Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Estrogen receptor status and...
BRCA1 and mammary epithelial...
Direct interactions of BRCA1 and...
BRCA1 and E-ER co-regulate cell...
ROS management by BRCA1 and E-ER
C-MYC and CYCLIN D1 as common...
Correlation of BRCA1 and E-ER in...
Correlation between BRCA1 and...
Summary
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(5):566-575. doi:10.7150/ijbs.8579 This issue Cite
Review
BRCA1 And Estrogen/Estrogen Receptor In Breast Cancer: Where They Interact?
Li Wang, Li-Jun Di ![]()
Faculty of health sciences, University of Macau, SAR of People's Republic of China.
Received 2014-1-14; Accepted 2014-3-24; Published 2014-5-14
Abstract
BRCA1 mainly acts as a tumor suppressor and BRCA1 mutation correlates with increased cancer risk. Although it is well recognized that BRCA1 related tumorigenesis is mainly caused by the increased DNA damage and decreased genome stability, it is not clear that why BRCA1 related patients have higher risk for cancer development mainly in estrogen responsive tissues such as breast and ovary. Recent studies suggested that BRCA1 and E-ER (estrogen and estrogen receptor) signaling synergistically regulate the mammary epithelial cell proliferation and differentiation. In this current presentation, we reviewed the correlation between mammary gland epithelial cell transformation and the status of BRCA1 and ER. Then the mechanisms of BRCA1 and E-ER interaction at both gene transcription level and protein-protein interaction level are discussed. Furthermore, the tumorigenic mechanisms are discussed by focusing on the synergistic effect of BRCA1 and E-ER on cell metabolism, ROS management, and antioxidant activity in mammary gland epithelial cells. Also, the possibility of cell de-differentiation promoted by coordinated effect between BRCA1 mutation and E-ER signal is explored. Together, the currently available evidences suggest that BRCA1 mutation and E-ER signal together, contribute to breast tumorigenesis by providing the metabolic support for cancer cell growth and even may directly be involved in promoting the de-differentiation of cancer-prone epithelial cells.
Keywords: BRCA1, estrogen, estrogen receptor, cell metabolism, mammary gland development, breast cancer, ROS, oxidative stress.
Introduction
The survival rate of breast cancer patients has been improved from around 75% in 1970' to over 90% in 2013 owing to the improvement in early diagnosis and more effective treatment. But this disease still kills over 40000 women in US alone each year. Therefore, breast cancer is still a big challenge for etiological study, diagnosis and treatment [1]. Breast cancer can be divided into those with identified genetic risk factors and those what are not. The former group accounts for only around 10% of total breast cancer cases. Among that, inherited gene mutations such as BRCA1 and BRCA2 mutation lead to over 90% of inherited breast cancer. The majority of breast cancers are sporadic caused by more complicated reasons such as the activation of mitogenic signaling pathways, loss of tumor suppressor expression epigenetically etc. The breast cancer can also be further defined as basal-like, luminal A, luminal B, and HER2 (human epidermal growth factor receptor 2) positive subtypes according to the gene expression profile of the tumor cells [2, 3]. Both luminal A and luminal B are ER positive and respond very well to the hormone therapies. The drugs targeting HER2 also significantly increase the therapeutic effect of HER2 positive tumors [2, 4]. But the treatment for basal-like subtype of breast cancer is still very inefficient. This type of breast cancer is the most aggressive subtype with worse outcome in clinical treatment and account for 15-20% of breast cancers. Also, basal-like breast cancer is frequently showed as PR (progesterone receptor), ER (estrogen receptor) and HER2 negative (or triple negative) and therefore, has no obligation to hormone therapy. The basal-like subtype of breast cancer is also highly heterogeneous. Within the basal-like subtype, a significant fraction of patients are BRCA1 mutation carriers [5, 6].
BRCA1 is a nuclear phosphoprotein that plays an important role by acting as a genome surveillance factor and forming numerous different complexes that are involved in various cellular processes, including DNA damage repair, cell cycle checkpoint control, apoptosis regulation and gene transcriptional regulation [7-12]. BRCA1 gene was firstly mapped to 17q21 and cloned in 1994 [13]. Mutation of BRCA1 associates with almost half of inherited breast cancer cases. While more than 90% of breast cancer cases are sporadic, half of them have decreased BRCA1 expression owing to the BRCA1 promoter hypermethylation. Homogenous deletion of Brca1 gene locus in mouse results in embryonic lethality. Therefore, most of BRCA1 mutation carriers have one allele of normal functional Brca1 gene expression. BRCA1 mutation carriers have much higher rate of developing cancer in hormone responsive tissues such as breast, ovary and prostate by comparing to other tissues [14-17]. Intriguingly, hormone therapy is not feasible to treat BRCA1 mutation related breast cancer because it generally shows basal-like characters and is triple negative.
In mammary tissue, estrogens are the primary female sex hormones and play important role in reproductive organs such as mammary tissues and ovary. Estrogen executes its function via binding to ERs, generating either genomic effects or non-genomic effects [18]. There are two types of ER expressed in mammalian cells including ERα and ERβ. But only ERα knockout affects mammary development in mice and leads to curtailed duct elongation, suggesting ERα has much significant roles in regulating breast development than ERβ [19-22]. Also, it is ERα that was well known for its interaction with BRCA1. So all the ER we discussed hereafter means ERα. ER regulates gene expression by binding to ERE (estrogen receptor elements) directly or binding to other transcriptional factors such as AP1 or SP1 [23]. The major effect of ER is stimulating cell proliferation, probably by up-regulating protein synthesis genes and cell cycle regulating genes such as c-Myc and Cyclin D1 in response to estrogen [24-26].
BRCA1 acts as the inhibitor of E-ER signaling by interacting and inhibiting ER or inhibiting downstream effectors of ER. The functional interaction between E-ER and BRCA1 ensures the quality of replicated genome DNA when the cells experience proliferation under mitogenic effect of E-ER. When BRCA1 is in absence or insufficient, the balance is break down and the cells start to accumulate genomic mutations, contributing to the oncogenic transformation of mammary epithelial cells. In this review, we would like to summarize the recent progresses in E-ER and BRCA1 related disciplines such as the status of both ER and BRCA1 in mammary gland epithelial cell transformation, interactions between E-ER and BRCA1 at gene transcriptional regulation level and protein-protein interaction level. Also, the tumorigenic mechanisms associated with BRCA1 and E-ER interactions will be discussed.
