Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(5):466-478. doi:10.7150/ijbs.8337 This issue Cite
Research Paper
Involvement of PAR-4 in Cannabinoid-Dependent Sensitization of Osteosarcoma Cells to TRAIL-Induced Apoptosis
Antonietta Notaro1, Selenia Sabella1, Ornella Pellerito2, Riccardo Di Fiore1, Anna De Blasio1, Renza Vento1, Giuseppe Calvaruso1*, Michela Giuliano1* ![]()
1. Laboratory of Biochemistry, Department of Biological, Chemical and Pharmaceutical Sciences and Technologies, University of Palermo, Polyclinic, Palermo, Italy.
2. Laboratory of Cellular and Developmental Genetics, Department of Molecular Biology, Medical Biochemistry and Pathology, Faculty of Medicine, Université Laval, Québec, Canada.
* The authors share senior coauthorship.
Received 2013-12-11; Accepted 2014-2-27; Published 2014-4-8
Abstract
The synthetic cannabinoid WIN 55,212-2 is a potent cannabinoid receptor agonist with anticancer potential. Experiments were performed to determine the effects of WIN on proliferation, cell cycle distribution, and programmed cell death in human osteosarcoma MG63 and Saos-2 cells. Results show that WIN induced G2/M cell cycle arrest, which was associated with the induction of the main markers of ER stress (GRP78, CHOP and TRB3). In treated cells we also observed the conversion of the cytosolic form of the autophagosome marker LC3-I into LC3-II (the lipidated form located on the autophagosome membrane) and the enhanced incorporation of monodansylcadaverine and acridine orange, two markers of the autophagic compartments such as autolysosomes. WIN also induced morphological effects in MG63 cells consisting in an increase in cell size and a marked cytoplasmic vacuolization. However, WIN effects were not associated with a canonical apoptotic pathway, as demonstrated by the absence of specific features, and only the addition of TRAIL to WIN-treated cells led to apoptotic death probably mediated by up-regulation of the tumor suppressor factor PAR-4, whose levels increased after WIN treatment, and by the translocation of GRP78 on cell surface.
Keywords: Cannabinoids, ER stress, autophagy, TRAIL, osteosarcoma cells, GRP78/PAR-4 complex.
Introduction
Osteosarcoma is the most common malignant bone tumor in childhood and adolescence. Although the tumor can initially respond to chemotherapy, in patients with metastatic disease the prognosis remains adverse [1]. Therefore, a better understanding of osteosarcoma biology should be helpful to optimize treatment strategies, or develop new chemotherapeutic agents.
Cannabinoids (CBs), the active constituents of Cannabis sativa, are known to exert a wide range of central and peripheral effects. Recently, numerous studies evidenced the role of cannabinoids in the regulation of cell death and survival, focusing the anti-proliferative effects of these compounds in various tumours [2-5]. Cannabinoids can trigger several different signalling pathways. Following the interaction with the specific CB1 and CB2 receptors, cannabinoids have been shown to induce accumulation of ceramide [6-7] or activation of c-Jun N-terminal kinase (JNK) and p38 MAPK [8], as well as, increase in calcium concentration, ROS production, inhibition of PKA or NF-kB and modulation of pro- and anti-apoptotic members of the Bcl-2 family [9-10].
Recently, it has been demonstrated that cannabinoids can also activate autophagic process, an evolutionarily conserved mechanism that occurs in all eukaryotic cells [11]. In the first phase of autophagy a portion of the cytoplasm and other intracellular organelles are sequestered in double-membrane structures called autophagosomes. This process requires the formation of a multiprotein complex associated with the conjugation of phosphatidylethanolamine to microtubule-associated protein light chain 3 (LC3) and the conversion of soluble form LC3-I to autophagic vesicle-associated LC3-II form. Then, the autophagosomes fuse with lysosomes to form autolysosomes where the sequestered contents are degraded by lysosomal hydrolases and recycled in the cytosol. However, the effects of these events on cancer cells are not well known. A large body of evidence indicates a role for autophagy in sustaining cell survival [12]. In contrast, cell death resulting from progressive cellular consumption has been attributed to unrestrained autophagy, which can replace the classical apoptotic pathway, or cooperate with it [13].
In a recent study, it has been demonstrated that in human glioma cells, the main experimental model for the studies on cannabinoid action, the natural cannabinoid delta(9)-tetrahydrocannabinol induces autophagy-mediated cell death through stimulation of ER stress [14]. However the exact role exerted by autophagy in cannabinoid action remains unclear.
The aim of the present study was to investigate the effects induced by cannabinoids in osteosarcoma cells and the molecular pathway by them activated. Results indicated that in this experimental model the synthetic cannabinoid WIN 55,212-2 (WIN) induces morphological changes which are not associated with the induction of cell death but with the triggering of ER stress and autophagic process. These events make WIN able to sensitizing TRAIL-resistant MG63 and Saos-2 cells to TRAIL-induced apoptosis. Moreover, we indicate that a key role in WIN action is played by the tumor suppressor protein PAR-4.
Materials and Methods
Reagents
R-[2,3-Dihydro-5-methyl-3[(4-morpholinyl)methyl] pyrrolo[1,2,3,-de]-1,4-benzoxazin-6-yl]-1-naphthalenyl methanone mesylate (WIN55,212-2), anandamide (ANA), meth-anandamide (MethANA), 3-methyl-adenine (3-MA) and BAPTA-AM were purchased from Sigma, soluble human recombinant TRAIL/APO2L was obtained from PeproTech (EC Ltd., London, UK), benzyloxy-carbonyl-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk) from Promega (Madison, WI). Stock solutions were prepared in DMSO and opportunely diluted in culture medium except for 3-MA which was prepared in ethanol. The final concentration of DMSO or ethanol never exceeded 0.04%, which is a concentration that was experimentally determined to have no discernible effect. All antibodies used were purchased from Santa Cruz Bio (Santa Cruz, CA, USA), except for anti-procaspase-3, procaspase-8, Beclin-1 and PARP (Cell Signalling, Beverly, MA, USA), anti-LC3 (Novus Biologicals, Cambridge, UK), p62 and Actin (Sigma, MI, Italy).
Cell cultures
Human osteosarcoma MG63 and Saos-2 cells were acquired from Interlab Cell Line Collection (ICLC, Genova, Italy). Cells were cultured at 37° C in Dulbecco's modified Eagle medium (DMEM), supplemented with 10% (v/v) heat-inactivated foetal bovine serum (FBS), 2.0 mM L-glutamine, and antibiotic, anti-mycotic solution (100 U/ml penicillin, 100 μg/ml streptomycin and 250 ng/ml amphotericin B, Sigma) in a humidified atmosphere containing 5% CO2. For the experiments, cells were seeded at 60-70% confluence. After overnight incubation, culture medium was replaced with fresh DMEM containing low percentage of FBS (2%) and cells were treated with the compounds. Control cells were cultured in the presence of vehicle alone.
