Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Infection by Various Pathogens
Atherosclerosis and...
Obesity and Insulin Resistance
Tumor
Asthma
Septic Complications
Conclusion and Perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(5):520-529. doi:10.7150/ijbs.8879 This issue Cite
Review
Macrophage Polarization in Inflammatory Diseases
Yan-Cun Liu1*, Xian-Biao Zou2*, Yan-Fen Chai1, Yong-Ming Yao2,3 ![]()
1. Department of Emergency Medicine, Tianjin Medical University General Hospital, Tianjin 300052, P.R.China;
2. Burns Institute, First Hospital Affiliated to the Chinese PLA General Hospital, Beijing 100048, P.R.China;
3. State Key Laboratory of Kidney Disease, the Chinese PLA General Hospital, Beijing 100853, P.R.China.
* contributed equally to the study.
Received 2014-2-19; Accepted 2014-4-8; Published 2014-5-1
Abstract
Diversity and plasticity are two hallmarks of macrophages. M1 macrophages (classically activated macrophages) are pro-inflammatory and have a central role in host defense against infection, while M2 macrophages (alternatively activated macrophages) are associated with responses to anti-inflammatory reactions and tissue remodeling, and they represent two terminals of the full spectrum of macrophage activation. Transformation of different phenotypes of macrophages regulates the initiation, development, and cessation of inflammatory diseases. Here we reviewed the characters and functions of macrophage polarization in infection, atherosclerosis, obesity, tumor, asthma, and sepsis, and proposed that targeting macrophage polarization and skewing their phenotype to adapt to the microenvironment might hold great promise for the treatment of inflammatory diseases.
Keywords: macrophage polarization, alternatively activated macrophage, signal pathways, inflammatory diseases, immune regulation.
Introduction
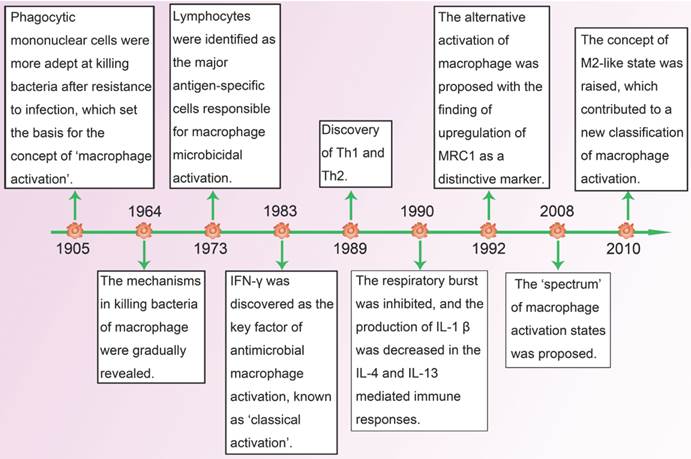
Macrophages were first identified by Elie Metchnikoff as phagocytic cells which helped to eliminate pathogens in both invertebrates and vertebrates. In 1905, his research findings suggested that macrophages from infected animals had elevated ability of killing bacteria, thereby proposing the basis of the concept of macrophage activation [1]. After six decades of efforts, the mechanisms with regard to killing bacteria of macrophages were gradually elucidated, but there were still no definite answers about how macrophages became more efficient bacterial killers. In 1973, North and his colleagues found that independent cellular factors could also promote resistance of infection without involvement of pathogens [2]. Almost at the same time, David indicated that lymphocytes were the major antigen-specific cells responsible for microbicidal activation of macrophages [3]. Soon after that, interferon (IFN)-γ, produced by lymphocytes, was identified as the first factor for interaction between macrophages and lymphocytes [4]. It transforms resting macrophages into active ones which have stronger antigen presenting capacity and complement mediated phagocytosis, and secrete more pro-inflammatory cytokines as well as toxic mediators. As the first type of antimicrobial macrophage activation was recognized, it became known as classically activated macrophages (CAM, also known as M1).
In 1989, when the heterogeneity in the helper T-cell compartment was subsequently reported, Mosrnann and Coffman reviewed the different functions and lymphokines secretion between two types of cloned helper T cells (Th), and proposed the concept of Th1 and Th2 [5]. One year later, Abramson and his colleagues recognized that interleukin (IL)-4, which was mainly produced by Th2 cells, could convert macrophages into a special activation state compared with IFN-γ induced activation in which respiratory burst was inhibited and major histocompatibility complex class II antigens (MHC-II) expression was increased [6]. With the discovery of up-regulation of macrophage mannose receptor (MRC1) as a specific marker of IL-4/IL-13-activated macrophages in 1992, which was coupled with the enhanced expression of MHC-II, the concept of alternatively activated macrophages (AAM, also known as M2) was first proposed [7]. In the following years, when the plasticity of macrophages in response to different environment was gradually studied, Mosser and Edwards reviewed the full spectrum of macrophage activation and pointed out that M1 and M2 were two terminals of the spectrum [8]. In addition to IL-4/IL-13, a great number of stimuli, such as antibody immune complexes together with lipopolysaccharide (LPS) or IL-1, transforming growth factor-β (TGF-β), glucocorticoids and IL-10, were found to have the ability of alternative activation of macrophages. As they shared properties with IL-4/IL-13-activated macrophages, a new functional state called M2-like phenotype [9] was proposed, and it held great promise for the research of macrophage activation in a dynamic microenvironment (Figure 1).
Timeline: advance in research of macrophage polarization.