Estrogen receptor status and epithelial cell transformation
ER mainly expresses in less than 25% of luminal epithelial cells and has no expression in basal or stromal cells [27]. It is interesting to observe that there is no correlation between the proliferating cell marker Ki67 and ER positive cells, suggesting that not all the ER positive cells actively proliferate. But more than 90% of mammary gland epithelial cell proliferation is contributed by luminal epithelial cells [28]. Therefore, it was proposed that the ER positive cells actually promote neighbor cells proliferating by secreting paracrine growth factors [29, 30]. Consistently, by exposing ER negative mammary epithelial cells to ER positive cells, these ER negative cells regain the proliferation and contribute to mammary gland development [19]. In BRCA1 related breast cancer, around 70~80% of the cases are ER negative and only less than 20% of cases are ER positive, while ER positive cases are more prevalent in sporadic breast cancer [31]. The ER+ BRCA1 related breast cancers are age dependent and the clinical characters are distinct from the basal features associated with ER- BRCA1 related breast cancers, leading to the suspicion that these cancers are incidental ductal carcinomas. But the slightly increased aggressive phenotype observed only in ER+ BRCA1 related tumors, not in the ER positive sporadic tumors, may also indicate an unknown mechanism [31, 32]. Nevertheless, ER positive cells also contribute to the tumorigenesis in ER- BRCA1 related tumors [31, 33-35]. In Li W. et al.'s study, the tumor growth, upon loss of BRCA1, associates with the increased ER positive mammary epithelial cells depending on the activation of MAPK/ERK pathway [34]. Jones LP. et al. reported that exogenous estrogen was able to stimulate the proliferation of the mammary epithelial cells and tumor growth in BRCA1 mutant mice [35]. A common observation in these studies is that ER positive cells gradually disappear along with the growth of tumor. Importantly, the approaches aiming to reduce the estrogen effect by either prophylactic oophorectomy or Tamoxifen decrease the breast cancer risk in BRCA1 mutation carriers, suggesting E-ER signaling is critical to promote the tumor growth [36, 37]. Thus, if the mammary epithelial cells that have oncogenic transformation are from ER negative cells, these ER negative cells may gradually outgrow the surrounding ER positive cells. ER positive cells may contribute to the oncogenic transformation of ER negative cells by providing mitogenic signaling stimulation. However, it could also be true that ER positive cells themselves experience oncogenic transformation and de-differentiation (see below), resulting in loss of ER expression. Actually, a recent study indicated that over-expression of stem cell specific transcriptional factors such as OCT-4, SLUG, and NANOG in MCF-7 cells reduces ER expression, increases the stem cell features, and enhances the cell mobility [38]. Interestingly, the same set of transcriptional factors promotes the oncogenic transformation of ER negative mammary epithelial cells MCF-10A [39], indicating the cells having oncogenic transformation could be irrelevant to their ER status. The detailed mechanism of ER status conversion in BRCA1 related tumors still needs to be further studied.
BRCA1 and mammary epithelial cell transformation
Although BRCA1 is mainly a well-known genome surveillance factor, it was also observed to be essential for proper mammary epithelial cell differentiation. In BRCA1 mutation carriers, blocking of progenitor cell differentiation may associate with increased cell proliferation as was observed in several mouse models [40-42]. Because BRCA1 related tumors show basal like features, the earlier studies mainly focused on the correlation between basal cells and BRCA1 related tumors. Actually, several studies observed that BRCA1 related tumors come from the transformation of expanded basal cells [10, 40, 43]. By analyzing the cell surface markers of in vitro cultured and basal cell generated mammary spheres, BRCA1 is found to be essential for the basal progenitor cell differentiation, because loss of BRCA1 blocks the differentiation process [40]. A dosage effect of BRCA1 in regulating the differentiation of mammary gland epithelial cells has also been proposed, suggesting gradually increased BRCA1 is critical to promote the appropriate differentiation of mammary epithelial progenitor cells [44]. However, several recent studies demonstrated that the BRCA1 related cancer is caused by the transformation of luminal progenitor cells with BRCA1 mutation [10, 33, 45, 46]. These experimental evidences have been obtained in both mouse models and human breast samples, highly suggesting that it is conservative that BRCA1 mutated luminal cells give rise to BRCA1 related tumors [47]. But why these tumors show more basal-like features such as K14+, p63+, ER- and other histological characters instead of luminal tumor features is unknown. However, the flexibility in luminal cell differentiation suggests that BRCA1 mutation may promote the conversion of luminal progenitor cells to basal-like progenitor cells, because most of the luminal progenitor cells have the ability to give rise to all types of mammary epithelial cells [47]. A recent study also observed that the differentiated luminal cells can be converted back to mammary stem cells driven by transcriptional factors only such as SOX9 and SLUG [48]. Consistent to this observation, Proia et al. observed that SLUG is a possible mediator of expansion of luminal progenitor cells promoted by BRCA1 mutation because BRCA1 indirectly represses Slug expression [45]. Actually, deficiency of BRCA1 associates with increased Slug expression in both breast cancer cells and human primary cells [45], suggesting that BRCA1 repressing of Slug may be critical to promote the differentiation of mammary epithelial cells. In contrast, a recent report found that SLUG represses BRCA1 by epigenetically silencing Brca1 loccus as a downstream effect of canonical WNT signaling. Repression of Brca1 expression by SLUG is essential for WNT mediated EMT (epithelial mesenchymal transition) [49]. EMT is defined as the malignant transformation of tumor cells and requires the reactivation of many genes that overlap significantly with the signature genes of stem cells [50]. Therefore, the genes that promote EMT may be able to promote other de-differentiation processes as well.
Direct interactions of BRCA1 and ER: protein-protein interactions and transcriptional regulations
There are several different ways for BRCA1 interacting with E-ER. For instance, BRCA1 is able to repress ER transactivation function by forming complex with ER directly. ER is also the substrate of BRCA1 ubiquitin ligase activity and can be mono-ubiquitinized. Mutation of the ubiquitinization sites abolished BRCA1 mediated inhibition of ER activity [51]. In addition, BRCA1 is able to inhibit P300 mediated ER acetylation [51-53], while acetylation of ER by P300 is essential for its transactivation function [54, 55]. Further work disclosed that BRCA1 also directly regulates the expression of p300 and quantitatively influences P300 [56]. Overall, BRCA1 negatively regulates the ER activity through P300 with multiple mechanisms involved. Another study reported that BRCA1 also represses the synergistic effect between ER and PR, leading to the repressed PR and probably its mitogenic effect [57]. Decreased BRCA1 expression also results in the ligand-independent ER activation, partially through activation of PI3K-AKT signaling [58], suggesting BRCA1 is also a repressor of PI3K-AKT signaling. Interestingly, inhibiting PI3K-Akt pathway eliminates Brca1 expression in triple negative breast cancer cells [59], further indicating that BRCA1 is required to restrict the PI3K-AKT signaling. These data suggested that BRCA1 is a critical guardian factor for mitogenic pathways such as the E-ER and PI3K-AKT.