Cell viability assay
Cell viability was determined by 3-[4,5-dimethylthiazolyl- 2] 2,5-diphenyl-tetrazolium bromide (MTT) (Sigma Aldrich, Milan, Italy) assay as previously reported [15]. Briefly, after treatment in 96-well plates (triplicate wells for each sample), MTT stock solution was added to each well (final concentration 1 mg/ml) for 2 h. The medium was then removed and lysis buffer (0.1 ml) was added to dissolve the formazan product. Finally, the absorbance at 570 nm (test wavelength) and at 630 nm (reference wavelength) was measured using an ELISA microplate reader (Dynex Technologies). Data reported in the figures are the means ± S.E. of at least four independent experiments involving triplicate assays.
For acridine orange/ethidium bromide staining (AO/EB), cells were incubated on 96-well plates, treated with the compounds and stained with a solution composed of 100 μg/ml acridine orange plus 100 μg/ml ethidium bromide. Cells were examined under a Leica DM IRB inverted microscope (Leica Microsystems Srl, Milano, Italy) equipped with epifluorescence optics and suitable filters for FITC and rhodamine detection. Viable cells appear with green nuclei and dead cells have bright orange chromatin. Images were photographed and captured by a computer-imaging system (Leica DC300F camera and Adobe Photoshop for image analysis).
Cell cycle analysis
To analyse cell cycle distribution, after treatment with the compounds, cells were harvested by trypsinization (0.025% trypsin-EDTA; Life Technologies Ltd, Monza, Italy) and resuspended in a hypotonic solution containing 25 μg/ml propidium iodide, 0.1% sodium citrate, 0.01% Nonidet P-40 and 10 μg/ml RNase A. The cell cycle phase distribution was evaluated by Coulter Epics XL flow cytometer (Beckman Coulter) using Expo32 software. Cell debris and aggregates were excluded by opportune gating. The percentage of cells in sub G0/G1 phase was considered as an index of DNA fragmentation. The red fluorescence was measured in the FL3 channel using a 620 nm BP filter. At least 1 x 104 cells per sample were analyzed and data were stored in list mode files.
The results showed in the figures are representative of four independent experiments.
Autophagy analysis
Autophagy was examined by staining cells with acridine orange (AO) (2 μg/ml in PBS) at 37°C for 15 min in the dark. Images of AO staining were immediately visualized by fluorescence microscopy. To quantify the number of acidic vesicles, after staining with AO the cells were washed with PBS and analyzed by flow cytometric assay.
The formation of acidic vesicular organelles was also detected by monodansylcadaverine (MDC) staining. Cells were stained with 0.05 mM MDC in PBS at 37°C for 15 min. Following the incubation, cells were washed three times with PBS and immediately analyzed under fluorescence microscope by using DAPI filter.
Western blotting analysis
Protein extracts were prepared by washing the cells in PBS and incubating for 20 min in ice-cold lysis buffer supplemented with protease inhibitor cocktail, as previously reported [16]. After sonication for three times for 10 s, proteins were quantified by Bradford method and equal amount of proteins (40 μg) was separated by SDS-PAGE and then electrotransferred to a nitrocellulose membrane for the detection with specific antibodies. The blots were developed using the alkaline phosphatase colorimetric or ECL chemiluminescent labeling systems. Optical densities of the bands were analyzed with Quantity One Imaging Software from Bio-Rad Laboratories. The correct protein loading was verified by means of both Ponceau red staining and immunoblotting for actin. The results showed in the figures are representative of almost four independent experiments with similar results.
Gene silencing using siRNA
Small interfering RNAs (siRNAs) against CHOP (siCHOP) (D-004819-01-0005 and D-004819-02-0005) and scrambled siRNA (siScr), as a negative nonsilencing control, were purchased from Dharmacon RNA Technologies (Chicago, IL USA).
Cells (105) were seeded in six well plates and cultured in antibiotic-free DMEM medium supplemented with 2.0 mM L-glutamine, to reach about 50% confluence. Then, cells were transfected with 100 nM siCHOP in the presence of 6.2 µl Metafectene Pro (Biontex Laboratories GmbH, Martinsried/Planegg, Germany) in a final volume of 1 ml serum-free medium. The reaction was stopped after 6 h replacing the culture medium with DMEM +10% FBS and then silenced cells were treated with the compounds after 24 h. In CHOP silencing experiments, data reported were obtained by using siRNA duplex D-004819-02-0005 which resulted more efficacious than D-004819-01-0005.
Detection of surface GRP78 in non-permeabilized cells
To analyse cell surface GRP78, after treatment with the compounds, cells were washed, fixed, and incubated overnight with GRP78 primary antibody (H-129, Santa Cruz) in PBS containing 0.1% BSA at 4°C. Subsequently, cells were washed with PBS, incubated with FITC-conjugated secondary antibody for 1 h at 4°C. As controls, cells were incubated with the secondary IgG alone. Cell nuclei were counterstained with cell-permeant dye Hoechst 33342. Cells were examined under a Leica DM IRB inverted microscope (Leica Microsystems Srl, Milano, Italy) equipped with epifluorescence optics and suitable filters for FITC. Images were photographed and captured by a computer-imaging system (Leica DC300F camera and Adobe Photoshop for image analysis).
To quantify the level of cell surface GRP78, cells were harvested by trypsinization and resuspended in PBS containing 0.1% BSA, adjusting cell concentration to 1x106 cells/ml. Then, the cells, without fixing to allow detection of GRP78 at the cell surface, were incubated with GRP78 antibody (H-129, Santa Cruz) or isotype control for 30 min at 4°C. Then, cells were washed twice in 1 ml PBS to remove excess antibody. Appropriate volume of FITC-conjugated secondary antibody was added and cell suspension was incubated for 30 min at 4°C in the dark. Cells were washed twice in 1 ml PBS, resuspended in 0.5 ml PBS and acquired by using Coulter Epics XL flow cytometry (Beckman-Coulter) and Expo 32 software.
Statistical analysis
Cell viability data were expressed as the mean ± S.E. and evaluated by Student's t-test. Differences were considered significant when the p value was less than 0.05.
Results
Cytotoxic and morphological effects induced by cannabinoids in MG63 osteosarcoma cells
To examine whether cannabinoids can influence osteosarcoma cell proliferation, MG63 cells were treated for different times with various doses of the endogenous cannabinoid anandamide (ANA), its stable analogue meth-anandamide (MethANA), or synthetic cannabinoid WIN55,212-2 (WIN). Fig. 1A shows the results obtained after 48 h of treatment on cell viability measured by means of MTT assay. At the highest dose employed (20 μM), only WIN induced a significant reduction of cell number (about 50%), while the other cannabinoids induced modest effects. Time course analysis of WIN effect showed that the reduction of cell number was already evident after 8 h of treatment, as reported in Fig. 1B.