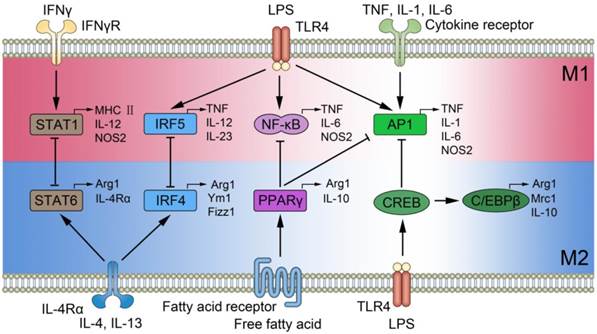
M1 phenotype macrophages express numerous pro-inflammatory mediators including tumor necrosis factor (TNF)-α, IL-1, IL-6, reactive nitrogen and oxygen intermediates, which have a strong microbicidal and tumoricidal activity; while M2 phenotype express molecules including resistin-like-α (also known as Fizz1), Arginase1 (Arg1), chitinase 3-like 3 (also known as Ym1), IL-10 and Mrc1 (also known as CD206), which are supposed to be involved in parasite infestation, tissue remodeling and tumor progression (immunoregulatory functions) [10]. M1 and M2 phenotype macrophages can be converted into each other in their specific microenvironment, and they are quite different with Th1 and Th2 [11]. Many key transcription factors are involved in macrophage polarization[12], like signal transducer and activator of transcription (STATs)[13], interferon-regulatory factor (IRFs)[14, 15], nuclear factor (NF)-κB [16], activator protein (AP) 1 [17], peroxisome proliferator-activated receptor (PPAR)-γ [18,19] and cAMP-responsive element-binding protein (CREB) [20], which interact with each other and regulate macrophages to certain phenotype in the various inflammatory diseases (Figure 2). Here we briefly review the polarization of macrophages and their functions in some typical inflammatory diseases.
Signal pathways of macrophage polarization. The figure illustrates several mechanisms underlying macrophage polarization and shows the feedback regulation between M1 and M2 signal pathways. Those include the activation of STAT1 mediated by IFN-γ receptor, increase in IRF5, NF-κB, as well as AP1 expression mediated by Toll-like receptor 4 (TLR4), enhanced AP1 expression mediated by cytokine receptor, activation of STAT6 and increased IRF4 mediated by IL-4 receptor, increased level of PPARγ mediated by fatty acid receptor, and enhanced expression in CREB by TLR4. The feedback regulation between M1 and M2 are implemented by STAT1-STAT6, IRF5-IRF4, NF-κB-PPARγ, AP1-CREB, and AP1-PPARγ, and they play essential roles in the initiation, development, and cessation of inflammatory diseases.
Infection by Various Pathogens
Bacteria
When tissues are challenged by pathogens, inflammatory monocytes in circulation are recruited and differentiated into macrophages, which keep a homeostatic status with the resident macrophages in the affected tissues. Generally, macrophages are deliberated to be polarized toward an M1 phenotype in the early stage of bacterial infection. When the pathogen associated molecular patterns (PAMPs) presented in bacteria are recognized by pathogen recognition receptors (such as Toll-like receptors, TLRs), macrophages are activated and produce a large amount of pro-inflammatory mediators including TNF-α, IL-1, and nitric oxide (NO), which kill the invading organisms and activate the adaptive immunity[11]. This mechanism has been considered to be involved in infection with Salmonella typhi, Salmonella typhimurium, Listeria monocytogenes [21], and the early phases of infection with Mycobacterium tuberculosis, Mycobacterium ulcerans, and Mycobacterium avium [22]. If macrophage-mediated inflammatory response cannot be quickly controlled, a cytokine storm is formed, thereby contributing to the pathogenesis of severe sepsis [23].
In order to counteract the excessive inflammatory response, macrophages undergo apoptosis or polarize to an M2 phenotype to protect the host from excessive injury and facilitate wound healing [24]. For example, microarray analysis and transcriptional profiling of peripheral blood cells showed that typical M1 genes and M1-related genes were replaced by M2 signature during treatment or convalescence in patients with typhoid fever [25]. LPS, large molecules in the outer membrane of gram-negative bacteria, play a critical pro-inflammatory role in acute infections. As the infection persists, host may present a LPS-tolerant state, and macrophages are polarized to M2 phenotype. A recent study has confirmed that the p50 subunit of NF-κB served as the key regulator of M2-driven LPS-tolerant state in this transformation [26]. As the excessive injury is reduced, however, M2 phenotype macrophages also induce an immunosuppressive state, resulting in a more susceptible situation to re-infection, thus relapse may occur or a carrier state may be found.
Virus
Macrophage polarization is also involved in virus infection, and M2 phenotype macrophages can suppress inflammation and promote tissue healing. Influenza virus augments the phagocytic function of human macrophages, which is a major feature of M2 phenotype, to clear the apoptotic cells and accelerate the resolution of inflammation [27]. In severe acute respiratory syndrome (SARS)-Cov infection, M2 phenotype macrophages are critical to regulate immune response and protect host from the long term progression to fibrotic lung disease by a STAT dependent pathway [28]. In addition, severe respiratory syncytial virus (RSV) induced bronchiolitis is closely associated with mixed M1 and M2 macrophages [29]. IL-4-STAT6 dependent M2 macrophage polarization can attenuate inflammation and epithelial damage, and cyclooxygenase-2 inhibitor, which increases expression of lung M2 macrophages, is proposed as a treatment strategy [29].
In acute virus infection, macrophage is recruited from the circulation in stimulation of chemokines and serves as a powerful killer of invading pathogens with the secretion of inflammatory mediators, such as TNF-α and inducible nitric oxide synthase [30]. However, the tissue-resident macrophage plays a completely different role in the virus infection. For instance, Kupffer cells (KCs) limit the severity of the virally infected liver through removing apoptotic hepatocytes in a manner dependent on scavenger receptors and thus play a major pathological role in chronic virus associated diseases [31]; alveolar macrophage is not only the monitor of clearing daily cellular debris, but also the initiator of a strong inflammation response to viral infection [32]. In addition, as the major target of HIV-1 infection, resident macrophage acts as viral reservoir which protects HIV-1 from hostile elements and induces HIV-1 associated neurological damage [33, 34].