E-ER regulates BRCA1 mainly through affecting its expression. It is well established that BRCA1 gene is an E-ER responsive gene. However, there are contradictory observations of whether ER directly regulates BRCA1 expression by binding to its promoter. Several genome wide studies on ER binding profile failed to find ER at BRCA1 promoter [60-64] . However, it deserves to point out that all these ER studies capture the immediate responses of cells within less than one hour after estrogen treatment. Actually, other studies observed the “delayed” response of Brca1 expression to estrogen [65-67]. It could be owing to the delay for new protein synthesis because the de novo protein synthesis inhibitor, cycloheximide is able to inhibit the BRCA1 gene expression in response to estrogen [65, 68, 69]. The peak Brca1 expression appears after twenty four hours treatment by estrogen in MCF-7 cells. So more likely, BRCA1 is not a first response gene to estrogen as described by Kininis et al [60]. However, it is still too early to rule out the possibility that ER could directly regulate Brca1 expression in a much delayed fashion after estrogen stimulation.
Interestingly, it was shown that BRCA1 is able to promote expression of Esr1, the coding gene for ER, via Oct-1 in breast cancer cells [70]. This data has been used to explain the exclusive association between ER negative character and BRCA1 related breast tumors [71]. But this observation is inconsistent with the repressive function of BRCA1 on ER mediated transactivation [51-53]. Moreover, this data can't fit into the novel observation that BRCA1 related breast tumor cells is originally from the luminal progenitor cells, because the luminal progenitor cells are not exclusively ER negative. Also, neoplastic transformation of mammary epithelial cells may be irrelevant to the ER status. So it is quite possible that BRCA1 is not the essential regulator of Esr-1 expression, although it may be a true activator. Moreover, it was found that BRCA1 actually represses the estrogen synthesis gene aromatase [72].
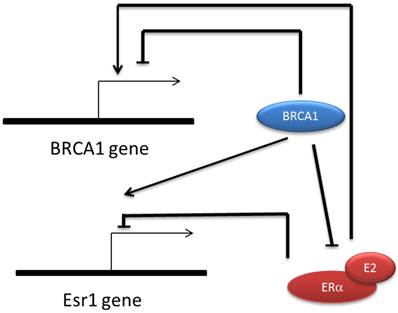
It was also found that both Brca1 and Esr1 are auto-regulated genes, ie. BRCA1 and ER repress their own promoter respectively [73, 74]. BRCA1 represses its own promoter by forming complex with E2F-RB and CtBP [65, 73]. ER repressing Esr1 expression is estrogen dependent and requires a co-repressor factor SIN3A which is an important adaptor for recruiting extra catalytic modules like histone methyl-transferase, DNA methyl-transferase, chromatin remodeling factors etc. [74]. Because BRCA1 and ER cause totally opposite effect on cell proliferation, the regulatory link between them ensures a balance that directs the appropriate cell growth. Also, the multi-channel regulatory connection between ER and BRCA1 further supports the role of BRCA1 in prohibiting cancer occurrence in estrogen responsive tissues (Fig.1).
Regulatory relationship between BRCA1 and ERα. BRCA1 transcription is repressed by its own protein but can be activated by E-ER indirectly. ESR1 expression is also repressed by its own protein and activated by BRCA1 directly. BRCA1 inhibit the transactivation function of ERα via several mechanisms including mediating the ubiquitinization of ER, competitively binding to p300 and transcriptionally repress p300 expression.
BRCA1 and E-ER co-regulate cell metabolism
Mitogenic function of E-ER relies on the presence of sufficient supply of nutrients such as glucose, because E-ER signaling also promotes the glycolysis and Krebs cycling [75]. A recent work, however, reported that estrogen up-regulates glycolysis via activation of PI3K-AKT signaling pathway, promotes cell proliferation under high glucose condition and represses Krebs cycle simultaneously [76, 77]. This is similar to the situation in proliferating cancer cells that consume glucose and rely on glycolysis over Krebs cycle in generating ATP, which is termed as “Warburg effect” [78]. However, when the extracellular glucose decreases, estrogen treatment activates mitochondria respiration via up-regulating PDH (pyruvate dehydrogenase) activity and repressing glycolysis [76], suggesting estrogen's effect on cell metabolism is adaptable and is under control of glucose availability. In the scenarios of cancer prone condition, glucose is frequently enriched. Estrogen probably promotes the cell proliferation by stimulating the anabolic metabolism. In fact, release of glycolysis proteins into plasma precedes the diagnosis of ER+ breast carcinoma [79], suggesting E-ER signaling promoted glycolysis is a very early event that associates with tumorigenesis. It was shown that the genes maximally induced by estrogen treatment after relatively long time (160 mins) incubation have the top hit of GO (gene ontology) term “cellular biosynthetic process” by ontology analysis [67]. These observations indicate E-ER signaling plays an important role in promoting tumor growth. But the E-ER signaling may also have its own risk management strategy because BRCA1 is responsive to E-ER signaling, and the response of BRCA1 needs to be mediated by CtBP and the cell metabolite NADH [65]. Estrogen was found to be able to activate tumor suppressor gene expression via manipulation of the cellular metabolism status globally [65]. Although BRCA1 function in regulating cell metabolism pathways has just been realized, several recent findings suggested that BRCA1 is a negative regulator of anabolic cell metabolism. BRCA1 has been shown to negatively regulate Igf-1 expression and mediate phosphorylated AKT degradation [80, 81]. Also, BRCA1 directly inhibits ACC (acetyl-CoA carboxylase) by interacting with it [82]. ACC catalyzes the converting of Acetyl-CoA to malonyl-CoA during fatty acid synthesis which is essential for tumor cell growth [83]. Since de novo fatty acid synthesis frequently associates with cancer cell growth, and probably the EMT, it suggests BRCA1 has novel tumor repressor function by controlling fatty acids metabolism. Thus, E-ER activated BRCA1 expression forms an important negative regulatory feedback that slows down the anabolic process promoted by E-ER.