Cytotoxic and morphological effects induced by cannabinoids in human osteosarcoma MG63 cells. (A) Effects of anandamide (ANA), meth-anandamide (MethANA) and WIN on MG63 cell viability. Cells were incubated for 48 h with different doses (5, 10 and 20 μM) of cannabinoids. (B) Time and dose-dependent effects of WIN on MG63 cell viability and effect of 100 μM z-VAD in cells treated with 5 μM WIN for 36 h. In A and B, cell survival was estimated by MTT assay, as reported in Materials and Methods, and expressed as the percentage of control cells. Data are the means ± S.E. of four independent experiments involving triplicate assays. **p<0.01 versus untreated cells. (C) Fluorescent microscopic evaluation of MG63 cells after acridine orange/ethidium bromide staining (AO/EB). Cells were treated with 5 μM ANA, MethANA and WIN for 36 h. Then, after AO/EB staining, viable cells appear with green nuclei, while dead cells show red nuclei. (Original magnification 200X). (D) Morphological effects induced by cannabinoids after 36 h of treatment evaluated by phase contrast microscopy. (Original magnification 200X). The inset shows a magnification of the cell indicated by the arrow. In C and D, the images are representative of three independent experiments.
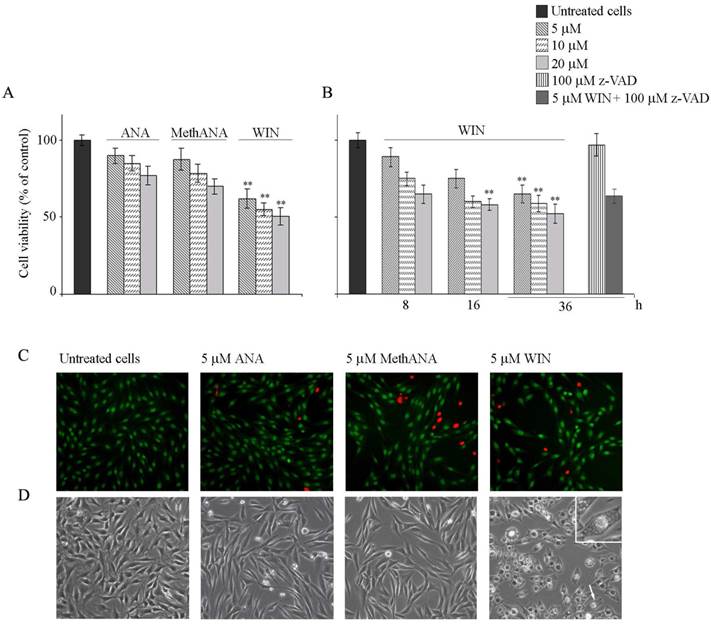
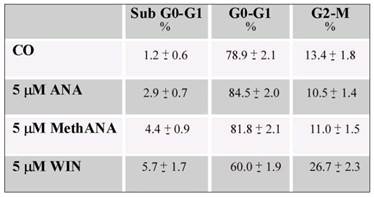
Next, we aimed to address whether the cell number reduction was a consequence of apoptotic cell death. For this purpose, cells were visualized under fluorescent microscopy after acridine orange/ethidium bromide staining. WIN treatment induced a non-significant induction of apoptosis; cells appeared with a diffuse green staining and only few red fluorescent nuclei, indicative of cell death, were observed (Fig. 1C). Also Hoechst 33258 staining seemed to indicate the absence of nuclear fragmentation (not shown). Moreover, the lack of cannabinoid-dependent MG63 cell death was confirmed by the analysis of cell distribution in the different phases of cell cycle which did not show relevant differences in the percentage of cells in sub G0/G1 phase of cell cycle indicative of DNA fragmentation. In the presence of WIN, we only observed an increase in the percentage of cells in G2/M phase which reached about 27% after 36 h of 5 μM WIN treatment (Table 1). Similar effects were obtained also after long periods of treatment (48 h) in the presence of WIN at the highest dose (20 μM) (Supplementary Fig. 1). Moreover, the addition of 100 μΜ z-VAD to WIN-treated cells did not induce any variation in cell viability with respect to cells treated with WIN alone, thus confirming the failure of WIN to induce apoptosis (Fig. 1B and supplementary Fig. 1).
Effects of cannabinoids on MG63 cell cycle distribution. Cells were treated with cannabinoids for 36 h. Both floating and attached cells were collected and processed for analysis of cell cycle distribution by flow cytometry following staining with propidium iodide. The percentage of cells in each phase of the cell cycle was calculated using Expo32 software. Data are means ± S.E. of three independent experiments.
Light microscopy analysis showed morphological aberrations in MG63 cells treated with cannabinoids. 5 μM ANA or MethANA induced the appearance of elongated, translucent and isolated cells. Differently, 5 μM WIN induced an increase in cell size, cytoplasmic contraction and the appearance of many vacuolar structures (Fig. 1D).
WIN triggers endoplasmic reticulum stress in MG63 osteosarcoma cells and induces the appearance of autophagic hallmarks
It is known that in many different experimental models cannabinoids act through a molecular mechanism that involves the induction of endoplasmic reticulum (ER) stress [14]. Thus, we were interested to study this relationship in our experimental model. Initially, in WIN-treated cells, we analysed the level of proteins which are considered ER stress markers such as CHOP, TRB3 and GRP78/Bip (GRP78). As shown in Fig. 2A, the level of CHOP, a transcription factor induced by cellular stresses such as protein misfolding in ER, time-dependently increased after treatment with 5 μM WIN. The effect was evident at 8 h of treatment reaching the maximum at 36 h. In the same time, also the level of TRB3, a target of CHOP, and GRP78, the main reticular HSP70, increased after WIN treatment in time-dependent manner.
WIN induces endoplasmic reticulum stress in osteosarcoma MG63 cells. (A) Time-dependent effect of 5 μM WIN and 5 ng/ml TRAIL on the level of CHOP, TRB3 and GRP78. The results were obtained by immunoblotting employing specific antibodies as reported in Materials and Methods. (B) Morphological effects exerted by the addition of BAPTA-AM to non-silenced or CHOP-silenced MG63 cells. Silencing of CHOP expression was carried out as reported in Materials and Methods. Cytoplasmic vacuolization was analysed by phase contrast microscopy after treatment for 24 h with 5 μM WIN and/or 5 μM BAPTA-AM. (Original magnification 200X). The images are representative of three independent experiments. (C) Effects of BAPTA-AM on the levels of CHOP and GRP78 proteins. Western blotting analysis was performed after treatment for 36 h with the compounds. In A and C, actin blots were included as a loading control.
