TRIzol Reagent (Invitrogen) following the manufacturer's instructions. Briefly, single-stranded cDNA synthesis was performed with 2 µg total RNA using an oligo(dT)18 primer and the Omniscript Reverse Transcriptase (Qiagen, Germany).
Real-time polymerase chain reaction (RT-PCR). Gene-specific primers for ELK3, ELK-1, MT1-MMP, MMP-1, MMP-2 and MMP-10 were designed and quantitative RT-PCR was performed as previously described [10]. The mRNA levels of TIMP-1, VE-cadherin and HDAC7 were determined by real-time PCR using the following primers: TIMP-1 sense 5'-AAT TCC GAC CTC GTC ATC AG-3', TIMP-1 antisense 5'-TGC AGT TTT CCA GCA ATG AG-3', VE-cadherin sense 5'-CAG CCC AAA GTG TGT GAG AA-3', VE-cadherin antisense 5'-CGG TCA AAC TGC CCA TAC TT-3', HDAC7 sense 5'-CCC AGC AAA CCT TCT ACC AA-3', and HDAC7 antisense 5'-AAG CAG CCA GGT ACT CAG GA-3'. Fluorescence-based real-time PCR was performed using the DNA Engine OPTICON®2 system (MJ Research, Waltham, MA) with SYBR Green I (Molecular Probes, Eugene, OR). The cycling conditions were as follows: 40 cycles of 95°C for 30 s, 50°C for 40 s and 72°C for 60 s. For quantification, each sample was normalized to human GAPDH as a reference. The results of the real-time PCR analysis were expressed as the mean value ± SE of triplicates from each sample.
MTT assay. For the MTT assays, the HUVECs were plated at a density of 1.5x103 cells per well in a 96-well plate and incubated with EC medium for the indicated times. Next, the medium was replaced with EC medium excluding cytokines (hEGF, VEGF, hFGF-B, R3-IGF-1) that either contained or lacked 20 ng/ml VEGF165 (R&D Systems, Inc., MN, USA). After another period of incubation for the indicated times, the medium was changed again and MTT solution (Sigma, M2128) was added. Following a 3 h incubation period, the medium and MTT solution were removed and dimethyl sulfoxide (DMSO) and glycine buffer (0.1 M glycine, 0.1 M NaCl pH 10.5 with 1 M NaOH) were added. The optical density of each well was measured at 450 nm using a microplate reader. The experiments were performed in triplicate.
Tube formation. The HUVECs were transfected with siRNA (ELK3 or negative control) for 24 h. Next, the transfected cells were trypsinized and seeded at 5x104 cells per well in a 48-well plate on Matrigel (BD Biosciences) that had polymerized for 30 min at 37°C. The HUVECs were incubated in EC medium (without cytokines) in the presence or absence of VEGF. The changes in cell morphology were photographed using Leica inverted microscope (x100 magnification) after incubation for 18 h.
In vivo Matrigel plug assay. Six-week-old male BALB/c mice (Orient Bio Inc, Sung-nam, Korea) were used for the Matrigel plug assay. All procedures of animal experiments were performed after the approval of Animal Care & Use Committee of Kyungpook National University. The mice were injected subcutaneously into their ventral area with 500 µl of Matrigel containing 20 units of heparin, 1 µM siScramble or siElk3 (ON-TARGETplus Mouse Elk3 siRNA, L-058294-01-0005) and in the presence or absence of 500 ng of VEGF. After 7 d, the mice were sacrificed, and the Matrigel plugs were excised from the skin and photographed. To quantify the formation of microvessels, hematoxylin and eosin (H&E) staining was performed and quantified manually using a microscope.
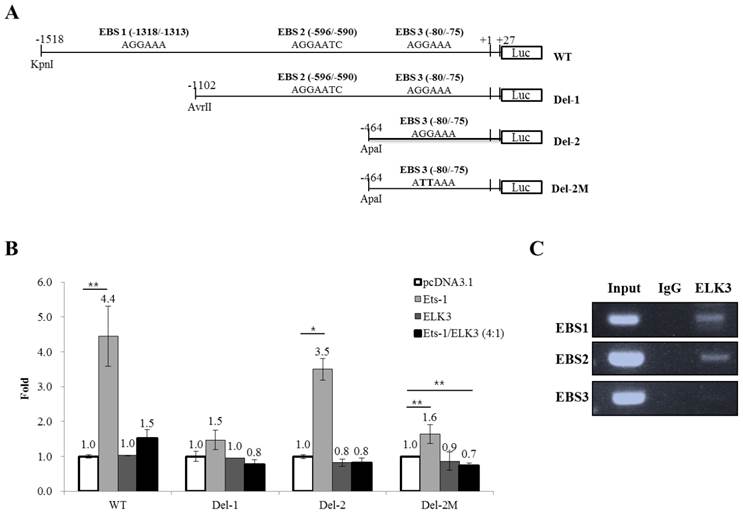
DNA construction. pcDNA3.1-Ets-1 and pcDNA3.1-Elk3 were constructed in our previous study [10]. A 1.5 kb region of the MT1-MMP promoter (WT) was generated by PCR amplification from human genomic DNA based on the GenBank™ accession number AB011056 (from 570 to 2114 bp, marked as -1518 to +27 bp in Fig. 4A). The fragment was cloned into the KpnI and NheI sites of the pGL3-Basic reporter vector. The MT1-MMP promoter deletion constructs Del-1 (-1102 to +27 bp) and Del-2 (-464 to +27 bp) were generated by serial deletion using AvrII (Del-1) or ApaI (Del-2) from the 5′ end of the 1.5 kb promoter and cloned into the pGL3-Basic vector. To produce the constructs that bear the specific mutations of two nucleotides (AGGAAA to ATTAAA) in the putative Ets binding site (EBS, -80 to -75 bp) of the Del-2 construct, a site-directed mutagenic two-step PCR strategy was utilized. For the first set of PCRs, a mutant primer was designed and used as the forward primer to create Del-2M; the Del-2 promoter construct was used as the template. In the second round of PCRs, the PCR products synthesized from the first round were used as the reverse primers; again, the Del-2 promoter construct was used as the template. The PCR products with the mutated binding sites were digested with KpnI and NheI and the resulting 491 bp fragments were ligated to pGL3-Basic. All of the generated constructs were confirmed by sequencing (SolGent, Daejeon, Korea).