Parasites
Both M1 and M2 phenotype macrophages are involved in parasite infestation, depending on the subtype and duration of parasite infestation models. In general, macrophages undergo a dynamic switch toward M2 phenotype. For instance, during the early stage of Taenia crassiceps infestation, it is characterized by responses of Th1-driven M1 phenotype macrophages, however, as infection goes to a late stage, Th2-driven IL-4-mediated M2 phenotype would become dominant with a decreased parasite burden [22]. In addition, different subtypes of parasite show different macrophage phenotypes during infection. Toxoplasma gondii has been shown to have three distinct clonal lineages, type I, type II, and type III, and they differ in virulence. Type I and III infected macrophages are alternatively activated through activation of STAT6 by Toxoplasma rhoptry kinase ROP16, but type II infected macrophages are classically activated through activation of NF-κB by Toxoplasma dense granule protein GRA15 [35]. Moreover, it is not a terminal differentiation for M2 phenotype in parasite infestation. In a recent study, utilizing a murine model of filarial infection demonstrated that macrophages which are exposed to Th2 cytokines and anti-inflammatory signals in vivo for a long time could still develop a classically activated phenotype in response to LPS or IFN-γ, and become antimicrobial through producing NO [36].
M2 phenotype macrophages are believed to play pivotal roles in regulating various pathologic features of helminth infestation, including suppression of T cell response, regulation of fibrosis, and formation of multinucleated giant cells in parasite-induced granulomas. Some important molecules are involved in this process. For example, in Leishmania infantum infection, Dectin-1 and mannose receptor, two kinds of C-type lectin receptors (CLRs) expressed on macrophages, respectively activate Sykp47phox and arachidonic acid-NADPH oxidase signaling pathways, and they are crucial for reactive oxygen species (ROS) production and also trigger Syk-coupled signaling for caspase-1 induced IL-1β release. In contrast, specific intercellular adhesion molecule-3-grabbing nonintegrin receptor 3 (SIGNR3), another kind of CLRs, helps parasite resilience through inhibition of the leukotriene (LTB) 4-IL-1β axis. Therefore, CLRs are key modulators for macrophage polarization, and are served as potential targets for prevention as well as treatment of Leishmania infantum infection [37]. Arg1 is not only an important marker of M2 phenotype macrophages but also a regulator of the immune response in parasite infestation. A study using myeloid cell restricted Arg1 deficiency Schistosoma mansoni infection model suggested that Arg1 appeared to be a key mediator for the development of helminth infestation through restraining both unrestricted Th2-mediated fibrotic pathology and intestinal damage associated with increased Th1/Th17 cytokines, nitric oxide synthase (NOS) 2 levels, and endotoxemia[38, 39]. However, it is not a generalized effect in the pathogenesis of parasite infestation. Arg1 blockade or deficiency in hematopoietic and endothelial cell lineages had little effect on response to acute orchronic infection by Trichuris muris[40].
Atherosclerosis and Cardiovascular Diseases
Atherosclerosis is a common type of degenerative disease of the vessel wall characterized by the accumulation of apolipoprotein B-lipoproteins in the inner lining of large and medium sized arteries [41]. It underlies the leading cause of death in developed countries and is likely soon to attain this status worldwide [42].
Monocytes and macrophages play essential roles in the development of atherosclerosis [43]. As the apolipoprotein B-lipoproteins accumulated, the endothelial cells become dysfunction and secrete a sum of chemokines, which interact with receptors on the circulating monocytes and promote them into the vessel wall. Those monocytes then transform into macrophages and take up cholesterol to give rise to a structure called atherosclerotic plaque [44]. As diseases develop, atherosclerotic plaque can grow larger, even become vulnerable and rupture, potentially resulting in a heart attack, stroke and even sudden cardiac death [41]. The fact that prevention of monocyte recruitment by blocking chemokines or their receptors could inhibit or slow down atherogenesis in mouse model of atherosclerosis, might provide strong support for the essential role of macrophages in the development of atherosclerosis[45].
In patients with unstable angina and myocardial infarction, the pro-inflammatory cytokines secreted by M1 phenotype macrophages were elevated, such as IL-6, with high levels predicating a poor outcome [46]. An in vitro study indicated that M1 phenotype macrophages could also induce smooth muscle cell proliferation and release of vasoactive molecules including NO, endothelins as well as eicosanoids, and they were important consequences for lipoprotein oxidation and cytotoxicity[47,48]. Early atherosclerotic plaques were infiltrated by M2 phenotype macrophages, however, along with the progression of the plaques, M1 phenotype macrophages gradually increased and occupied a major position [47], thereby more likely leading to an acute atherothrombotic vascular accident.
Both pro-atherosclerotic and anti-atherosclerotic functions have been demonstrated for M2 macrophages in atherosclerosis. IL-4 dependent macrophages expressed CD36, and they promoted oxidized low-density lipoprotein uptake and were abnormally high in patients with symptomatic atherosclerotic carotid plaques [49]. 15-lipoxygenase expressed by M2 macrophages linked to formation of foam cell and was critical for the development of atherosclerotic plaques [50]. TGF-β, secreted by M2 phenotype macrophages, inhibited the recruitment of inflammatory cells and was associated with a significant protection against atherosclerosis [51]. Efferocytosis, engulfing the apoptotic macrophages to prevent the formation of secondary necrosis, was defective in advanced atherosclerotic plaque and contributed to the formation of a vulnerable plaque [52].