Then, since it has been largely demonstrated the relationship between ER stress and cytoplasmic vacuolization [17], we investigated the effects of silencing the expression of the ER stress marker CHOP. After confirming the efficiency of CHOP silencing (not shown), we evaluated cell morphology in WIN-treated CHOP-silenced cells. As shown in Fig. 2B, CHOP silencing completely counteracted intracellular vacuolar formation induced by WIN, being cell morphology very similar to that observed in control cells. To further demonstrate the dependence of WIN-induced vacuolization by ER stress, we employed BAPTA-AM, a chelator of intracellular calcium. The addition of BAPTA-AM to WIN-treated cells counteracted cytoplasmic vacuolization both in non-silenced and CHOP-silenced MG63 cells (Fig. 2B). In the same way, BAPTA-AM also counteracted WIN-dependent increase in expression level of ER stress markers CHOP and GRP78 (Fig. 2C), confirming the dependence of WIN-induced morphological effect on ER stress.
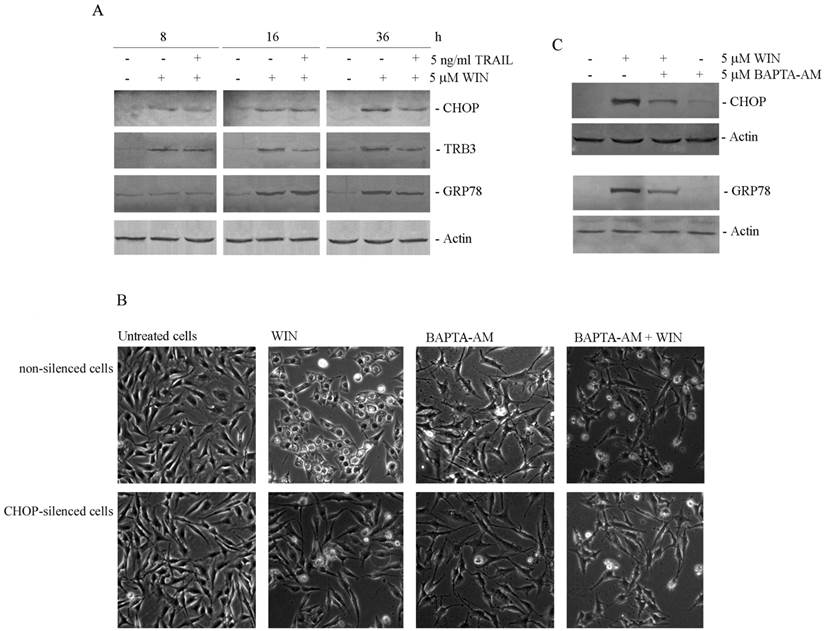
Then, we analysed whether the induction of ER stress was correlated with the activation of autophagic pathway. The microtubule-associated protein 1 light chain 3 (LC3), a marker of the presence of autophagosomes [18], is activated by a cleavage of the cytosolic form LC3-I to LC3-II, and recruited to double layered membrane of autophagosomes [19]. Immunoblotting analysis showed that the band corresponding to LC3-II increased in cells exposed to WIN in time-dependent manner reaching the maximum after 16 h of treatment (Fig. 3A). Differently, no variation was found in the level of Beclin1, a marker for autophagosome formation. The analysis of p62, a multifunctional protein involved in selective autophagy and considered as a marker to study autophagic flux [20], showed a progressive reduction of protein level, indicative of protein degradation.
WIN causes the appearance of autophagic hallmarks in MG63 cells. (A) Time-dependent effect of 5 μM WIN employed alone or in combination with 5 ng/ml TRAIL on the level of non-lipidated (LC3-I) and lipidated (LC3-II) forms of LC3, Beclin1 and p62. The levels of autophagic specific markers were analysed after 8, 16, 36 h of treatment. Cell lysates were analysed by immunoblotting employing specific antibodies as reported in Materials and Methods. Actin blots were included as a loading control. (B-C) Evaluation of autophagic vacuoles induced by WIN treatment, and effect of the autophagic inhibitor, 3-methyladenine (3-MA). MG63 cells were treated for 16 h with 5 μM WIN in the presence or absence of 5 mM 3-MA. After treatment, cells were incubated with MDC (0.05 mM) (B) or AO (2 μg/ml) (C) for 15 min at 37 °C and analyzed using a fluorescent microscope as described in Materials and Methods. (Original magnification 400X). The images are representative of three independent experiments. (D) Flow cytometric examination of red fluorescence emission (FL3) in WIN (red line) and WIN/3MA (green line) -treated cells with respect to control untreated cells (blue line). After incubation with the compounds, cells were treated with 2 μg/ml AO for 15 min at 37 °C. The plot is representative of three independent experiments.
The autophagic response to WIN was also studied by using monodansylcadaverine (MDC), a marker for the acidic compartment of autolysosomes [21]. As shown in Fig. 3B, treatment of MG63 cells with 5 μM WIN for 16 h caused the accumulation of punctate fluorescent structures, distributed within the cytoplasm or localized in the perinuclear regions. Acidic organelles (AVOs) were also detected after staining the cells with acridine orange (AO), a weak base that moves freely across biological membranes when uncharged. In AO-stained cells, the cytoplasm and nucleolus fluoresce bright green and dim red, whereas acidic compartments fluoresce bright red [22]. Fluorescent microscopic analysis showed that the number of acidic vesicles in WIN-treated MG63 cells increased compared to untreated cells (Fig. 3C). Data were confirmed by flow cytometric examination which evidenced the increase in the intensity of red fluorescence in WIN-treated cells (Fig. 3D).
In order to confirm the involvement of the autophagic pathway, 5 mM 3-methyladenine (3-MA), an inhibitor of the early stages of autophagy [23] was added to WIN-treated cells. In these conditions, a marked reduction of the number of fluorescent spots was already observed after 16 h of treatment (Fig. 3B, C and D).
WIN/TRAIL combined treatments induce apoptotic cell death mediated by PAR-4 protein
In a previous study, we demonstrated the ability of WIN to sensitize hepatocellular carcinoma cells to TRAIL-induced apoptosis [24]. Thus, we were interested to investigate the effects of the addition of different doses of TRAIL to cannabinoids in MG63 cells. MTT assay confirmed that MG63 cells are resistant to TRAIL (10 ng/ml), while the addition of TRAIL to WIN-treated cells resulted in a marked time-dependent reduction of cell viability which reached about 80% of the control value after 36 h (Fig. 4A). Differently, ANA/TRAIL or MethANA/TRAIL combinations (also at high doses) were almost ineffective in increasing the effects induced by cannabinoids alone (Fig. 4A).