ELK3 transcriptional regulation of MT1-MMP. (A) The MT1-MMP promoter constructs. The 1.5 kb promoter region of the human MT1-MMP gene containing the 3 Ets binding sites (EBS) was subcloned into the pGL3 vector. The serial deletion constructs of the MT1-MMP promoter were produced by restriction enzyme digestion and ligation. The mutant construct of the MT1-MMP promoter was produced by two-step PCR. (B) Luciferase assays were performed to analyze the transcriptional regulation of MT1-MMP in HUVECs. ETS-1 and/or ELK3 were transfected with the 1.5 kb MT1-MMP promoter construct, the deletion constructs, the mutant construct, or an empty vector. The promoter activity was measured as described in the Methods and Materials. The data are presented as means ± SE. (C) Chromatin immunoprecipitation was performed using an anti-ELK3 antibody and the HUVECs lysates. EBS1, EBS2 and EBS3 site-specific PCR primers were used to detect ELK3 binding to the EBSs. *p < 0.01; **p < 0.05 compared to in pcDNA 3.1 transfected cells.
Luciferase reporter assays. The HUVECs, 293 cells and L-132 cells were seeded into 48-well plates and cultured overnight until they reached 60-80% confluence. A total of 0.2 µg of DNA (reporter and/or expression construct) per well were transiently transfected in triplicate using Effectene (Qiagen). After transfection for 6 h, the medium was changed and cultured with new medium for an additional 24 h. The luciferase activity was evaluated using the Luciferase Assay kit (Promega) following the manufacturer's instructions.
Isolation of protein and Western blot analysis. The cultured cells were washed twice with cold phosphate-buffered saline (PBS) and lysed with RIPA buffer containing a protease inhibitor mixture (diluted 1:30) (Roche, Mannheim, Germany) on ice for 20 min. The protein concentrations of the cell lysates were measured using the Bradford method; then, 20 μg of protein were separated by 12% SDS-PAGE for Sin3A and ELK3 analysis. After electrophoresis, the proteins were transferred to a nitrocellulose membrane (Whatman, Germany). Next, the membrane was incubated with monoclonal anti-Sin3A (Santa Cruz Biotechnology, sc-994) (1:1000 dilution), anti-ELK3 (Santa Cruz Biotechnology, sc-17860) (1:1000 dilution) or anti-β-actin antibody (1:3000 dilution) overnight at 4°C, then incubated with anti-rabbit IgG antibody (1:2000 dilution, secondary antibody against anti-Sin3A) or anti-goat IgG antibody (1:2000 dilution, secondary antibody against anti-ELK3 and anti-β-actin) for 1 h at room temperature. The detection of immunoreactive proteins was performed using the Pierce ECL Plus Western Blotting Substrate kit (Thermo Scientific, USA). GelQuant.ELK3 software used for quantification was provided by biochemlabsolutions.com
Chromatin immunoprecipitation (ChIP) assay. The HUVECs were plated at approximately 70% confluence in EGM-2 medium. The next day, formaldehyde was added to the HUVECs to a final concentration of 1% and incubated for 30 min at room temperature to achieve crosslinking. After the incubation period, glycine was added to a final concentration of 125 mM. After 5 min, the cells were washed with cold PBS and scraped in 1 ml of PBS. The cell pellet was lysed in 600 µl of SDS lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris-HCl, pH 8.1) containing protease inhibitors (Roche Diagnostics mixture tablet). The lysed cells were sonicated for 10 rounds of 10 pulses and the resulting pellet was cleared by centrifugation; 20 µl was retained as an input control. The cleared lysates were resuspended in IP buffer (167 mM NaCl, 16.7 mM Tris-HCl, pH 8.1, 1.2 mM EDTA, 0.01% SDS and 1% Triton X-100) and immunoprecipitated with or without antibody (2 µg of anti-ELK3 antibody, Santa Cruz) by rotating overnight at 4°C. The immune complexes were recovered with protein A/G-agarose (Santa Cruz) and washed in sequence with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1 and 150 mM NaCl), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1 and 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% NP40, 1% deoxycholate, 1 mM EDTA and 10 mM Tris-HCl, pH 8.1) and TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The immunoprecipitated DNA and input DNA were extracted by the addition of 100% ethanol followed by reverse crosslinking. The DNA was purified and served as the template for PCR. The following primers were used: ESE1 (-1318 to -1313 bp) forward 5'- CTG AGG TCT CCA TGG TTC TG -3', ESE1 reverse 5'- TGG GAG GTA TAT GGT GTG TG- 3', ESE2 (-596 to -590 bp) forward 5'- CAG ATC CCA CGG CCT TGT AC -3', ESE2 reverse 5'- GTA GAC AAC TGT CTA TGT CC- 3', ESE3 (-80 to -75 bp) forward 5'- CAG ATG GCA GCC TGC ACC AC -3' and ESE3 reverse 5'- CTT CTG CTT AGT CGG CGA AC- 3'.