Given the crucial roles of varied phenotypes of macrophages in promotion and progression of atherosclerosis, intervention with macrophage polarization may provide a novel therapeutic opportunity to atherosclerosis and cardiovascular diseases.
Obesity and Insulin Resistance
Obesity and its attendant metabolic disorder challenge the public health of modern society worldwide. Nearly 75% adults in America are overweight, and more than one-third of them are obese [53]. Furthermore, the persistent increase in obesity, especially in children, will halt or even decrease the life expectancy of America within the first half of this century [54]. As a disease with metabolic disturbance, obesity could lead to insulin resistance, glucose intolerance, dyslipidemia as well as hypertension. In addition, recent studies had uncovered that obesity was involved in cancers, hepatic and renal failure, thrombotic disease, and many infectious diseases [55]. Those findings suggest that obesity should pay more attentions than ever before.
Substantial evidences demonstrate that obesity is a chronic low-grade inflammatory disease [56]. An important initiator of the inflammatory reaction to obesity is adipose tissue, which is consisting of adipocytes, preadipocytes, endothelial cells and immune cells (e.g., macrophages and lymphocytes). In obesity, adipocytes can release pro-inflammatory mediators, such as CC chemokine ligand (CCL)-2, TNF-α, free fatty acids (FFAs), instead of leptin and adiponectin which promote insulin sensitivity in normal state [57]. Those pro-inflammatory mediators induce the recruitment and activation of adipose tissue macrophage (ATM). The activated ATM secrets pro-inflammatory cytokines and forms the inflammatory circuit which blocks the insulin action of adipocytes and leads to insulin resistance [58].
Analysis of ATM in obese mice revealed that these cells predominantly showed M1 phenotype, which was activated through TLR4/NF-κB and c-jun amino-terminal kinase (JNK) 1 signaling pathways [59]. They secreted pro-inflammatory cytokines with the participation of adipocytes, contributing to insulin resistance. Nevertheless, adipose tissue in lean animals also contains a moderate number of macrophages with a state of alternatively activated. In addition, a recent animal study demonstrated that F4/80+CD11c-CD301+ macrophages with M2-like characteristics were increased in adipose tissue during chronic weight loss. Those cells might play an essential role in lipolysis and tissue homeostasis by preventing inflammation and promoting insulin sensitivity [60]. Nguyen and colleagues recently confirmed a role for alternatively activated ATM in the orchestration of response to cold [61]. Adaptation to cold temperature promoted the ATM to the alternative activation state, which secreted catecholamines to induce the expression of thermogenic genes including PPARγ coactivator 1a (Ppargc1a), uncoupling protein 1 (Ucp1) and acyl-CoA synthetase long-chain family member 1 (Acsl1) in brown adipose tissue, and the lipolysis in white adipose tissue was also increased [61]. By this way, ATM keeps the cellular functions and physiological processes in the normal state when confronted with cold environments.
Macrophage polarization and switching in obesity together with insulin resistance can be modulated by life style, diet, humoral mediators and transcription factors, as Chinetti-Gbaguidi and Staels have revealed [62]. Investigation of the intrinsic relationships between various molecules and the corresponding receptors, which were activated in different orientations of macrophages, can lead to a possible therapeutic application in obesity and insulin resistance.
Tumor
It is widely accepted that smouldering inflammation is an essential cause of many cancers, and has been suggested as the 7th hallmark of cancer [63]. Similar to other chronic inflammation, cancer-related inflammation is also mediated by inflammatory mediators (chemokines, cytokines, and prostaglandins) and inflammatory cells, constituting a microenvironment for the initiation, growth and metastasis of cancers.
Tumor-associated macrophages (TAM), the major component of those inflammatory cells infiltrated in cancer, play critical roles in the formation of cancer. In the stage of tumor initiation, TAM releases a large amount of inflammatory mediators to create an inflammatory environment and promote tumor growth. When tumors progress to malignancy, TAM contributes to the angiogenesis, invasion, metastasis of tumors, and decreases the anti-tumor immunity. As tumors become metastatic, TAM infiltrates the target tissues and makes beds for the arrival of tumor cells. In addition, some TAM even changes its phenotype to help extravasation, survival, and subsequent growth of tumor cells [64].
It has been demonstrated that there is a strong link between poor prognosis and increased TAM in thyroid, lung, and hepatocellular tumors [64-66]. A recent experiment showed that an increased number of TAM was strongly associated with a shortened survival in classical Hodgkin's lymphoma patients [67]. Therefore, the population of TAM could be a biomarker for the risk stratification in tumors [68].
Although the previous hypotheses proposed an M1 role played by TAM in incipient and regressing tumors and necrotic areas of progressing tumors [68, 69], some recent studies supported an M2-like role according to its biological characters [70]. TAM is initially poor in producing NO and reactive oxygen intermediates which are the hallmarks of M1 phenotype macrophages. Then, in response to M1 signals including LPS and TNF-α, TAM shows a defective NF-κB activation together with low levels of IL-12, IL-1β, TNF-α as well as IL-6. Finally, markers of M2 such as Arg1, Ym1, FIZZ1, and MRC1 are all expressed on TAM. Nevertheless, a wide accepted view is that different infiltrating leukocytes, mediators, and signals in tumor microenvironments interact with TAM, forming a series of distinct phenotypes of TAM, in which M1/M2 designations represent two extreme ends of a scale [64].