WIN/TRAIL combined treatments induce apoptotic cell death. (A) Effects of cannabinoids and TRAIL on MG63 cell viability. Cells were treated with ANA, MethANA, WIN employed alone or in combination with TRAIL for 36 h at the indicated concentrations. (B) Morphological changes induced by WIN/TRAIL combined treatment in MG63 cells. Cells were treated for 36 h with 5 μM WIN and/or 5 ng/ml TRAIL and analysed by phase contrast microscopy. (Original magnification 200X). The images are representative of three independent experiments. (C) Effects of WIN/TRAIL combined treatment on MG63 cell cycle distribution. Flow cytometric analysis was carried out on propidium iodide stained cells treated for 36 h with WIN and/or TRAIL. The percentage of cells in each phase of the cell cycle was calculated using Expo32 software. (D) Effects of the addition of 5 mM 3-MA or 100 μM z-VAD to WIN or WIN/TRAIL treated cells for 36 h. In A and D, MG63 cell viability was estimated by MTT assay as reported under Materials and Methods and expressed as the percentage of control value. Data are the means ± S.E. of four independent experiments involving triplicate assays. **, p<0.01 versus untreated cells. (E) Western blotting analysis of caspase-8, pro-caspase-3 and PARP levels in MG63 cells. After treatment for 36 h with 5 μM WIN and/or TRAIL, cell lysates were analysed via immunoblotting using specific antibodies as reported in Materials and Methods. Actin blots were included as a loading control.
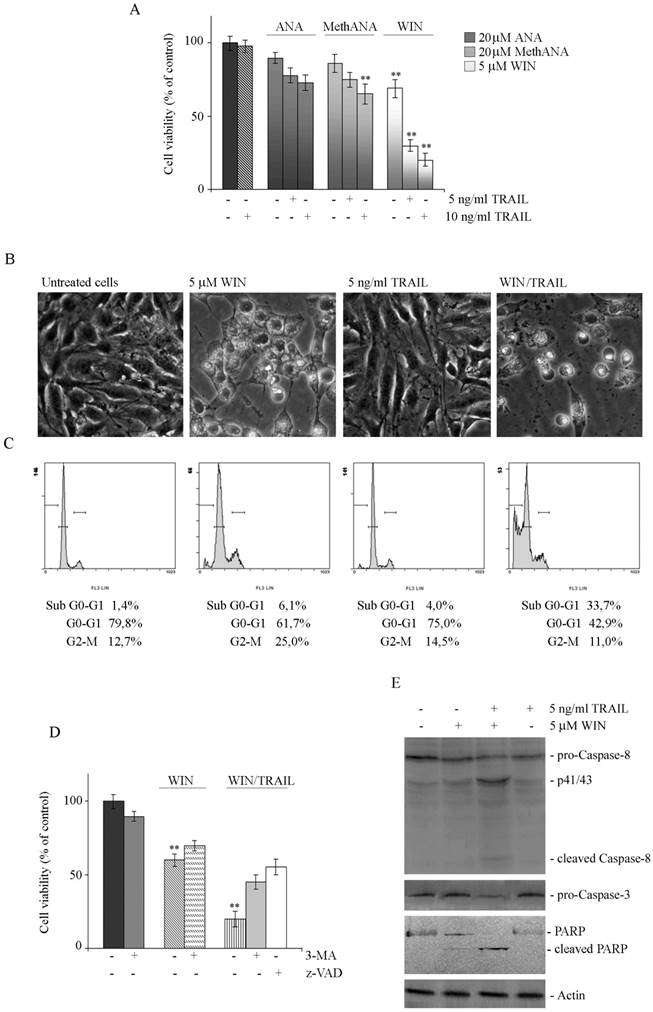
The reduction of viability induced by WIN/TRAIL combined treatment was accompanied by induction of cell death as demonstrated by light microscopic analysis which evidenced the appearance of the typical features of apoptosis consisting in rounded, detached cells floated in the medium (Fig. 4B). Apoptotic pathway was also confirmed by the analysis of cell cycle distribution. As shown in Fig. 4C in WIN/TRAIL treated cells the percentage of cells confined in sub G0/G1 phase of cell cycle increased, reaching a percentage of about 34% at 36 h of treatment.
The addition of 100 μM z-VAD to WIN/TRAIL treated cells counteracted cytotoxicity; in fact, after 36 h of treatment, the percentage of residual viability was comparable to that observed in cells treated with WIN alone (Fig. 4D). Western blotting analysis of the level of caspase proteases evidenced that WIN induced only a modest activation of caspase-8 while after WIN/TRAIL combined treatment we observed a marked reduction in the inactive, pro-enzymatic forms of caspase-8 and -3 and the concomitant appearance of the lower molecular weight, active fragments of caspase-8, indicative of activation of the extrinsic apoptotic pathway (Fig. 4E). The activation of executioner caspase-3 was also confirmed by the concomitant caspase-dependent cleavage of Poly-(ADP-ribose) polymerase (PARP), a target of caspase-3, which was evident in WIN/TRAIL treated cells (Fig. 4E). Taken together, data indicated that WIN was able to sensitize MG63 cells to TRAIL, inducing a canonical caspase-dependent apoptotic pathway.
Western blotting analysis showed that in WIN/TRAIL treated cells ER stress and autophagic protein markers remained at high levels (similar to those observed in the presence of WIN alone) after short incubation time points while after prolonged treatment we observed a modest decrease in the levels of proteins which can probably be dependent on the high percentage of dead cells (Fig. 2A and 3A).
To better investigate the contribution of autophagic process in cell death induced by WIN/TRAIL combined treatment we analyzed the effect of the addition of the autophagic inhibitor 3-MA on viability of WIN/TRAIL-treated cells. As shown in Fig. 4D, after 36 h of treatment with 5 mM 3-MA, the cytotoxic effects induced by WIN/TRAIL combined treatment were reduced.
Finally, we wondered what was the link between the ability of WIN to induce ER stress and the sensitization of MG63 cells to TRAIL action. Some evidence indicates that the tumor suppressor protein PAR-4 (Prostate Apoptosis Response-4, also known as PAWR) is able to activate apoptotic pathway by interacting with GRP78 and inducing its translocation on cell surface [25] where GRP78 behaves as a receptor for soluble ligands to induce apoptosis [26]. To explore the possible involvement of this pathway, we firstly analyzed the expression level of PAR-4 in WIN-treated cells. As shown in Fig. 5A, the level of PAR-4 increased after 8 and 16 h of treatment with WIN and further in WIN/TRAIL treated cells, indicating an actual involvement of this factor in the early phases of apoptotic process. In the presence of TRAIL alone, no variation in the level of PAR-4 was observed (not shown). Then, we detected the presence of GRP78 on cell membrane of non-permeabilized WIN-treated MG63 cells by immunofluorescence microscopy. As shown in Fig. 5B (upper panel), a marked increase in green fluorescence, indicative of an enhanced GRP78 expression on cell surface, was observed after WIN treatment for 24 h. The results were quantified by cytofluorimetric analysis that indicated an increase of about 2.5-fold in green fluorescence (lower panel).