ROS management by BRCA1 and E-ER
The direct consequence of abnormal cell metabolism is the rising ROS in cancer cells. Estrogen was shown to be an important contributor to ROS (reactive oxygen species). ROS is generated in many of the biological processes such as the NADPH oxidation and oxidative-phosphorylation coupled respiration. Increased ROS associates with increased macromolecules damages, especially the damages to DNA. Therefore, ROS was considered as a fundamental reason for cellular senescence and oncogenic transformation. The primary reason for estrogen promoted ROS is because of mitogenic function of estrogen that is most likely ER dependent. For instance, E-ER increases the cellular anabolic metabolism by up-regulating c-Myc [24]. However, it was also shown that estrogen promotes ROS in ER independent manner by directly affecting mitochondria [84].
BRCA1 is also a regulator of ROS. BRCA1 repressing of ROS is part of the integrative function of BRCA1 as a genome surveillance factor. BRCA1 regulates ROS via regulating NRF2, a critical transcriptional factor of several important anti-oxidant genes such as Are, Nqo1, and Hmox1 etc. [85, 86]. A recent study discovered that PI3K-AKT activation promotes a novel function of NRF2 that redirects the cell metabolism from aerobic to anaerobic respiration [87]. This study disclosed that ROS management and cell metabolism is interconnected by NRF2 in proliferating cells. Since proliferating cells prefer to tolerate relative higher amount of ROS, NRF2 participation in regulating cell metabolism may be beneficial to boost the threshold that cells can tolerate. Because part of E-ER non-genomic signaling is through PI3K-AKT activation, it indicates that E-ER probably activates NRF2 function in redirecting the cell metabolism pathways as well, which compromises the ROS pressure brought by the mitogenic effect of estrogen. In view of the important role of BRCA1 in maintaining NRF2 stability and its antioxidant ability [88], it is predictable that loss of BRCA1 may also increase the cellular oxidant stress, which will further exaggerate the risk of DNA damages.
Recent findings suggested that combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer by inducing genomic instability [89, 90]. The combined effect is more significant than application of any one of these inhibitors respectively. The reason is because PI3K inhibition increases DNA damage and further decreases the DNA damage repair ability in BRCA1 dificient cells. Since PI3K-AKT is able to activate NRF2, it is possible that increased PI3K-AKT compromises the risk of damage brought by the ROS in BRCA1 defect cells, through NRF2 mediated antioxidant pathway [87]. In fact, PI3K is frequently activated owing to the constitutive mutation or losing of PTEN, the repressor of PI3K activation. Thus, these studies indicate that PI3K-AKT signaling is an important contributor to the BRCA1 related breast cancer by increasing the antioxidant activities. So in BRCA1 mutant cancer cells, NRF2 can be activated by PI3K-AKT but loss its stability owing to the BRCA1 deficiency, hinted that NRF2 might be quickly and constantly degraded and regenerated.
Rising ROS benefits the self-renewal of neural stem cells [91]. Also, ROS plays a role in WNT initiated intestinal epithelial cells de-differentiation and converting of these cells to tumor initiation cells [92]. Therefore, these data may provide a potential mechanism that ROS can be beneficial to the cancer cell proliferation. Since E-ER signaling function also partially depends on activation of PI3K-AKT, the prevalence of abnormal activation of PI3K-AKT in BRCA1 related breast cancer suggested that NRF2 may be important in E-ER activated breast proliferating cells. In support of this speculation, several recent studies identified NRF2 activation induced by E-ER signaling in non-breast cells [93, 94]. Hence, it is quite possible that E-ER is also able to promote Nrf2 expression in breast cells.
C-MYC and CYCLIN D1 as common targets of BRCA1 and E-ER
BRCA1 and E-ER also regulate cell metabolism via their downstream targets in common such as CYCLIN D1 and C-MYC. Both factors are important regulator of cell proliferation and over expression of these factors are frequently associated with tumor cell growth. It was known that E-ER signaling promotes expression of Cyclin D1 and c-Myc by transcriptionally binding to their cis-regulatory elements [24-26]. Both CYCLIN D1 and C-MYC are well known for their function in cell cycle regulation and acting as oncogenes, but they are also identified to be important regulators of cell metabolism by recent studies [95-97]. CYCLIN D1 was shown to repress expression of Nrf-1 (nuclear respiratory factor 1), an activator of multiple mitochondria genes [96]. In addition, CYCLIN D1 was also found to repress the mitochondria function and aerobic glycolysis in cancer cells [97]. This is actually consistent with the cancer cell features that the growth of the cells prefers to use anaerobic glycolysis as the major energetic pathway instead of aerobic respiration in mitochondria. C-MYC was found to regulate expression of amino acid transporters Slc1a5, Asct2 and glutaminase Gls. C-MYC also promotes the conversion of glutamine to glutamate and the absorption of glutamate as anaplerotic substance for TCA cycle. Moreover, increased glutamine consumption is important to produce glutathione, an important antioxidant molecule in most of the proliferating cells. So, E-ER signaling may promote the cell proliferation with simultaneously stimulation of the anaplerotic metabolism that not only provides the carbon source but also increases the antioxidant capacity.
BRCA1 may limit the E-ER impact on cell metabolism via regulating CYCLIN D1 and C-MYC as well. On the one hand, CYCLIN D1 is able to competitively bind ER at the same domain recognized by BRCA1. This binding releases ER from being repressed by BRCA1 at ER target gene promoters [98]. On the other hand, Murray et al. showed that CYCLIN D1 is probably the target of BRCA1/BARD1 ubiquitin ligase activity and may be degraded in a cell cycle dependent manner [70]. BRCA1 repression of both ER and CYCLIN D1 mainly relies on protein-protein interaction. This is a relatively faster response and may be critical to inhibit ER and CYCLIN D1 promoted cell cycling when cells experience emergency condition such as oxidative stress. In fact, BRCA1 also interacts with C-MYC and represses the transcription activator function of C-MYC [99]. In contrast, E-ER promotes Cyclin D1 mainly through transcriptionally up-regulating Cyclin D1 expression, in a similar mechanism as BRCA1 promoted Esr-1 expression. Likewise, C-MYC activates BRCA1 mainly through transcriptionally activating Brca1 gene expression. Obviously, transcriptional regulation of these genes is relatively slow and requires more supporting mechanisms such as the availability of transcription machinery. So these processes take more time and provide a bigger window for being monitored and controlled.