TAMs vary in distinct phenotypes in the different stages of tumors. Previous data showed an M1-like role for TAM in the initiation and promotion of tumors with the activation of NF-κB signal pathway [71]. In liver tumors, macrophages (known as Kupffer cells) engulf and kill circulating tumor cells in an M1-like manner, and the depletion of Kupffer cells in rats enhances metastasis [72]. However, TAM is transformed into M2-like phenotype in the development of tumors, and NF-κB signal pathway is down-regulated [73]. During the stage of invasion and migration, TAM is converted into M2 phenotype upon the stimulation of IL-4 secreted by CD4+ T cells and tumor cells, and functions as the key to the gate of invasion of tumor cells [74]. Mechanistically, TAM migrates and synthesizes epidermal growth factor (EGF) under the stimulation of colony-stimulating factor (CSF)-1 secreted by tumor cells, in turn activating the migration of tumor cells [75]. More interestingly, TAM and tumor cells migrate in lock-step, and both migration and chemotaxis of them could be inhibited by either the EGF or CSF-1 signaling pathways [64]. Matrix metalloproteinases 2 and matrix metalloproteinases 9 secreted by TAM could impair the matrix, and help tumor cells escape from the basement membrane, which is another important regulator of invasion of tumor cells [76]. In a word, the distinct phenotypes of macrophages regulate the activity of tumor cells in the different stages of tumors, which provide an attractive target for the treatment in the future.
TAM polarization is also affected by the spatial location in tumor environment. For example, TAM in hypoxic zones of solid tumors may express Arg1, a marker of M2 phenotype, but this phenomenon is not found in areas near functional vasculature [77]. The high expression of Arg1 in hypoxic zones is related to hypoxia-inducible factor (HIF)-1 and HIF-2, which regulate the suppressive capabilities of TAM and contribute to the TAM recruitment in models of inflammatory hepatocellular and colon carcinoma[78, 79]. Similarly, cytokines secreted by tumor-infiltrating leukocytes in the microenvironment play a pivotal role in the polarization of TAM. For instance, IL-4/IL-13 secreted by tumor-infiltrating Th2 CD4+ T cells contributes to the formation of M2 phenotype. In addition, the intracellular molecules, such as high mobility group box 1 protein (HMGB1), can migrate to the outer space of TAM, bind to the TLRs, and promote the transformation of a pro-inflammatory phenotype in the development of tumors [80].
As a bidirectional modulator of immune response, TAM could definitely be a target for the treatment of tumors. TAM modulates the efficiency of chemotherapy, radiation therapy, and vascular-targeted therapies with monoclonal antibodies and immunotherapy in different stages of various tumors [81]. The fact that M1 phenotype plays an anti-tumor role in the initiation of tumors, while M2 phenotype promotes the development and metastasis of tumor give a new direction for the treatment of tumors. Reeducating TAM to an anti-tumor M1 phenotype from the pro-tumor M2 phenotype in the development and metastasis stages represent a pivotal target for the immunotherapy. A recent study indicated that histidine-rich glycoprotein (HRG) could induce macrophage polarization and vessel normalization through the down-regulation of placental growth factor (PlGF), and inhibition of tumor growth as well as metastasis [82]. As NF-κB signal pathway plays an important role in the polarization of macrophages and correlates with the prognosis of tumor [73], targeting NF-κB also means a novel direction for the treatment in the future. In brief, the recognition of mechanisms underlying multiple roles played by TAM in different stages of various tumors provides a basis for macrophage-centered therapeutic strategies.
Asthma
Asthma is a chronic inflammatory disease which is wide-spread in both developing and developed countries. Disorder in the regulation of phenotypes of pulmonary macrophages contributes to the pathogenesis of asthma [83]. It is widely accepted that M2 phenotype macrophages take a leading role in asthma, which they are beneficial for tissue repairing and restoration of homeostasis in the microenvironment of lung tissue. However, excessive M2 macrophages may increase cell recruitment and mucus secretion, and result in airway hyper-responsiveness. Moreira and colleagues transferred M2 phenotype macrophages into the lungs of fungus-induced asthmatic mice, then, both the inflammatory response and collagen deposition were enhanced, in turn accelerating the pathophysiological process of asthma [84]. With a boom in research of macrophage polarization, growing evidences support that M1 phenotype macrophages contribute to the development of asthma. In severe forms of asthma, especially in patients resistant to glucocorticoid therapy, macrophages are shown to become an M1 phenotype, which produces a great amount of pro-inflammatory mediators, including TNF-α, IL-1β, NO, exacerbates the lung injury and accelerate the airway remodeling [85]. For instance, NO produced by M1 phenotype leads to oxidative DNA damage and inflammation, enhances mucus production, and amplifies the lung injury in murine model of allergen-induced airway disease [86]. To sum up, regulation of the balance of macrophage polarization might provide a new way for the resolution of clinical asthma.
Septic Complications
Macrophage polarization is involved in the pathogenesis of severe sepsis, which represents an uncontrolled inflammatory response caused by infection or acute insults. Clinical or laboratory evidence for sepsis being an immunosuppressive disorder has been proposed by Hotchkiss [87]: apoptosis of the immune cells, relative increase in regulatory T cells [88] as well as myeloid derived suppressor cells [89], thereby weakening the defense to pathogens. Subsequently a secondary infection occurs which acting as the leading cause of death in patients with severe sepsis or septic shock. As a kind of fugitive cells which change their phenotypes in the various microenvironments, macrophages probably participate in the development of immunosuppressive state in septic response by polarizing to an M2 phenotype. Porta and colleagues found that LPS-tolerant macrophages shared the same characters with M2 macrophages, and p50 subunit of NF-κB was the key regulator to the process [26]. Therefore, uncovering the mechanisms in the polarization of macrophages in different stages of sepsis and regulating the balance of it may hold great promise for the treatment of septic complications.