WIN treatment induces PAR-4 upregulation and cell surface GRP78 translocation. (A) Time dependent effect of WIN or WIN/TRAIL combined treatment on the level of PAR-4 protein. After treatment with 5 μM WIN employed alone or in combination with 5 ng/ml TRAIL, cell lysates were analysed by immunoblotting using a specific antibody as reported in Materials and Methods. (B) Effects of WIN treatment on surface GRP78 levels. Upper panel: Determination of surface GRP78 by immunofluorescence. Cells were treated for 24 h with 5 μM WIN, incubated with anti-GRP78 antibody followed by FITC-conjugated secondary antibody and analyzed using an inverted fluorescent microscope as described in Materials and Methods. Nuclei were counterstained with Hoechst 33342 (blue). Lower panel: Cytometric analyses showing cell surface expression of GRP78 in MG63 cells. The open histograms indicate isotype control, filled histograms indicate the expression of GRP78 in untreated and WIN-treated cells. (C) PAR-4 level in CHOP-silenced cells. Silencing of CHOP expression was carried out as reported in Materials and Methods. After WIN treatment cell lysates were analysed by immunoblotting using specific antibody as reported in Materials and Methods. In A and C, actin blots were included as a loading control. (D) Effects of WIN/TRAIL combined treatment in CHOP silenced cells. Silencing of CHOP expression was carried out as reported in Materials and Methods. Cells were treated with 5 μM WIN and/or 5 ng/ml TRAIL for 24 h. MG63 cell viability was estimated by MTT assay as reported under Materials and Methods and expressed as the percentage of control value. Data are the means ± S.E. of four independent experiments involving triplicate assays. **, p<0.01 versus control untreated cells.
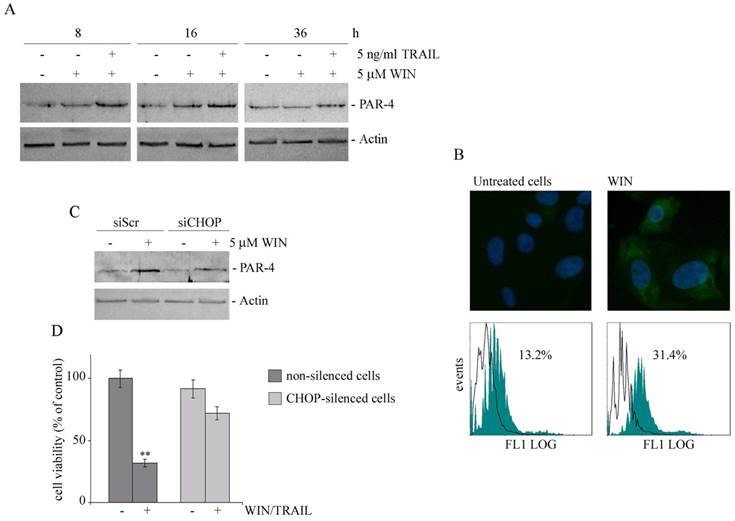
To analyze the relationship between ER stress, PAR-4 expression and cytotoxicity induced by WIN/TRAIL combined treatment, we analyzed the level of PAR-4 in CHOP silenced cells. As shown in Fig. 5C, in siCHOP WIN-treated cells the level of PAR-4 markedly lowered, indicating a plausible dependence of PAR-4 level on CHOP activity. Lastly, in CHOP silenced cells we also observed a significant recovery of cell viability (Fig. 5D).
Effects of WIN or WIN/TRAIL combined treatment in human osteosarcoma Saos-2 cells
Then, to evaluate whether the effects induced by WIN in MG63 cells could be reproduced in other osteosarcoma cell lines we performed some experiments in Saos-2 cells.
Fig. 6A shows the effects on cell viability induced in Saos-2 cells by anandamide (ANA), meth-anandamide (MethANA), or WIN55,212-2 (WIN) after 48 h of treatment. It is possible to observe that also in this cell line WIN was the most efficacious drug among the employed cannabinoids although overall its effects were slightly less evident than in MG63 cells. Moreover, time course analysis confirmed the results obtained in MG63 cells (Fig. 6B).
Effects of WIN and WIN/TRAIL treatment on Saos-2 cells. (A) Effects of anandamide (ANA), Meth-anandamide (MethANA) and WIN on Saos-2 cell viability. Cells were incubated for 48 h with different doses (5, 10 and 20 μM) of cannabinoids. (B) Time and dose-dependent effects of WIN on Saos-2 cell viability. In A and B, cell survival was estimated by MTT assay, as reported in Materials and Methods, and expressed as the percentage of control cells. Data are the means ± S.E. of three independent experiments involving triplicate assays. **, p<0.01 versus untreated cells. (C) Effect of WIN and/or WIN/TRAIL combined treatment on the level of CHOP and GRP78 proteins. (D) Time-dependent effect of WIN on the level of Beclin-1 and LC3 protein. (E) Effects of WIN or WIN/TRAIL combined treatment on Saos-2 cell viability. Cells were treated with WIN (5 μM) employed alone or in combination with TRAIL at the indicated concentrations. The effect of caspase inhibitor z-VAD (100 μM) at 36 h of treatment is also shown. Cell viability was estimated by MTT assay as reported in Materials and Methods and expressed as the percentage of control value. Data are the means ± S.E. of three independent experiments involving triplicate assays. **, p<0.01 versus control untreated cells. (F) Effect of WIN and/or WIN/TRAIL combined treatment on the level of PAR-4 protein. For western blotting analysis, after treatment with the compounds, cell lysates were analysed using specific antibodies as reported in Materials and Methods. Actin blots were included as a loading control.