Correlation of BRCA1 and E-ER in regulating mammary epithelial cell differentiation
ER was shown to be involved in regulating the cell differentiation process such as the EMT [100]. It was observed that re-introduction of ER in ER- breast cancer cells is able to inhibit EMT by inhibiting REL-B (NF-kB subunit) synthesis via repressing the expression of FRA-2 and AP-1, the main transcriptional factors of RelB gene, in an estrogen independent manner [100]. ER expression is also correlated with the reversing of the invasive feature of these cancer cells, suggesting ER is a repressor of EMT. Another independent study showed the important role of ER in repressing EMT via repressing Slug expression [101]. Slug is an important factor to promote EMT and is also essential to maintain the undifferentiating status of mammary epithelial stem/progenitor cell. Thus, reintroduction of ER to ER- breast cancer cells may promote the differentiation program which is in against of the EMT. Controversially, estrogen was shown to promote EMT by repressing E-cadherin in both normal and epithelial breast cancer cells and ovarian cancer cells in an ER dependent way [102, 103]. The inconsistency may be determined by the ligand binding status of ER because a separate and independent study explained that estrogen binding is able to alter ER from activator to repressor of E-cadherin [104]. Consequently, the ligand binding status of ER determines its role in regulating EMT [104]. These data suggested that estrogen is a dominant promoter of cell proliferation and dedifferentiation by activating the E-ER signaling pathway. Although the EMT and oncogenic transformation are two distinct processes, both of them show active proliferation and de-differentiation represented by activation of stem cell-related pathways [50]. Therefore, it is possible that the aberrant elevated estrogen and subsequent activation of ligand-bound ER may promote the de-differentiation in oncogenic cells.
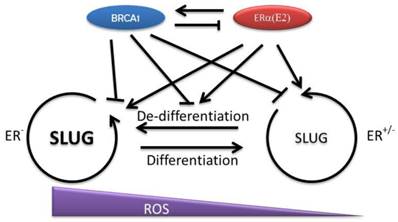
Since BRCA1 negatively regulates SLUG, a factor that is essential to maintain the pluripotency of mammary epithelial stem/progenitor cells, it is reasonable to suspect that the function of BRCA1 is to pave the way for differentiation by repressing stem cell related transcriptional factors [48]. But in BRCA1 mutation circumstances, the overlapped effects of estrogen and BRCA1 mutation may significantly promote the cell proliferation and the expression of the factors, such as SLUG and SOX9 that promote de-differentiation [48]. A recent study suggested that intestinal epithelial cells can de-differentiate upon NF-κB activation, which results in oncogenic transformation [92]. The mediator of NF-κB activation is Wnt signaling. Thus, cell proliferation and cell de-differentiation may be the prerequisite to turn on the neoplastic transformation in mammary epithelial cells with BRCA1 mutation as well. This idea is appealing because this will explain the increased breast cancer risk in BRCA1 deficient patients when being over-exposed to estrogen. Moreover, absence of BRCA1 reduces the ability of cells to deal with the oxidative stress partially raised by E-ER. Therefore, the cells that experience the de-differentiation may have much higher opportunity to be exposed to oxidative damages that accelerate the tumorigenesis (Fig. 2).
Interaction of BRCA1 and E-ER and their regulation of mammary epithelial cell differentiation. The differentiation of mammary epithelial progenitor cells is restricted by several key regulators such as SLUG and SOX9. BRCA1 may contribute to the cell differentiation by repressing SLUG in both multi-potent progenitor cells and committed progenitor cells. However, E-ER signaling may promote cell proliferation and increase these stem cell related factors, which start to turn on the related gene transcription programs and promote cell de-differentiation. In normal condition, the balance between BRCA1 and E-ER is important to maintain a homeostatic SLUG level and ensures the appropriate differentiation program. Under BRCA1 deficiency condition, the cells start to gain stem cell features. When this process is coupled with increased cellular damages, such as ROS related damages, the opportunity of oncogenic transformation increases.
Correlation between BRCA1 and E-ER in other metabolic disorders
The connection between obesity and breast cancer risk in postmenopausal women has been well established, probably because of the contribution of adipocytes in estrogen production in mammary gland of these patients [105]. Such correlation between weight gain and breast cancer risk may be also true for BRCA1 mutation carriers in postmenopausal period [106, 107]. But BRCA1 associated breast cancer tends to occur in premenopausal women [108]. In fact, it is still controversial whether there is correlation between the weight gain and cancer risk in premenopausal patients. In sporadic breast cancer patients, BRCA1 promoter methylation leads to increased invasive breast cancer risk mainly in premenopausal period [109]. Thus, it is not surprise that loss of BRCA1 also leads to increased breast cancer risk mainly in premenopausal patients. The prevalence of cancer risk in premenopausal women under the condition of BRCA1 insufficiency, highly suggested the important role of BRCA1 in preventing tumorigenesis at the earlier stage of life time. The leading cause of BRCA1 repression in sporadic breast cancer is still not clear. The recently identified molecular mechanism of CtBP repression of BRCA1 expression suggested the potential role of metabolic molecule, NADH, in BRCA1 repression [65]. In fact, NADH status has important effect on cancer risk and NADH level in cancer cells is generally higher owing to the glycolysis dependent growth of cancer cells [65, 110, 111].
Diabetes also has significant correlation to breast cancer risk in postmenopausal women. Since diabetes patients are characterized by hyperinsulinemia, it is possible that the crosstalk between the E-ER signaling pathway and PI3K-AKT signaling pathway contributes to the increased breast cancer risk [18, 112], because PI3K-AKT pathway is one of the major downstream pathways activated by insulin and insulin receptor. Another important reason is that over 80% of type II diabetic patients are obese. Thus, fat tissue associated overproduction of estrogen, and other fat tissue specific factors, may contribute to breast cancer risk as well. Consequently, BRCA1 expression may be affected in diabetes patients similar as in obese patients.
Summary
The evidences showing the direct inhibitory effect of BRCA1 on ER suggested BRCA1 is a major antagonistic factor of E-ER signaling pathway. Since ER is an important transcriptional factor, the inhibitory effect of BRCA1 on ER is expected to have much profound impact on the ER directed transcriptional programs. For example, the cell metabolism related aspects may be affected in a much more complicated way than we realized under BRCA1 deficient condition. The way to decipher these affected genes regulated by ER is to genome widely identify these genes and functionally annotate these gene expression programs. In fact, genome wide profiling of ER binding has been repeatedly studied by several labs and relatively consistent data have been obtained. But these studies are limited to the estrogen treatment condition only. None of these studies investigated the binding change of ER upon BRCA1 deficiency [60-64]. Therefore, it is important to further define the ER binding profile under BRCA1 deficiency condition, which will be critical to understand the details of E-ER involvement in tumorigenesis in BRCA1 related patients.