Conclusion and Perspective
The concept of macrophage polarization was first defined in 1992, almost 80 years after Metchnikoff's initial description of macrophage activation [1, 7]. The alternatively activated macrophages secrete anti-inflammatory mediators, promote tissue repair, and regulate immune response in the initiation, development, and cessation of many inflammatory diseases through signal pathways including STATs, interferon regulatory factors (IRFs), NF-κB, AP1, PPARγ and CREB [12]. The important areas for future work should focus on exploring the full spectrum of macrophage polarization, uncovering the mechanisms with regard to the transformation of polarization, and finding the targets to the therapies of inflammatory diseases [22].
Nevertheless, several problems obstruct the research progress of macrophage polarization. Firstly, cellular markers that distinguish the phenotypes of macrophages during inflammatory diseases need further studies to confirm. For example, some recent studies found that it was not appropriate to identify TAM through the expression of CD68 as it was also expressed by other stromal populations, so the conclusions drawn from CD68 as the marker of TAM should be further evaluated [77, 90]. Secondly, it is usually not convenient to acquire fresh macrophages of human, so most of human studies now are conducted to perform from cell lines, which are not exactly perfect and hinder the transformation to the clinical trials. Thirdly, marked differences exist between mouse and human macrophages. For example, Arg1, marker of mouse M2 macrophages, is not expressed by in vitro polarized human macrophages [91]. Thus, it is difficult for the preclinical development of therapies based on mouse models if the differences between mouse and human could not be fully investigated [92].
Therapeutic targeting of macrophage polarization is in its infancy [22]. CCL2 inhibitors (a human study in vitro) and anti-CSF-1 antibodies (a study in mouse models) were shown to promote M2-like skewing of macrophage function to suppress tumor growth [93, 94], although their primary effects were blockers of monocyte and macrophage recruitment. PPARγ is a key molecule of M2 polarization in diabetes and obesity [95], and PPARγ inhibitors could decrease M2 polarization and improve insulin sensitivity in mouse adipose tissue which is the basic pathophysiological process of obesity and diabetes [96]. Through down-regulation of PIGF, it was reported that HRG polarized mouse TAM to M1-like and inhibited tumor growth as well as metastasis [82]. CD40 agonist also plays a potential role as a M1 promoter and increases a remarkable anti-tumor effect in pancreatic carcinoma in mice and humans [97].
Macrophage is an attractive therapeutic target, but it is necessary to understand their phenotypes according to their characteristics, anatomical location and origins (yolk sac, fetal liver, bone marrow)[11]. The microenvironment can regulate the phenotype and function of macrophages from their progenitors, lineage-specific differentiation to the fully differentiated stage [98]. Though organs contained resident tissue macrophages could be replenished after infection and other forms of tissue injury, the mechanisms that control initiation and differentiation of macrophage subsets remain mysterious. Future research should pay more attention to the anatomical location and the microenvironment of macrophage to restore the balance status.
In summary, transforming macrophages into a proper phenotype to regulate the initiation, development, and ending of inflammatory diseases by targeting molecules in signal pathways and local microenvironment will be an attractive area for the therapies of various inflammatory diseases.
Acknowledgements
This study was supported, in part, by grants from the National Natural Science Foundation (Nos. 81130035, 81372054, 81272090, 81121004), the National Basic Research Program of China (No. 2012CB518102), and the Medical Research Foundation of Chinese PLA (Nos. AWS11J008, BWS12J050).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Metchnikoff E. Immunity in the Infectious Diseases. New York: Macmillan. 1905
2. North RJ. Cellular mediators of anti-Listeria immunity as an enlarged population of short lived, replicating T cells. Kinetics of their production. The Journal of experimental medicine. 1973;138:342-55
3. David JR. Lymphocyte mediators and cellular hypersensitivity. The New England journal of medicine. 1973;288:143-9 doi:10.1056/NEJM197301182880311
4. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. The Journal of experimental medicine. 1983;158:670-89
5. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annual review of immunology. 1989;7:145-73 doi:10.1146/annurev.iy.07.040189.001045
6. Abramson SL, Gallin JI. IL-4 inhibits superoxide production by human mononuclear phagocytes. The Journal of immunology. 1990;144:625-30
7. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. The Journal of experimental medicine. 1992;176:287-92
8. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews immunology. 2008;8:958-69 doi:10.1038/nri2448
9. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature immunology. 2010;11:889-96 doi:10.1038/ni.1937
10. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593-604 doi:10.1016/j.immuni.2010.05.007
11. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445-55 doi:10.1038/nature12034
12. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nature reviews immunology. 2011;11:750-61 doi:10.1038/nri3088
13. Ohmori Y, Hamilton TA. IL-4-induced STAT6 suppresses IFN-gamma-stimulated STAT1-dependent transcription in mouse macrophages. The Journal of immunology. 1997;159:5474-82
14. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y. et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nature immunology. 2010;11:936-44 doi:10.1038/ni.1920
15. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature immunology. 2011;12:231-8 doi:10.1038/ni.1990
16. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nature immunology. 2011;12:695-708 doi:10.1038/ni.2065
17. Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Annals of the rheumatic diseases. 2011;70(Suppl 1):i109-12 doi:10.1136/ard.2010.140533
18. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L. et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116-20 doi:10.1038/nature05894
19. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S. et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell metabolism. 2007;6:137-43 doi:10.1016/j.cmet.2007.06.010
20. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N. et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17475-80 doi:10.1073/pnas.0908641106