Statistical analysis. The data are presented as means ± SE. The statistical significance of the results was assessed using one-way ANOVA followed by Student's t-test. P values less than 0.05 was considered statistically significant. * p< 0.01 compared with control, ** p< 0.05 compared with control.
Results
The effect of ELK3 knockdown on HUVECs during VEGF-induced angiogenesis
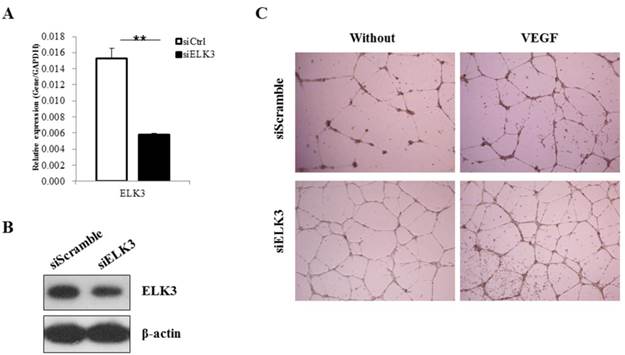
ELK3 knockdown was performed to elucidate the role of ELK3 in endothelial cells during VEGF-induced angiogenesis. The efficiency of ELK3 siRNA (siELK3) was estimated using quantitative real-time PCR after transfection of the HUVECs. Although siRNA transfection into the HUVECs was not efficient, we optimized the transfection of ELK3-specific siRNA and achieved a decrease of approximately 60% in ELK3 mRNA expression (Figure 1A) and protein expression (Figure 1B). In addition, the other two ELK3 siRNA also led to significant decrease of ELK3 expression and increased tube formation function (Additional file 1: Supplementary Figure 1). ELK3 knockdown enhanced the tube formation of HUVECs in the absence and presence of VEGF (Figure 1C). These results suggest that ELK3 have an inhibitory effect on the functional tube formation in HUVECs.
The effect of ELK3 knockdown on VEGF-induced angiogenesis. (A) The efficiency of ELK3 knockdown was evaluated by quantitative RT-PCR. ELK3 knockdown in HUVECs was accomplished using ELK3 siRNA as described in the Materials and Methods section. The data are presented as means ± SE. (B) ELK3 protein expression was confirmed after the knockdown using ELK3 specific siRNA. After 48 hr, proteins were extracted and loaded on 10% SDS-PAGE gel. Specific band for ELK3 was detected using ELK3 antibody. β-Actin was used as a control. (C) The effect of ELK3 knockdown on VEGF-induced tube formation. HUVECs transfected with siELK3 were cultured in medium with or without VEGF (20 ng/ml) on Matrigel and photographed 18 h after incubation. **p < 0.05 compared siCtrl.
The role of ELK3 in VEGF-induced vessel formation in vivo
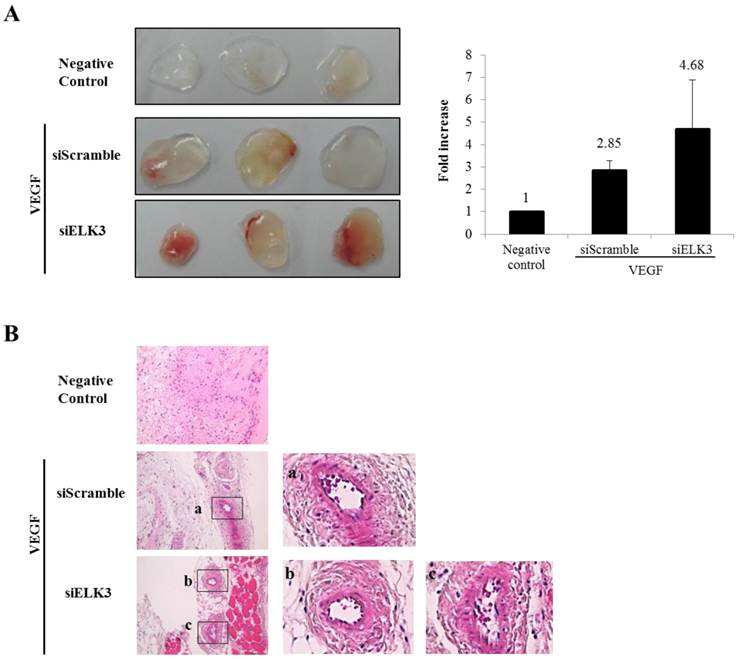
ELK3 knockdown increased the tube formation of HUVECs stimulated by VEGF. To confirm the inhibitory effect of ELK3 in vivo, the Matrigel plug assay was performed to see whether ELK3 knock-down enhances VEGF-induced blood vessel formation. The Matrigel plug was implanted after soaking with either the siRNA scrambled control (siScramble) or siELK3 along with VEGF. Heparin was present in all Matrigel plugs including the negative control. The Matrigel plugs with siELK3 significantly enhanced blood vessel formation compared to siScramble (Figure 2A). Hematoxylin and eosin (H&E) staining showed the newly formed vessel-like structures in the Matrigel plugs (Figure 2B). These results indicate that ELK3 also functions as a repressor of VEGF-induced angiogenesis in vivo.