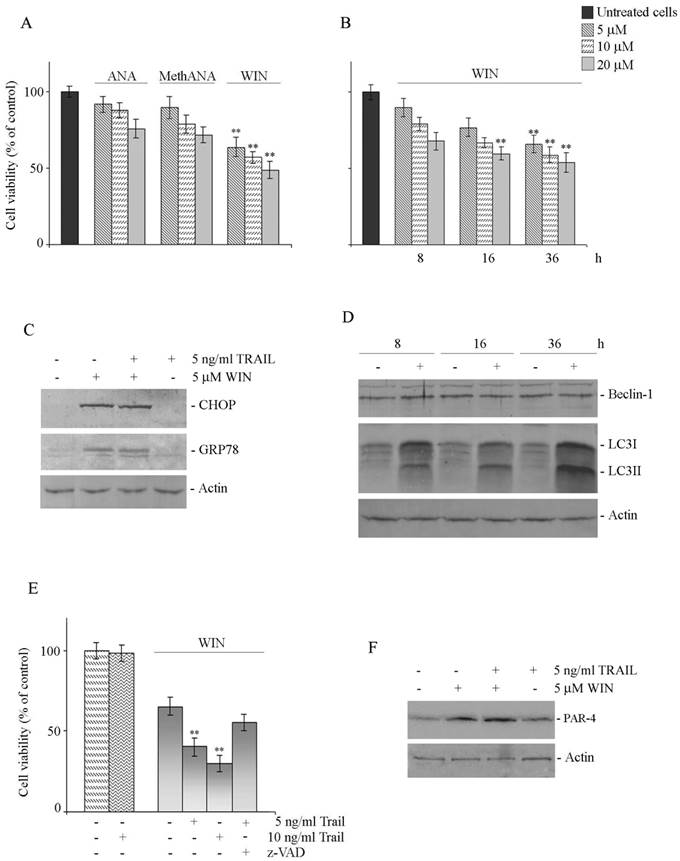
Similarly to MG63 cells, in Saos-2 cells WIN and WIN/TRAIL combined treatment induced ER stress and autophagy as demonstrated by the increase in the levels of CHOP and GRP78 (Fig. 6C) and in those of LC3 (Fig. 6D) while, also in these cells, it was not observed any variation in the level of Beclin-1 (Fig. 6D). WIN/TRAIL combined treatment resulted in a marked reduction of Saos-2 cell viability which was almost entirely counteracted by the addition of 100 μM z-VAD indicating an involvement of caspase activities in this event (Fig. 6E). Finally, in WIN or WIN/TRAIL treated cells we demonstrated a clear increase in the level of the tumor suppressor protein PAR-4 (Fig. 6F) thus suggesting a common mechanism that accounts for the effects of the drug in osteosarcoma cells.
Discussion
In this study we explored the effects induced by cannabinoids in osteosarcoma MG63 and Saos-2 cells. We found that among the three cannabinoids tested (the endogenous cannabinoid anandamide, its stable analogue meth-anandamide, and the synthetic cannabinoid WIN), only WIN caused a clear reduction in cell number and morphological alterations that, however, were not accompanied by induction of cell death. In particular, very interesting was the observation that WIN treatment induced cytoplasmic vacuolization in MG63 cells clearly visible under light microscopy. This event, which is normally calcium-dependent, may originate from multiple intracellular organelles such as endoplasmic reticulum, endosomes and lysosomes and can be interpreted as a specific phase of autophagic process [27]. Moreover, it is well known the relationship between cell vacuolization and ER stress and between cannabinoids and ER stress induction. Wasik et al. reported that in mantle cell lymphoma (MCL) cells WIN induces cytoplasmic vacuolization which originates from ER and is associated to programmed cell death of type III [28]. The observed up-regulation of CHOP, TRB3 and GRP78, the main ER stress markers, in MG63 cells supported the involvement of ER stress in WIN-induced vacuolization which was also confirmed by the loss of vacuolar structures observed both in CHOP-silenced WIN treated cells and in cells co-treated with WIN and BAPTA-AM, a chelator of intracellular calcium. Interestingly, the addition of BAPTA-AM also counteracted WIN-induced up-regulation of ER-stress markers, strongly sustaining the hypothesis that the increase in intracellular calcium level can be an early event in WIN pathway.
Frequently, ER stress is related to autophagic process [14]. Depending on the cell context, autophagy can counterbalance ER stress induction for cell survival or switch the cytoprotective function into cell death promoting pathway. The results obtained in osteosarcoma cells showed that WIN treatment induced the activation of LC3, an autophagosome marker, by promoting the cleavage of its cytosolic form LC3-I to LC3-II. The involvement of autophagy was also confirmed by the augmented accumulation of acidic intracellular compartments. Moreover, the observation that the level of p62 protein, a marker of autophagic degradation, decreased after long periods of WIN treatment, together with the appearance of acidic vacuoles suggested that WIN triggered a complete autophagic response, differently from that observed in MCL cells [28].
In spite of the induction of ER stress and autophagy, low doses of WIN were not capable to trigger significant cell death in osteosarcoma cell lines. However, such as previously demonstrated in hepatocellular carcinoma cells [24], WIN sensitized MG63 and Saos-2 cells to apoptosis induced by TRAIL, a promising anticancer agent [29-30]. Data reported in this paper indicate that the addition of low doses of TRAIL to WIN-treated cells was able to inhibit osteosarcoma cell growth (about 80% of cell viability reduction after 36 h of treatment) which was accompanied by the typical morphological and biochemical features of apoptotic cell death, as confirmed by the employment of the caspase inhibitor z-VAD. Also in this case, the endogenous cannabinoids showed a different behaviour than WIN; the addition of TRAIL to anandamide or meth-anandamide, in fact, did not modify the effects induced by the cannabinoids employed alone. Presently, we cannot explain the different response of MG63 cells to these combined treatments and new experiments are in progress to verify a potential connection between the induction of cytosolic vacuolization and the sensitization to TRAIL.
The observation that the addition of autophagy inhibitor 3-MA counteracted the cytotoxicity induced by WIN/TRAIL combined treatment suggested an involvement of autophagic process in WIN/TRAIL induced cell death. It has been reported that following TRAIL exposure cancer cells can induce a cytoprotective autophagy that blunts apoptosis and TRAIL antitumoral effect can be enhanced in the presence of autophagy inhibitors [31]. However, mounting evidence has suggested that autophagy can also be considered a caspase-dependent or -independent death pathway. In a recent paper Jin et al [32] observed that in papillary thyroid cancer cells autophagy inhibition desensitized cells to TRAIL-induced apoptosis even if the authors did not propose any hypothesis to explain the mechanism.
The crosstalk between apoptosis and autophagy in inducing cell death is strongly sustained by the observations that several molecules required for autophagy also play a key role in the regulation of apoptosis and viceversa [33-34]. In this connection, it has been demonstrated that the autophagosomal membrane can serve as a platform for the intracellular activation of caspase-8 thus permitting to initiate caspase cascade and apoptotic cell death [35]. In accordance to this evidence, our preliminary observations (data not shown) seem to indicate that in WIN/TRAIL treated cells the addition of autophagy inhibitor 3-MA counteracted the activation of caspase-8. This evidence could explain the role of autophagy in WIN-induced sensitization to TRAIL. However, further investigations are necessary to confirm this relationship.
After establishing the sensitization to TRAIL action induced by WIN, we further identified PAR-4 as a probable downstream factor. PAR-4, a tumor suppressor protein initially identified in prostate cancer cells, is characterized by the ability to selectively induce apoptosis in cancer cells through both an intracellular and an extrinsic pathway [36]. Although PAR-4 is often down-regulated in cancer, its re-expression sensitizes cancer cells, but not normal cells, to death following to oncogene inhibition or chemotherapy. It has been demonstrated that PAR-4 activates the translocation of Fas and FasL to the cell membrane and, at the same time, it inhibits the pro-survival transcriptional factor NF-κB [37-38]. Recently, it has been reported a relationship between PAR-4 and GRP78. In an elegant paper, Burikhanov et al [25] proposed a novel regulatory loop that involves the translocation of GRP78 to cell surface mediated by intracellular PAR-4 and its successive interaction with extracellular PAR-4. This mechanism can correlate ER stress with the activation of extrinsic apoptotic pathway. In osteosarcoma cells we observed a marked increase in PAR-4 level after WIN or WIN/TRAIL combined treatment and a concomitant translocation of GRP78 to cell surface. Differently from the Authors, we did not observe any increase in PAR-4 level in the presence of TRAIL alone, suggesting that the enhancement of PAR-4 level is related to ER stress induced by WIN. This hypothesis was confirmed by the reduction in PAR-4 level observed following to the silencing of ER stress marker CHOP which is accompanied by the recovery of cytotoxicity induced by WIN/TRAIL combined treatment.