BRCA1 is also an important transcriptional factor, but very few studies addressed the BRCA1 binding profile at the genome level. Because comparison of ER and BRCA1 binding profile is a valuable channel to understand the interactive connection between these two factors, lack of BRCA1 genome wide binding data further impedes the detailed analysis of the opposite functions of ER and BRCA1. In recent released ENCODE data, BRCA1 binding profile in human ES cells has been included. So far, this is the only public available genome wide binding analysis of BRCA1 and can be a reference when BRCA1 binding profiles are studied in mammary gland epithelial cells and breast cancer cells.
Another intriguing issue associated with BRCA1 related tumorigenesis for long time is the difficulties in defining the tumor initiation cells precisely during mammary gland development, probably owing to the dynamic nature of this process and the challenges in tracking this process. The recent discovery defining these tumor initiation cells as cancer stem cells provides a new opportunity to carefully identify such cells by taking advantage of the well-defined cancer stem cell markers. More importantly, the newly emerged single cell isolation, manipulation and subsequent analysis techniques also provide the possibility to closely follow up the oncogenic transformation of the mammary epithelial cells that are BRCA1 deficient. By combining with the single cell exon sequencing, the transcriptome analysis may provide significant insight to the cell transformation event at the single cell level.
Involvement of both BRCA1 and E-ER in regulating cell metabolism pathways provides a new angle to review the BRCA1 related tumorigenesis mechanisms. Specifically, the BRCA1 function in prohibiting fatty acid synthesis by inhibiting ACC further strengthens its tumor repressor function by serving as the “caretaker” for the synthesis of the most important cell structural component, the membrane system, because fatty acids are the major substance to build up all the membrane. However, the function of BRCA1 in directly regulating cell metabolism shouldn't be limited to only one unique target. Therefore, further investigation of BRCA1 function in regulating cell metabolism pathways, either via direct protein-protein interactions as observed on the ACC inhibition or via its transcriptional target genes, should be necessary.
The most important observation that correlated to BRCA1 gene clinically is the abundance of BRCA1 silencing caused by promoter methylation. Whether or not this hypermethylation is caused by E-ER directly or indirectly, alleviation of BRCA1 promoter methylation may be an important therapeutic opportunity or preventive strategy for breast cancer. The hypermethylation of BRCA1 promoter is an epigenetic event and recent studies suggested that epigenetic modifications can be regulated by the cell metabolism status. Since E-ER signaling stimulates the cell proliferation and promotes the anabolic metabolism, there is a potential connection between the E-ER effect and BRCA1 silencing. This connection leads to the suspicion that increased physical activity and diet control may be effective in reducing the breast cancer risk. However, there is no direct evidence to support this hypothetical explanation yet. So, further studies are required to investigate the mechanism of BRCA1 silencing and clinically correlation studies are also necessary to establish the causing effect of metabolism status on BRCA1 silencing.
Acknowledgements
Funding support: SRG2013-00044-FHS from University of Macau.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69-90 doi:10.3322/caac.20107
2. Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812-23 doi:10.1016/S0140-6736(11)61539-0
3. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52 doi:10.1038/35021093
4. Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. The New England journal of medicine. 2009;360:790-800 doi:10.1056/NEJMra0801289
5. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121:2750-67 doi:10.1172/JCI45014
6. Metzger-Filho O, Tutt A, de Azambuja E, Saini KS, Viale G, Loi S. et al. Dissecting the heterogeneity of triple-negative breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:1879-87 doi:10.1200/JCO.2011.38.2010
7. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416-26 doi:10.1093/nar/gkl010
8. Yarden RI, Papa MZ. BRCA1 at the crossroad of multiple cellular pathways: approaches for therapeutic interventions. Mol Cancer Ther. 2006;5:1396-404 doi:10.1158/1535-7163.MCT-05-0471
9. Dine J, Deng CX. Mouse models of BRCA1 and their application to breast cancer research. Cancer Metastasis Rev. 2013;32:25-37 doi:10.1007/s10555-012-9403-7
10. Buckley NE, Mullan PB. BRCA1--conductor of the breast stem cell orchestra: the role of BRCA1 in mammary gland development and identification of cell of origin of BRCA1 mutant breast cancer. Stem Cell Rev. 2012;8:982-93 doi:10.1007/s12015-012-9354-y
11. Deng CX, Wang RH. Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet. 2003;1(R):113-23
12. Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes & development. 2003;17:201-13 doi:10.1101/gad.1050003
13. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
14. Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA. et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28:266-71 doi:10.1038/90108
15. Kim SS, Cao L, Lim SC, Li C, Wang RH, Xu X. et al. Hyperplasia and spontaneous tumor development in the gynecologic system in mice lacking the BRCA1-Delta11 isoform. Mol Cell Biol. 2006;26:6983-92 doi:10.1128/MCB.00796-06
16. Bachelier R, Xu X, Wang X, Li W, Naramura M, Gu H. et al. Normal lymphocyte development and thymic lymphoma formation in Brca1 exon-11-deficient mice. Oncogene. 2003;22:528-37 doi:10.1038/sj.onc.1206208
17. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37-43 doi:10.1038/8743
18. Alexaki VI, Charalampopoulos I, Kampa M, Vassalou H, Theodoropoulos P, Stathopoulos EN. et al. Estrogen exerts neuroprotective effects via membrane estrogen receptors and rapid Akt/NOS activation. FASEB J. 2004;18:1594-6 doi:10.1096/fj.04-1495fje
19. Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A. 2006;103:2196-201 doi:10.1073/pnas.0510974103
20. Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104:14718-23 doi:10.1073/pnas.0706933104
21. Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358-417
22. Bocchinfuso WP, Lindzey JK, Hewitt SC, Clark JA, Myers PH, Cooper R. et al. Induction of mammary gland development in estrogen receptor-alpha knockout mice. Endocrinology. 2000;141:2982-94
23. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J. et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905-31 doi:10.1152/physrev.00026.2006
24. Wang C, Mayer JA, Mazumdar A, Fertuck K, Kim H, Brown M. et al. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Molecular endocrinology. 2011;25:1527-38 doi:10.1210/me.2011-1037
25. Dubik D, Shiu RP. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene. 1992;7:1587-94
26. Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R. et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol. 2004;24:7260-74 doi:10.1128/MCB.24.16.7260-7274.2004