The effects of ELK3 on VEGF-induced blood vessel formation in Matrigel plugs in vivo. Six-week-old BALB/c mice were injected with 0.5 ml of Matrigel containing 500 ng of VEGF and 20 units of heparin along with siScramble or siELK3. The Negative Control contains heparin alone; siScramble contains heparin, VEGF and 1 μM scrambled siRNA; and siELK3 contains heparin, VEGF, and 1 μM ELK3-specific siRNA. After 7 d, the mice were sacrificed and the intact Matrigel plugs were removed from the skin and photographed to determine vessel formation. Knockdown of ELK3 enhanced VEGF-induced angiogenesis in vivo. (A) The Matrigel plugs were photographed. (B) The excised Matrigel plugs were fixed with 4% paraformaldehyde and then the plugs were sectioned and stained with H&E. Scale bar=100 μm. (a), (b) and (c) are presented as magnified images.
mRNA expression levels of angiogenic factors following ELK3 knockdown in HUVECs
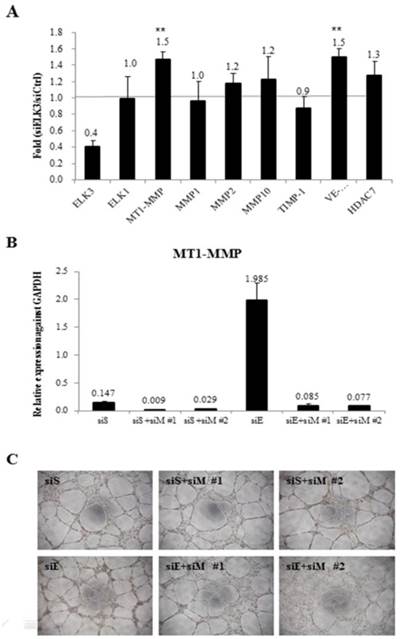
To investigate the target genes of ELK3 in relation with angiogenesis, the mRNA expression levels of several angiogenic factors were determined using quantitative RT-PCR after ELK3 knockdown in HUVECs (Figure 3A). Elk expression was evaluated to determine if the functional compensation by a member of the same subfamily of ETS transcription factors occurs following ELK3 knockdown. While Elk expression was not altered by ELK3 knockdown, the mRNA expression levels of the angiogenic factors MT1-MMP and VE-cadherin were upregulated an average of 1.5-fold following ELK3 knockdown (Figure 3A). It was confirmed whether upregulated MT1-MMP led to increase tube formation in ELK3 knockdown using two MT1-MMP specific siRNA. The mRNA expression of MT1-MMP was efficiently decreased by siRNAs treatment (Figure 3B). Then, MT1-MMP knockdown in the ELK3 knockdowned cells resulted in the decrease tube formation suggesting that MT1-MMP transcriptional repression is required for ELK3-mediated anti-angiogenesis effect (Figure 3C).
The mRNA expression levels of specific angiogenic factors and the effect of MT1-MMP knockdown in ELK3-knockdown endothelial cells. (A) The transfection of siELK3 resulted in a 40% decrease in ELK3 expression. We investigated whether ELK3 knockdown altered the expression of specific angiogenesis-related factors. The mRNA levels of MT1-MMP, MMP1, MMP2, MMP10, TIMP-1, VE-cadherin and HDAC7 in siELK3-transfected cells were evaluated by comparing to siScramble-transfected HUVECs using quantitative RT-PCR. The experiments were performed in triplicate and the data are presented as means ± SE. **p < 0.05 compared siS. (B) Specificity of siRNA for knockdown of MT1-MMP was confirmed by analyzing the mRNA expression of MT1-MMP. (C) Tube formation was carried out after siScramble (siS), siS+siMT1-MMP (siM)#1 or siS+siM#2 knockdown, and siELK3 (siE) alone, siE+siM#1, or siE+siM#2 knockdown.