21. Shaughnessy LM, Swanson JA. The role of the activated macrophage in clearing Listeria monocytogenes infection. Frontiers in bioscience: a journal and virtual library. 2007;12:2683-92
22. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of clinical investigation. 2012;122:787-95 doi:10.1172/JCI59643
23. Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The Pathogenesis of Sepsis. Annual review of pathology: mechanisms of disease. 2011;6:19-48 doi:10.1146/annurev-pathol-011110-130327
24. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews immunology. 2011;11:723-37 doi:10.1038/nri3073
25. Thompson LJ, Dunstan SJ, Dolecek C, Perkins T, House D, Dougan G. et al. Transcriptional response in the peripheral blood of patients infected with Salmonella enterica serovar Typhi. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22433-8 doi:10.1073/pnas.0912386106
26. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D. et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14978-83 doi:10.1073/pnas.0809784106
27. Hoeve MA, Nash AA, Jackson D, Randall RE, Dransfield I. Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PloS one. 2012;7:e29443. doi:10.1371/journal.pone.0029443
28. Page C, Goicochea L, Matthews K, Zhang Y, Klover P, Holtzman MJ. et al. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. Journal of virology. 2012;86:13334-49 doi:10.1128/JVI.01689-12
29. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC. et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal immunology. 2010;3:291-300 doi:10.1038/mi.2010.6
30. Liddiard K, Rosas M, Davies LC, Jones SA, Taylor PR. Macrophage heterogeneity and acute inflammation. European journal of immunology. 2011;41:2503-8 doi:10.1002/eji.201141743
31. Sitia G, Iannacone M, Aiolfi R, Isogawa M, van Rooijen N, Scozzesi C. et al. Kupffer cells hasten resolution of liver immunopathology in mouse models of viral hepatitis. PLoS pathogens. 2011;7:e1002061. doi:10.1371/journal.ppat.1002061
32. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nature reviews immunology. 2014;14:81-93 doi:10.1038/nri3600
33. Tan J, Sattentau QJ. The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends in microbiology. 2013 doi:10.1016/j.tim.2013.05.001
34. Herbein G, Varin A. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology. 2010;7:33. doi:10.1186/1742-4690-7-33
35. Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L. et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell host & microbe. 2011;9:472-83 doi:10.1016/j.chom.2011.04.015
36. Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. The Journal of immunology. 2009;182:3084-94 doi:10.4049/jimmunol.0803463
37. Lefevre L, Lugo-Villarino G, Meunier E, Valentin A, Olagnier D, Authier H. et al. The C-type lectin receptors Dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity. 2013;38:1038-49 doi:10.1016/j.immuni.2013.04.010
38. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM. et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS pathogens. 2009;5:e1000371. doi:10.1371/journal.ppat.1000371
39. Herbert DR, Orekov T, Roloson A, Ilies M, Perkins C, O'Brien W. et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. The Journal of immunology. 2010;184:6438-46 doi:10.4049/jimmunol.0902009
40. Bowcutt R, Bell LV, Little M, Wilson J, Booth C, Murray PJ. et al. Arginase-1-expressing macrophages are dispensable for resistance to infection with the gastrointestinal helminth Trichuris muris. Parasite immunology. 2011;33:411-20 doi:10.1111/j.1365-3024.2011.01300.x
41. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341-55 doi:10.1016/j.cell.2011.04.005
42. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G. et al. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948-54 doi:10.1161/CIRCULATIONAHA.109.192666
43. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161-6 doi:10.1126/science.1230719
44. Fuster JJ, Fernandez P, Gonzalez-Navarro H, Silvestre C, Nabah YN, Andres V. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovascular research. 2010;86:254-64 doi:10.1093/cvr/cvp363
45. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends in cardiovascular medicine. 2008;18:228-32 doi:10.1016/j.tcm.2008.11.004
46. Kirbis S, Breskvar UD, Sabovic M, Zupan I, Sinkovic A. Inflammation markers in patients with coronary artery disease--comparison of intracoronary and systemic levels. Wiener klinische Wochenschrift. 2010;122(Suppl 2):31-4 doi:10.1007/s00508-010-1343-z
47. Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M. et al. Macrophage plasticity in experimental atherosclerosis. PloS one. 2010;5:e8852. doi:10.1371/journal.pone.0008852
48. Tsimikas S, Miller YI. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Current pharmaceutical design. 2011;17:27-37
49. Handberg A, Skjelland M, Michelsen AE, Sagen EL, Krohg-Sorensen K, Russell D. et al. Soluble CD36 in plasma is increased in patients with symptomatic atherosclerotic carotid plaques and is related to plaque instability. Stroke, a journal of cerebral circulation. 2008;39:3092-5 doi:10.1161/STROKEAHA.108.517128
50. Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T. et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024-31 doi:10.1161/01.CIR.0000143628.37680.F6
51. Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R. et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circulation research. 2001;89:930-4
52. Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. Journal of leukocyte biology. 2009;86:1089-95 doi:10.1189/jlb.0209115
53. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA: the journal of the American Medical Association. 2010;303:235-41 doi:10.1001/jama.2009.2014
54. Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J. et al. A potential decline in life expectancy in the United States in the 21st century. The New England journal of medicine. 2005;352:1138-45 doi:10.1056/NEJMsr043743
55. Grundy SM. Metabolic complications of obesity. Endocrine. 2000;13:155-65 doi:10.1385/ENDO:13:2:155
56. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL. et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine. 2011;17:179-88 doi:10.1038/nm.2279
57. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell metabolism. 2012;15:432-7 doi:10.1016/j.cmet.2011.11.013
58. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 2010;72:219-46 doi:10.1146/annurev-physiol-021909-135846
59. Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nature clinical practice endocrinology & metabolism. 2008;4:619-26 doi:10.1038/ncpendmet0976
60. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R. et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. The Journal of clinical investigation. 2010;120:3466-79 doi:10.1172/JCI42845
61. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T. et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104-8 doi:10.1038/nature10653
62. Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Current opinion in lipidology. 2011;22:365-72 doi:10.1097/MOL.0b013e32834a77b4
63. Mantovani A. Cancer: Inflaming metastasis. Nature. 2009;457:36-7 doi:10.1038/457036b
64. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51 doi:10.1016/j.cell.2010.03.014
65. Wei Q, Fang W, Ye L, Shen L, Zhang X, Fei X. et al. Density of tumor associated macrophage correlates with lymph node metastasis in papillary thyroid carcinoma. Thyroid: official journal of the American Thyroid Association. 2012 doi:10.1089/thy.2011-0452
66. Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T. et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surgery today. 2012;42:1-7 doi:10.1007/s00595-011-0058-8
67. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T. et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. The New England journal of medicine. 2010;362:875-85 doi:10.1056/NEJMoa0905680
68. Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA. et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta neuropathologica. 2013;125:159-68 doi:10.1007/s00401-012-1056-7
69. Wang B, Li Q, Qin L, Zhao S, Wang J, Chen X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC immunology. 2011;12:43. doi:10.1186/1471-2172-12-43
70. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG. et al. Macrophage polarization in tumour progression. Seminars in cancer biology. 2008;18:349-55 doi:10.1016/j.semcancer.2008.03.004
71. Gordon S. Alternative activation of macrophages. Nature reviews immunology. 2003;3:23-35 doi:10.1038/nri978
72. Heuff G, Oldenburg HS, Boutkan H, Visser JJ, Beelen RH, Van Rooijen N. et al. Enhanced tumour growth in the rat liver after selective elimination of Kupffer cells. Cancer immunology, immunotherapy: CII. 1993;37:125-30
73. Pollard JW. Trophic macrophages in development and disease. Nature reviews immunology. 2009;9:259-70 doi:10.1038/nri2528
74. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL. et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes & development. 2010;24:241-55 doi:10.1101/gad.1874010
75. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER. et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer research. 2004;64:7022-9 doi:10.1158/0008-5472.CAN-04-1449
76. Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H. et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nature genetics. 2007;39:467-75 doi:10.1038/ng1997
77. Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends in immunology. 2012;33:119-26 doi:10.1016/j.it.2011.12.001
78. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG. et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer research. 2010;70:7465-75 doi:10.1158/0008-5472.CAN-10-1439
79. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ. et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. The Journal of clinical investigation. 2010;120:2699-714 doi:10.1172/JCI39506
80. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nature reviews Rheumatology. 2012;8:195-202 doi:10.1038/nrrheum.2011.222
81. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer cell. 2013;23:277-86 doi:10.1016/j.ccr.2013.02.013
82. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C. et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer cell. 2011;19:31-44 doi:10.1016/j.ccr.2010.11.009
83. Moreira AP, Hogaboam CM. Macrophages in allergic asthma: fine-tuning their pro- and anti-inflammatory actions for disease resolution. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2011;31:485-91 doi:10.1089/jir.2011.0027
84. Moreira AP, Cavassani KA, Hullinger R, Rosada RS, Fong DJ, Murray L. et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. The Journal of allergy and clinical immunology. 2010;126:712-21 doi:10.1016/j.jaci.2010.06.010
85. Kim YK, Oh SY, Jeon SG, Park HW, Lee SY, Chun EY. et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. The Journal of immunology. 2007;178:5375-82
86. Naura AS, Zerfaoui M, Kim H, Abd Elmageed ZY, Rodriguez PC, Hans CP. et al. Requirement for inducible nitric oxide synthase in chronic allergen exposure-induced pulmonary fibrosis but not inflammation. The Journal of immunology. 2010;185:3076-85 doi:10.4049/jimmunol.0904214
87. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. The Lancet infectious diseases. 2013;13:260-8 doi:10.1016/S1473-3099(13)70001-X
88. Jiang LN, Yao YM, Sheng ZY. The role of regulatory T cells in the pathogenesis of sepsis and its clinical implication. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2012;32:341-9 doi:10.1089/jir.2011.0080
89. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM. et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. The Journal of experimental medicine. 2007;204:1463-74 doi:10.1084/jem.20062602
90. Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2796-801 doi:10.1073/pnas.1104303108
91. Raes G, Van den Bergh R, De Baetselier P, Ghassabeh GH, Scotton C, Locati M. et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. The Journal of immunology. 2005;174:6561-2
92. Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. Journal of leukocyte biology. 2011;89:557-63 doi:10.1189/jlb.0710409
93. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. The Journal of biological chemistry. 2009;284:34342-54 doi:10.1074/jbc.M109.042671
94. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. The Journal of immunology. 2007;178:5245-52
95. Lu M, Sarruf DA, Talukdar S, Sharma S, Li P, Bandyopadhyay G. et al. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nature medicine. 2011;17:618-22 doi:10.1038/nm.2332
96. Charo IF. Macrophage polarization and insulin resistance: PPARgamma in control. Cell metabolism. 2007;6:96-8 doi:10.1016/j.cmet.2007.07.006
97. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612-6 doi:10.1126/science.1198443
98. Liu G, Yang H. Modulation of macrophage activation and programming in immunity. Journal of cellular physiology. 2013;228:502-12 doi:10.1002/jcp.24157
Author contact
![]() Corresponding author: Yong-Ming Yao, MD, PhD, Department of Microbiology and Immunology, Burns Institute, First Hospital Affiliated to the Chinese PLA General Hospital, Beijing 100048, People's Republic of China. Tel: (+86)1066867394; Fax: (+86)1068989955; E-mail: c_ffcom.
Corresponding author: Yong-Ming Yao, MD, PhD, Department of Microbiology and Immunology, Burns Institute, First Hospital Affiliated to the Chinese PLA General Hospital, Beijing 100048, People's Republic of China. Tel: (+86)1066867394; Fax: (+86)1068989955; E-mail: c_ffcom.