27. Ricketts D, Turnbull L, Ryall G, Bakhshi R, Rawson NS, Gazet JC. et al. Estrogen and progesterone receptors in the normal female breast. Cancer research. 1991;51:1817-22
28. Perusinghe NP, Monaghan P, O'Hare MJ, Ashley S, Gusterson BA. Effects of growth factors on proliferation on basal and luminal cells in human breast epithelial explants in serum-free culture. In Vitro Cell Dev Biol. 1992;28A:90-6
29. Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer research. 1997;57:4987-91
30. Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast cancer research and treatment. 1999;53:217-27
31. Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT, Ghadirian P. et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 2004;10:2029-34
32. Tung N, Wang Y, Collins LC, Kaplan J, Li H, Gelman R. et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: clinical risk factors and pathologic features. Breast cancer research: BCR. 2010;12:R12. doi:10.1186/bcr2478
33. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R. et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403-17 doi:10.1016/j.stem.2010.07.010
34. Li W, Xiao C, Vonderhaar BK, Deng CX. A role of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. Oncogene. 2007;26:7204-12 doi:10.1038/sj.onc.1210527
35. Jones LP, Tilli MT, Assefnia S, Torre K, Halama ED, Parrish A. et al. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene. 2008;27:794-802 doi:10.1038/sj.onc.1210674
36. Narod SA, Brunet JS, Ghadirian P, Robson M, Heimdal K, Neuhausen SL. et al. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet. 2000;356:1876-81
37. Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van't Veer L, Garber JE. et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. The New England journal of medicine. 2002;346:1616-22 doi:10.1056/NEJMoa012158
38. Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I. et al. Effects of estrogen on the proportion of stem cells in the breast. Breast cancer research and treatment. 2011;129:23-35 doi:10.1007/s10549-010-1169-4
39. Nishi M, Sakai Y, Akutsu H, Nagashima Y, Quinn G, Masui S. et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2013 doi:10.1038/onc.2012.614
40. Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD. et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105:1680-5 doi:10.1073/pnas.0711613105
41. Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:9176-81
42. Burga LN, Tung NM, Troyan SL, Bostina M, Konstantinopoulos PA, Fountzilas H. et al. Altered proliferation and differentiation properties of primary mammary epithelial cells from BRCA1 mutation carriers. Cancer research. 2009;69:1273-8 doi:10.1158/0008-5472.CAN-08-2954
43. Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A. et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104:12111-6 doi:10.1073/pnas.0702969104
44. Ginestier C, Liu S, Wicha MS. Getting to the root of BRCA1-deficient breast cancer. Cell Stem Cell. 2009;5:229-30 doi:10.1016/j.stem.2009.08.007
45. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M. et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149-63 doi:10.1016/j.stem.2010.12.007
46. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH. et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907-13 doi:10.1038/nm.2000
47. Shehata M, Teschendorff A, Sharp G, Novcic N, Russell A, Avril S. et al. Phenotypic and functional characterization of the luminal cell hierarchy of the mammary gland. Breast cancer research: BCR. 2012;14:R134. doi:10.1186/bcr3334
48. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F. et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015-28 doi:10.1016/j.cell.2012.02.008
49. Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A. 2012;109:16654-9 doi:10.1073/pnas.1205822109
50. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15 doi:10.1016/j.cell.2008.03.027
51. Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG. et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Molecular endocrinology. 2010;24:76-90 doi:10.1210/me.2009-0218
52. Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5794-9 doi:10.1073/pnas.0610887104
53. Fan S, Wang J, Yuan R, Ma Y, Meng Q, Erdos MR. et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354-6
54. Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C. et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem. 2001;276:18375-83 doi:10.1074/jbc.M100800200
55. Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Molecular endocrinology. 2006;20:1479-93 doi:10.1210/me.2005-0531
56. Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA. et al. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer research. 2002;62:141-51
57. Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA. et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Molecular endocrinology. 2006;20:14-34 doi:10.1210/me.2004-0488
58. Ma Y, Hu C, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Molecular endocrinology. 2007;21:1905-23 doi:10.1210/me.2006-0397
59. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A. et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036-47 doi:10.1158/2159-8290.CD-11-0348
60. Kininis M, Isaacs GD, Core LJ, Hah N, Kraus WL. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol Cell Biol. 2009;29:1123-33 doi:10.1128/MCB.00841-08
61. Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC. et al. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090-104 doi:10.1128/MCB.00083-07
62. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ. et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33-43 doi:10.1016/j.cell.2005.05.008
63. Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M. et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS genetics. 2007;3:e87. doi:10.1371/journal.pgen.0030087
64. Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN. et al. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. Embo J. 2009;28:1418-28 doi:10.1038/emboj.2009.88
65. Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406-13 doi:10.1038/nsmb.1941
66. Gudas JM, Nguyen H, Li T, Cowan KH. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995;55:4561-5
67. Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT. et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622-34 doi:10.1016/j.cell.2011.03.042
68. Spillman MA, Bowcock AM. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene. 1996;13:1639-45
69. Marks JR, Huper G, Vaughn JP, Davis PL, Norris J, McDonnell DP. et al. BRCA1 expression is not directly responsive to estrogen. Oncogene. 1997;14:115-21 doi:10.1038/sj.onc.1200808
70. Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE. et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. Journal of the National Cancer Institute. 2007;99:1683-94 doi:10.1093/jnci/djm207
71. Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin Cancer Res. 2009;15:1514-8 doi:10.1158/1078-0432.CCR-08-0640
72. Hu Y, Ghosh S, Amleh A, Yue W, Lu Y, Katz A. et al. Modulation of aromatase expression by BRCA1: a possible link to tissue-specific tumor suppression. Oncogene. 2005;24:8343-8 doi:10.1038/sj.onc.1208985
73. De Siervi A, De Luca P, Byun JS, Di LJ, Fufa T, Haggerty CM. et al. Transcriptional autoregulation by BRCA1. Cancer research. 2010;70:532-42 doi:10.1158/0008-5472.CAN-09-1477