ELK3 regulation on the transcription of MT1-MMP
We then investigated whether ELK3 regulates the transcription of MT1-MMP. The MT1-MMP promoter has 3 ETS binding site (EBS): EBS1 (-1318 to -1313), EBS2 (-596 to -590) and EBS3 (-80 to -75). The 1.5 kb MT1-MMP promoter (WT) and serially deleted MT1-MMP promoter constructs (Del-1 and Del-2) were cloned into pGL3-Basic vector (Figure 4A). In addition, a mutant construct, Del-2M, was produced at the EBS3 site by substituting the “GGA” of the EBS core sequence with “TTA”. Overexpression of ETS-1, a well-established positive transcription factor, upregulated the promoter activity of MT1-MMP, especially the promoter constructs containing three EBS sites (WT) and EBS3 site (Del-2). The Del-1 construct (EBS1-deleted) was not activated by ETS-1 expression. In addition, the deletion of EBS2 (Del-2), leaving EBS3 alone, reversed the stimulatory effect of ETS-1. Together, these data indicate that EBS3 mediates positive response to ETS-1 and EBS2 may be involved in the suppression of MT1-MMP promoter activity. When the EBS1 site is lost but the EBS2 site is intact (Del-1), the EBS3 response to ETS-1 is lost probably by the suppressive effect of the factors bound to EBS2 site. Thus, it is speculated that EBS1 site might relieve the suppressive effect of EBS2 site on the EBS3 site for the transcription activated by ETS-1. The increased activity of the Del-2 MT1-MMP promoter triggered by ETS-1 was reduced close to basal levels by the mutation of the EBS3 core sequence (Del-2M). This experiment also implies that EBS3 is a binding site of ETS-1 for the activation of MT1-MMP by ETS-1.
ELK3 did not directly regulate the activity of the MT1-MMP promoter, but it almost completely abolished the positive activity of the MT1-MMP promoter triggered by ETS-1 (Figure 4B). This result implies that ELK3 inhibits the transcriptional activity of ETS-1 on the MT1-MMP promoter either by binding to the EBSs or by interacting with ETS-1 and thus blocking ETS-1 from binding to the EBSs.
The abolishing effect of ELK3 was observed either in the presence (Figure 4B, WT & Del-1) or absence (Del-2) of EBS2 site. However, deletion of EBS2 site recovered the ETS-1 transactivity, suggesting negative factors might exert on EBS2. To investigate if ELK3 binds to the EBSs of the MT1-MMP promoter, chromatin immunoprecipitation assays were performeded using the anti-ELK3 antibody and primers designed for each EBS site. We found that ELK3 binds to EBS1 and EBS2 but not EBS3 (Figure 4C). Combined with the promoter assay, these data suggest that EBS1 and EBS3 are preferential binding sites for ETS-1 activation of MT1-MMP. Additionally, the binding of ELK3 to EBS1 and EBS2 exert an inhibitory effect on the MT1-MMP promoter. However, it is possible that ELK3 also inhibits ETS-1 activity through a direct protein-protein interaction because ETS-1 transactivity on the EBS3 alone (Del-2 promoter) was also inhibited by ELK3 co-transfection (Figure 4B), despite our finding that ELK3 does not bind to EBS3 (Figure 4C).
ELK3 functions as a repressor by recruiting Sin3A co-repressor in HUVECs
ELK3 is known to be a direct transcriptional repressor. However, we did not observe the direct transcriptional repressor activity of ELK3 on the MT1-MMP promoter in HUVECs (Figure 4B). Therefore, we hypothesized that the indirect effect of ELK3 on the MT1-MMP promoter in HUVECs depends on cell context. We first tested the effect of ELK3 on the MT1-MMP promoter in several cell lines (Figure 5A). ETS-1 overexpression resulted in increased MT1-MMP promoter activity in all tested cell lines: HEK-293 (human embryonic kidney cells), L-132 (human embryonic pulmonary epithelial cells) and HUVECs. However, ELK3 reduced the transcriptional activation of MT1-MMP in the HEK-293 and L-132 cells but not in the HUVECs (Figure 5A). This result has suggested that ELK3, as a repressor, differently act on the HUVECs endothelial cells compared to other cell types such as epithelial cells.
The Sin3A co-repressor regulates ELK3 transcription in HUVECs. (A) The MT1-MMP promoter activity in several cell lines: 293 (human embryonic kidney cells), L-132 (human embryonic pulmonary epithelial cells) and HUVECs (human umbilical vein endothelial cells). The 1.5 kb MT1-MMP promoter was co-transfected with an empty vector (pcDNA3.1), ETS-1 or ELK3 cDNA. After 24 h, the luciferase activity was measured. (B) The protein expression levels of Sin3A in 293 cells, L-132 cells and HUVECs. The proteins were isolated from the 3 cell lines and separated by SDS-PAGE to detect Sin3A. β-actin was used as an internal control. The density of bands was measured by GelQuant.ELK3 software (lower table) (C) Confirmation of Sin3A protein expression in HUVECs following ELK3 overexpression by Western blotting. The ELK3 expression plasmid was transfected into the HUVECs. ELK3 overexpression was confirmed by Western blotting. Sin3A protein expression decreased in response to ELK3 overexpression in HUVECs. The intensity of bands was accessed by GelQuant.ELK3 software (lower table) (D) Sin3A overexpression in HUVECs almost completely abolished ELK3 expression. The intensity of bands was evaluated by GelQuant.ELK3 software (lower table). *p < 0.01 in comparison between ETS-1 and ELK3 transfected cells.