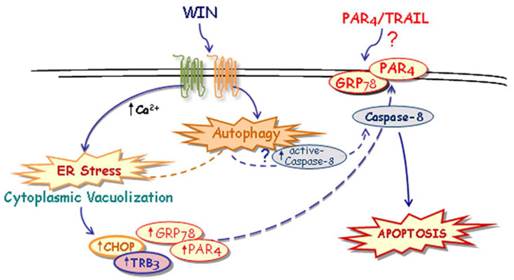
Taken together, although some important questions remain to be further clarified, here we hypothesize a novel mechanism, described in Fig. 7, through which WIN induces ER stress-dependent activation of autophagy and, in the same time, increases PAR-4 level and GRP78 translocation on cell surface. These events, together with autophagy-mediated activation of caspase-8, could contribute to the formation of a membrane microdomain and be responsible, directly or indirectly, for the sensitization to TRAIL action.
Suggested model of the mechanism of WIN/TRAIL-induced apoptosis on osteosarcoma MG63 cells.
Supplementary Material
Supplementary Figure 1Acknowledgements
The present study was partially funded by European Regional Development Fund, European Territorial Cooperation 2007-2013, CCI 2007 CB 163 PO 037, OP Italia-Malta 2007-2013; Italian Ministry of Education, University and Research (MIUR) ex-60%, 2012; MIUR-PRIN, contract number 2008P8BLNF (2008).
Conflict of Interests
The authors declare that they have no conflict of interest.
References
1. Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007;26:1-18
2. Bifulco M, Laezza C, Pisanti S, Gazzerro P. Cannabinoids and cancer: pros and cons of an antitumour strategy. Br J Pharmacol. 2006;148:123-35
3. Flygare J, Sander B. The endocannabinoid system in cancer-potential therapeutic target? Semin Cancer Biol. 2008;18:176-89
4. Sarfaraz S, Adhami VM, Syed DN, Afaq F. et al. Cannabinoids for cancer treatment: progress and promise. Cancer Res. 2008;68:339-42
5. Pisanti S, Bifulco M. Endocannabinoid system modulation in cancer biology and therapy. Pharmacol Res. 2009;60:107-16
6. Velasco G, Galve-Roperh I, Sànchez C. et al. Cannabinoids and ceramide: two lipids acting hand-byhand. Life Sci. 2005;77:1723-31
7. Galve-Roperh I, Sànchez C, Cortés ML. et al. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nature Med. 2000;6:313-9
8. Sarker KP, Biswas KK, Yamakuchi M. et al. ASK1-p38 MAPK/JNK signaling cascade mediates anandamide-induced PC12 cell death. J Neurochem. 2003;85:50-61
9. Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549-63
10. Pozzoli G, Tringali G, Vairano M. et al. Cannabinoid agonist WIN55,212-2 induces apoptosis in cerebellar granule cells via activation of the CB1 receptor and downregulation of bclxL gene expression. J Neurosci Res. 2006;83:1058-65
11. Yang Z, Klionsky DJ. An Overview of the Molecular Mechanism of Autophagy. Curr Top Microbiol Immunol. 2009;335:1-32
12. Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745-7
13. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891-906
14. Salazar M, Carracedo A, Salanueva IJ. et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359-72
15. Rubino S, Portanova P, Girasolo A. et al. Synthetic, structural and biochemical studies of polynuclear platinum(II) complexes with heterocyclic ligands. Eur J Med Chem. 2009;44:1041-8
16. Calvaruso G, Giuliano M, Portanova P. et al. G. Hsp72 controls bortezomib-induced HepG2 cell death via interaction with pro-apoptotic factors. Oncol Rep. 2007;18:447-50
17. Cheng CH, Shu KH, Chang HR, Chou MC. Cyclosporine-induced tubular vacuolization: the role of Bip/Grp78. Nephron Exp Nephrol. 2012;122:1-12
18. Kabeya Y, Mizushima N, Ueno T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720-8
19. Fujita N, Itoh T, Omori H. et al. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Mol Biol Cell. 2008;19:2092-100
20. Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32:431-6
21. Biederbick A, Kern HF, Elsässer HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3-14
22. Paglin S, Hollister T, Delohery T, Hackett N. et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439-44
23. Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889-92
24. Pellerito O, Calvaruso G, Portanova P. et al. The synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer binding protein homologous protein (CHOP)/death receptor 5 (DR5) axis. Mol Pharmacol. 2010;77:854-63
25. Burikhanov RK, Zhao Y, Goswami A. et al. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377-88
26. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181-8
27. Aki T, Nara A, Uemura K. Cytoplasmic vacuolization during exposure to drugs and other substances. Cell Biol Toxicol. 2012;28:125-31
28. Wasik AM, Almestrand S, Wang X. et al. WIN55,212-2 induces cytoplasmic vacuolation in apoptosis-resistant MCL cells. Cell Death Dis. 2011;2:e225
29. Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628-33
30. Zhang XD, Nguyen T, Thomas WD. et al. Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett. 2000;482:193-9
31. He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8:1811-21
32. Jin SM, Jang HW, Sohn SY, Kim NK, Joung JY, Cho YY, Kim SW, Chung JH. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine. 2014;45:256-62
33. Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy. “Is it your turn or mine”? Apoptosis. 2011;16:321-333
34. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing. Cross-talk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-52
35. Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455-68
36. Hebbar N, Wang C, Rangnekar VM. Mechanisms of apoptosis by the tumor suppressor Par-4. J Cell Physiol. 2012;227:3715-21
37. Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res. 2001;61:7255-63
38. Lee TJ, Jang JH, Noh HJ, Park EJ, Choi KS, Kwon TK. Overexpression of Par-4 sensitizes TRAIL-induced apoptosis via inactivation of NF-kappaB and Akt signaling pathways in renal cancer cells. J Cell Biochem. 2010;109:885-95
Author contact
![]() Corresponding author: Prof. Michela Giuliano, tel +39-91-6552473, fax +39-91-6552449; Email michela.giulianoit.
Corresponding author: Prof. Michela Giuliano, tel +39-91-6552473, fax +39-91-6552449; Email michela.giulianoit.