74. Ellison-Zelski SJ, Solodin NM, Alarid ET. Repression of ESR1 through actions of estrogen receptor alpha and Sin3A at the proximal promoter. Mol Cell Biol. 2009;29:4949-58 doi:10.1128/MCB.00383-09
75. Neeman M, Degani H. Early estrogen-induced metabolic changes and their inhibition by actinomycin D and cycloheximide in human breast cancer cells: 31P and 13C NMR studies. Proc Natl Acad Sci U S A. 1989;86:5585-9
76. O'Mahony F, Razandi M, Pedram A, Harvey BJ, Levin ER. Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Molecular endocrinology. 2012;26:2058-70 doi:10.1210/me.2012-1191
77. Ko BH, Paik JY, Jung KH, Lee KH. 17beta-estradiol augments 18F-FDG uptake and glycolysis of T47D breast cancer cells via membrane-initiated rapid PI3K-Akt activation. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2010;51:1740-7 doi:10.2967/jnumed.110.074708
78. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33 doi:10.1126/science.1160809
79. Amon LM, Pitteri SJ, Li CI, McIntosh M, Ladd JJ, Disis M. et al. Concordant release of glycolysis proteins into the plasma preceding a diagnosis of ER+ breast cancer. Cancer research. 2012;72:1935-42 doi:10.1158/0008-5472.CAN-11-3266
80. Kang HJ, Yi YW, Kim HJ, Hong YB, Seong YS, Bae I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis. 2012;3:e336. doi:10.1038/cddis.2012.78
81. Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, Powell SN. et al. Negative Regulation of AKT Activation by BRCA1. Cancer research. 2008;68:10040-4 doi:10.1158/0008-5472.CAN-08-3009
82. Moreau K, Dizin E, Ray H, Luquain C, Lefai E, Foufelle F. et al. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J Biol Chem. 2006;281:3172-81 doi:10.1074/jbc.M504652200
83. Chajes V, Cambot M, Moreau K, Lenoir GM, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer research. 2006;66:5287-94 doi:10.1158/0008-5472.CAN-05-1489
84. Felty Q, Xiong WC, Sun D, Sarkar S, Singh KP, Parkash J. et al. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005;44:6900-9 doi:10.1021/bi047629p
85. Kang HJ, Hong YB, Kim HJ, Rodriguez OC, Nath RG, Tilli EM. et al. Detoxification: a novel function of BRCA1 in tumor suppression? Toxicol Sci. 2011;122:26-37 doi:10.1093/toxsci/kfr089
86. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F. et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416-29 doi:10.1093/nar/gks409
87. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66-79 doi:10.1016/j.ccr.2012.05.016
88. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B. et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013;210:1529-44 doi:10.1084/jem.20121337
89. Yi YW, Kang HJ, Kim HJ, Hwang JS, Wang A, Bae I. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances antitumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol Carcinog. 2013;52:667-75 doi:10.1002/mc.21905
90. Kimbung S, Biskup E, Johansson I, Aaltonen K, Ottosson-Wadlund A, Gruvberger-Saal S. et al. Co-targeting of the PI3K pathway improves the response of BRCA1 deficient breast cancer cells to PARP1 inhibition. Cancer letters. 2012;319:232-41 doi:10.1016/j.canlet.2012.01.015
91. Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD. et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8:59-71 doi:10.1016/j.stem.2010.11.028
92. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25-38 doi:10.1016/j.cell.2012.12.012
93. Zhang T, Liang X, Shi L, Wang L, Chen J, Kang C. et al. Estrogen Receptor and PI3K/Akt Signaling Pathway Involvement in S-(-)Equol-Induced Activation of Nrf2/ARE in Endothelial Cells. PloS one. 2013;8:e79075. doi:10.1371/journal.pone.0079075
94. Chen CS, Tseng YT, Hsu YY, Lo YC. Nrf2-Keap1 antioxidant defense and cell survival signaling are upregulated by 17beta-estradiol in homocysteine-treated dopaminergic SH-SY5Y cells. Neuroendocrinology. 2013;97:232-41 doi:10.1159/000342692
95. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110-21 doi:10.1016/j.cmet.2011.12.009
96. Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J. et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 2006;103:11567-72 doi:10.1073/pnas.0603363103
97. Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M. et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006;26:5449-69 doi:10.1128/MCB.02074-05
98. Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y. et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer research. 2005;65:6557-67 doi:10.1158/0008-5472.CAN-05-0486
99. Wang Q, Zhang H, Kajino K, Greene MI. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene. 1998;17:1939-48 doi:10.1038/sj.onc.1202403
100. Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F. et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nature cell biology. 2007;9:470-8 doi:10.1038/ncb1559
101. Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B. et al. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29:1451-62 doi:10.1038/onc.2009.433
102. Oesterreich S, Deng W, Jiang S, Cui X, Ivanova M, Schiff R. et al. Estrogen-mediated down-regulation of E-cadherin in breast cancer cells. Cancer research. 2003;63:5203-8
103. Park SH, Cheung LW, Wong AS, Leung PC. Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Molecular endocrinology. 2008;22:2085-98 doi:10.1210/me.2007-0512
104. Cardamone MD, Bardella C, Gutierrez A, Di Croce L, Rosenfeld MG, Di Renzo MF. et al. ERalpha as ligand-independent activator of CDH-1 regulates determination and maintenance of epithelial morphology in breast cancer cells. Proc Natl Acad Sci U S A. 2009;106:7420-5 doi:10.1073/pnas.0903033106
105. Cleary MP, Grossmann ME. Minireview: Obesity and breast cancer: the estrogen connection. Endocrinology. 2009;150:2537-42 doi:10.1210/en.2009-0070
106. Manders P, Pijpe A, Hooning MJ, Kluijt I, Vasen HF, Hoogerbrugge N. et al. Body weight and risk of breast cancer in BRCA1/2 mutation carriers. Breast cancer research and treatment. 2011;126:193-202 doi:10.1007/s10549-010-1120-8
107. Kotsopoulos J, Olopado OI, Ghadirian P, Lubinski J, Lynch HT, Isaacs C. et al. Changes in body weight and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast cancer research: BCR. 2005;7:R833-43 doi:10.1186/bcr1293
108. Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. Journal of the National Cancer Institute. 2002;94:1365-72
109. Xu X, Gammon MD, Zhang Y, Bestor TH, Zeisel SH, Wetmur JG. et al. BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast cancer research and treatment. 2009;115:397-404 doi:10.1007/s10549-008-0075-5
110. Byun JS, Gardner K. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol. 2013;182:1055-64 doi:10.1016/j.ajpath.2013.01.009
111. Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S. et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun. 2013;4:1449. doi:10.1038/ncomms2438
112. Bowers LW, Cavazos DA, Brenner AJ, Hursting SD, Maximo IX, Degraffenried LA. Obesity enhances nongenomic estrogen receptor crosstalk with the PI3K/Akt and MAPK pathways to promote in vitro measures of breast cancer progression. Breast cancer research: BCR. 2013;15:R59. doi:10.1186/bcr3453
Author contact
![]() Corresponding author: Li-Jun Di, Av. Padre Tomas Pereira, Taipa, Macau SAR. Email: lijundimo.
Corresponding author: Li-Jun Di, Av. Padre Tomas Pereira, Taipa, Macau SAR. Email: lijundimo.