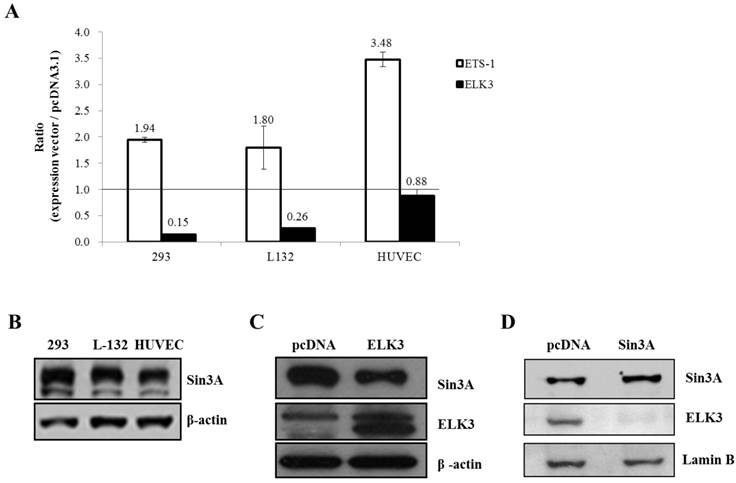
It is possible that specific co-repressors differentially contribute to ELK3 regulation on MT1-MMP transcription in different cell types. Among the candidate co-repressors (NCoRI, CtBP and Sin3A), the expression of Sin3A was lower in the HUVECs than in the HEK-293 and L-132 cells when analyzed by quantitative RT-PCR (data not shown) and western blotting (Figure 5B). This result suggests that the low-level expression of Sin3A in the HUVECs compared to the other cell lines may restrict the inhibitory effect of ELK3 on MT1-MMP transcription. In addition, ELK3 overexpression resulted in the down-regulation of Sin3A protein expression (Figure 5C). Conversely, Sin3A overexpression also led to decreased ELK3 protein expression in HUVECs (Figure 5D). These results imply that the expression of ELK3 and Sin3A are reciprocally repressed in HUVECs. Furthermore, these findings also suggest that the transcriptional repressor activity of ELK3, which requires Sin3A as a co-repressor, is greatly attenuated by low levels of Sin3A in HUVECs while the inhibitory effect of ELK3 on ETS-1 activity is maintained. Therefore, it is indicative that the ELK3 inhibitory effect on the angiogenesis might not be due to direct transcriptional repressor effect but its indirect suppressive effect on the ETS-1's positive angiogenic activity.
Discussion
In this study, the roles of ELK3 on the angiogenesis were investigated. Our results showed that ELK3 knockdown led to reinforced tube formation increased Matrigel angiogenesis induced by VEGF. ELK3 inhibited MT1-MMP promoter activity induced by ETS-1 in HUVECs. Our results also demonstrated that Sin3A functions as a co-repressor with ELK3 on the MT1-MMP promoter in HUVECs.
The ELK3 expression was originally detected in murine adult heart tissues and fibroblast cell lines [18]. In human cell lines, we and other group previously showed that ELK3 expression was the highest in the HUVECs compared to other cell types and ELK3 was detected predominantly in the HUVECs along with the ETS transcription factors ERG and GABPα [10, 22]. The localization of ELK3 expression was restricted to the vascular endothelial cells during development and the disruption of ELK3 expression in mice resulted in dilated lymphatic vessels and fatality [19, 23]. These studies strongly suggested that ELK3 has important functions in endothelial cells and led us to study more specific roles of ELK3 in HUVECs.
ELK3 was initially identified as a member of the ETS family transcription factors in Ras-transformed mouse cells that were used to search for downstream targets of the Ras signaling pathway [18]. ELK3 was identified as a transcriptional repressor but, in a later study, it also acted as a transcriptional activator of Ras [18, 28, 29]. The co-transfection of ELK3 with Ras resulted in the up-regulated activity of the VEGF promoter, suggesting that VEGF is a target gene of ELK3 [21]. Additionally, ELK3 is phosphorylated by VEGF through the FGF2-Ras-MAPK pathway [10, 18, 21, 28-31]. Our previous study also demonstrated that ELK3 is translocated to the cytosol by the treatment of VEGF ([10]. In this study, VEGF was used to induce angiogenesis and reveal the function of ELK3 in the HUVECs. ELK3 knockdown reinforced tube formation in the HUVECs (Figure 1B) following VEGF treatment and enhanced VEGF-induced blood vessel formation in vivo (Figure 2). Our data imply that ELK3 acts as a repressor of VEGF-induced angiogenesis.
Several genes including Egr-1, VEGF, PAI-1 and HIF1α were reported as ELK3 target genes by forming ternary complexes [21, 23, 32, 33]. In this study, we investigated the effect of ELK3 knockdown on a subset of angiogenesis-related factors to determine if ELK3 regulates the expression of these genes. We observed that the expression of MT1-MMP and VE-cadherin were upregulated by ELK3 knockdown in the HUVECs (Figure 3). However, it appears that the transcriptional regulation of ELK3 on MT1-MMP is indirect because ELK3 itself has no or little effect on the MT1-MMP promoter activity but ELK3 inhibited the promoter activity elevated by the ETS-1. MT1-MMP promoter activity was not significantly affected by the transfection of ELK3 alone, but co-transfection of ELK3 and ETS-1 almost completely blocked ETS-1 transcriptional activation on the MT-MMP promoter (Figure 4). This inhibitory activity of ELK3 is accomplished through the activity of co-repressors. It was reported that ELK3/NET suppressed the CTα promoter-luciferase activity during the activation of CTα transcription by ETS-1 [34]. Thus, it is also plausible that ELK3 affects the transcriptional regulation of ETS-1 on other target gene promoter including MT1-MMP. ELK3 binding to CtBP, a co-repressor marked by its CtBP inhibition domain (CID), plays an important role as a link between CtBP and Ras functions [35]. It was also shown that the recruitment of the co-repressor Sin3A to the ETS transcription factor PU.1 was responsible for PU.1 repressor activity [34]. One possible reason why ELK3 does not directly inhibit MT1-MMP promoter activity is the reciprocal inhibition of ELK3 and Sin3A: enforced ELK3 expression resulted in reduced Sin3A expression (Figure 5C), and constitutive Sin3A expression almost completely ablated ELK3 expression (Figure 5D). We also demonstrated that ELK3 has a minimal direct inhibitory effect on MT1-MMP promoter activity in HUVECs compared to other cells such as HEK-293 and L-132 cells, while ETS-1 effectively activated the MT1-MMP promoter in HUVECs (Figure 5A). These observations may also be due to the reciprocal inhibition between ELK3 and Sin3A in HUVECs (Figure 5C & D). The inhibitory effect of ELK3 on the MT1-MMP was due to its inhibitory effect on the ETS-1 activity not by direct suppression of MT1-MMP promoter. This is partly due to the fact that the co-repressor Sin3A level is low in endothelial cells and thus does not work efficiently with Sin3A for direct transcriptional suppression in HUVECs. Instead, it seems that ELK3 binds to ETS-1 and inhibits it. Our data also showed that overexpression of Sin3A inhibit the ELK3 expression, and thus reduces the ELK3 activity. Therefore, it is plausible that low expression of Sin3A in endothelial cells maintains the ELK3 expression to some extent and thus can suppress the ETS-1 activity when a suppressive signal comes to endothelial cells.
In conclusion, this study demonstrated ELK3 knockdown enhanced on VEGF-induced angiogenesis through partially increasing the transcription activity of ETS-1 on MT1-MMP. Our study revealed that ELK3 is a repressor whose activity is modulated by a co-repressor Sin3A. However, ELK3 may have different effects on the regulation of target genes depending on the levels of co-repressors in different cell types; in endothelial cells, ELK3 repress the angiogenesis by inhibiting the activity of ETS-1 transcription factor rather than directly suppressing a target promoter such as MT1-MMP, since the expression level of co-repressor Sin3A is low in these cells.
Abbreviations
ETS: E26 Transformation-Specific Factor; ELK3: ETS-like transcription factor-3; EBS: ETS domain binding sites; ICAMs: intracellular adhesion molecules; JEX: JNK-induced export; MMPs: matrix metalloproteinases; TIMP: tissue inhibitor of metalloproteinases.
Supplementary Material
Additional File 1Supplementary Figure 1.
Acknowledgements
We thank Hye-Sim Cho for animal care. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2012M3A9C6049716 & 20110019355) and the Research Settlement Fund for new faculty of Seoul National University, Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Folkman J. Angiogenesis. Annual review of medicine. 2006;57:1-18
2. Carmeliet P. Angiogenesis in health and disease. Nature medicine. 2003;9:653-60
3. Jain RK. Molecular regulation of vessel maturation. Nature medicine. 2003;9:685-93
4. Sato Y. Role of ETS family transcription factors in vascular development and angiogenesis. Cell structure and function. 2001;26:19-24
5. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews. 2003;3:401-10
6. Sementchenko VI, Watson DK. Ets target genes: past, present and future. Oncogene. 2000;19:6533-48
7. Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827-37
8. Karim FD, Urness LD, Thummel CS, Klemsz MJ, McKercher SR, Celada A. et al. The ETS-domain: a new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes & development. 1990;4:1451-3
9. Nye JA, Petersen JM, Gunther CV, Jonsen MD, Graves BJ. Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes & development. 1992;6:975-90
10. Heo SH, Choi YJ, Ryoo HM, Cho JY. Expression profiling of ETS and MMP factors in VEGF-activated endothelial cells: role of MMP-10 in VEGF-induced angiogenesis. Journal of cellular physiology. 2010;224:734-42
11. Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nature reviews. 2002;2:657-72
12. Rundhaug JE. Matrix metalloproteinases and angiogenesis. Journal of cellular and molecular medicine. 2005;9:267-85
13. Randi AM, Sperone A, Dryden NH, Birdsey GM. Regulation of angiogenesis by ETS transcription factors. Biochemical Society transactions. 2009;37:1248-53
14. Kita D, Takino T, Nakada M, Takahashi T, Yamashita J, Sato H. Expression of dominant-negative form of Ets-1 suppresses fibronectin-stimulated cell adhesion and migration through down-regulation of integrin alpha5 expression in U251 glioma cell line. Cancer research. 2001;61:7985-91
15. Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M. et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Molecular and cellular biology. 2000;20:5643-52
16. Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. The EMBO journal. 1997;16:4374-83
17. Wang LC, Swat W, Fujiwara Y, Davidson L, Visvader J, Kuo F. et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes & development. 1998;12:2392-402
18. Giovane A, Pintzas A, Maira SM, Sobieszczuk P, Wasylyk B. Net, a new ets transcription factor that is activated by Ras. Genes & development. 1994;8:1502-13
19. Ayadi A, Suelves M, Dolle P, Wasylyk B. Net, an Ets ternary complex transcription factor, is expressed in sites of vasculogenesis, angiogenesis, and chondrogenesis during mouse development. Mechanisms of development. 2001;102:205-8
20. Giovane A, Sobieszczuk P, Mignon C, Mattei MG, Wasylyk B. Locations of the ets subfamily members net, elk1, and sap1 (ELK3, ELK1, and ELK4) on three homologous regions of the mouse and human genomes. Genomics. 1995;29:769-72
21. Zheng H, Wasylyk C, Ayadi A, Abecassis J, Schalken JA, Rogatsch H. et al. The transcription factor Net regulates the angiogenic switch. Genes & development. 2003;17:2283-97
22. Hollenhorst PC, Jones DA, Graves BJ. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic acids research. 2004;32:5693-702
23. Ayadi A, Zheng H, Sobieszczuk P, Buchwalter G, Moerman P, Alitalo K. et al. Net-targeted mutant mice develop a vascular phenotype and up-regulate egr-1. The EMBO journal. 2001;20:5139-52
24. Nagase H, Woessner JF Jr. Matrix metalloproteinases. The Journal of biological chemistry. 1999;274:21491-4
25. Itoh Y, Seiki M. MT1-MMP: an enzyme with multidimensional regulation. Trends in biochemical sciences. 2004;29:285-9
26. Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer letters. 2003;194:1-11
27. Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33-45
28. Ducret C, Maira SM, Lutz Y, Wasylyk B. The ternary complex factor Net contains two distinct elements that mediate different responses to MAP kinase signalling cascades. Oncogene. 2000;19:5063-72
29. Maira SM, Wurtz JM, Wasylyk B. Net (ERP/SAP2) one of the Ras-inducible TCFs, has a novel inhibitory domain with resemblance to the helix-loop-helix motif. The EMBO journal. 1996;15:5849-65
30. Wasylyk C, Zheng H, Castell C, Debussche L, Multon MC, Wasylyk B. Inhibition of the Ras-Net (Elk-3) pathway by a novel pyrazole that affects microtubules. Cancer research. 2008;68:1275-83
31. Wasylyk C, Criqui-Filipe P, Wasylyk B. Sumoylation of the net inhibitory domain (NID) is stimulated by PIAS1 and has a negative effect on the transcriptional activity of Net. Oncogene. 2005;24:820-8
32. Buchwalter G, Gross C, Wasylyk B. The ternary complex factor Net regulates cell migration through inhibition of PAI-1 expression. Molecular and cellular biology. 2005;25:10853-62
33. Gross C, Dubois-Pot H, Wasylyk B. The ternary complex factor Net/Elk-3 participates in the transcriptional response to hypoxia and regulates HIF-1 alpha. Oncogene. 2008;27:1333-41
34. Sugimoto H, Okamura K, Sugimoto S, Satou M, Hattori T, Vance DE. et al. Sp1 is a co-activator with Ets-1, and Net is an important repressor of the transcription of CTP:phosphocholine cytidylyltransferase alpha. The Journal of biological chemistry. 2005;280:40857-66 doi:10.1074/jbc.M503578200
35. Criqui-Filipe P, Ducret C, Maira SM, Wasylyk B. Net, a negative Ras-switchable TCF, contains a second inhibition domain, the CID, that mediates repression through interactions with CtBP and de-acetylation. The EMBO journal. 1999;18:3392-403
Author contact
![]() Corresponding author: Je-Yoel Cho, D.V.M., Ph.D. Department of Veterinary Biochemistry, College of Veterinary Medicine, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, Korea. Phone: +82-2-880-1268 Fax: +82-2-886-1268 E-mail: jeychoac.kr.
Corresponding author: Je-Yoel Cho, D.V.M., Ph.D. Department of Veterinary Biochemistry, College of Veterinary Medicine, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, Korea. Phone: +82-2-880-1268 Fax: +82-2-886-1268 E-mail: jeychoac.kr.