Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):861-872. doi:10.7150/ijbs.8521 This issue Cite
Research Paper
Chondrocyte-specific Knockout of Cbfβ Reveals the Indispensable Function of Cbfβ in Chondrocyte Maturation, Growth Plate Development and Trabecular Bone Formation in Mice
Mengrui Wu1,2, Yi-Ping Li1, 2 ![]() , Guochun Zhu2, Yun Lu2, Yiping Wang1,2, Joel Jules2, Matthew McConnell2, Rosa Serra3, Jian-Zhong Shao1, Wei Chen2
, Guochun Zhu2, Yun Lu2, Yiping Wang1,2, Joel Jules2, Matthew McConnell2, Rosa Serra3, Jian-Zhong Shao1, Wei Chen2 ![]()
1. Institute of Genetics, Life Science College, Zhejiang University, Hangzhou, Zhejiang, 310058, People's Republic of China.
2. Department of Pathology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
3. Department of Cell, Development, and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Received 2014-1-7; Accepted 2014-4-22; Published 2014-7-29
Abstract
Despite years of research into bone formation, the mechanisms by which transcription factors specify growth plate development and trabecular bone formation remain unclear and the role of hypertrophic chondrocytes in trabeculae morphogenesis is controversial. To study the role of Core binding factor beta (Cbfβ) in postnatal cartilage development and endochondral bone formation, we generated chondrocyte-specific Cbfβ-deficient mice (Cbfβf/fCol2α1-Cre mice) using floxed alleles of Cbfβ (Cbfβf/f) and Cre driven by the Collagen 2α1 promoter (Col2α1-Cre). Cbfβf/fCol2α1-Cre mice evaded developmental and newborn lethality to survive to adulthood and displayed severe skeletal malformation. Cbfβf/fCol2α1-Cre mice had dwarfism, hypoplastic skeletons, defective bone mineralization, shortened limbs, shortened sternum bodies, and un-calcified occipital bones and hyoid bones. In the long bone cartilage, the resting zone was elongated, and chondrocyte proliferation and hypertrophy were impaired in Cbfβf/fCol2α1-Cre mice, which led to deformation of the growth plates. Primary spongiosa formation was delayed, diaphysis was shortened and trabecular bone formation was almost absent in the mutant mice. In addition, lamellar bone formation in the secondary spongiosa was also impaired. However, osteoclast formation in the trabecular bone was not affected. Cbfβ deficiency led to down-regulation of chondrocyte-regulating genes [i.e, patched (Ptc1), Cyclin D1 and Indian hedgehog (Ihh)] in the cartilage. Interestingly, the expression of Runx2 and Runx3 was not changed in the cartilage of the mutants. Collectively, the results revealed that Cbfβ is crucial for postnatal skeletal development and endochondral bone formation through its function in growth plate development and chondrocyte proliferation and differentiation. This study also revealed that chondrocyte maturation, mediated by Cbfβ, was critical to trabecular bone morphogenesis. Significantly, these findings provide insight into the role of Cbfβ in postnatal skeletogenesis, which may assist in the development of new therapies for osteoporosis.
Keywords: Cbfβ, Runx, Indian hedgehog (Ihh), skeletal development, chondrocyte proliferation and hypertrophy, endochondral bone formation.
Introduction
Core binding factors (Cbfs) are heterodimeric transcription factors comprised of the Cbf-alpha (Cbfα) and Cbf-beta (Cbfβ) subunits. The Cbfα subunits are encoded by the runt-related transcription factors (Runxs), which contain three members: Runx1, Runx2, and Runx3 [1]. Unlike the Cbfα subunits, the Cbfβ subunit is encoded by a single gene. The Cbfβ subunit is a non-DNA-binding protein that associates with the Runx proteins to mediate their DNA-binding affinities. Runx/Cbfβ heterodimeric transcription complexes play crucial roles in various developmental processes [2], including the skeletal system by in part mediating gene expression. Furthermore, Runx/Cbfβ complexes are implicated in many diseases including cleidocranial dysplasia (CCD), an autosomal-dominant disease resulting predominantly from Runx2 deficiency that is characterized by short stature, patent fontanelles, defective clavicles, and defective teeth [3].
Runx1 regulates development of certain parts of the skeleton (i.e, sternum morphogenesis, certain skull elements mineralization) [4, 5]. Runx1 has also been shown to promote chondrocyte proliferation and commitment of mesenchymal stem cells (MSCs) to the chondrocyte lineage [6]. Runx2 is a master regulator of osteoblast differentiation and hence plays an important role in skeletal development [7-9]. Runx2-deficient (Runx2-/-) mice died soon after birth and exhibited severe skeletal defects from blocked intramembranous and endochondral ossification [7-9]. The role of Runx2 in osteoblasts was buttressed by the finding that calvarial cells derived from Runx2-/- embryos could differentiate into adipocytes and chondrocytes but not into osteoblasts [10]. Finally, Runx3 cooperates with Runx2 to regulate chondrocyte proliferation and hypertrophy [11].
The Cbfβ subunit is also vital for skeletal development. Cbfβ-deficient (Cbfβ-/-) embryos died from an absence of fetal liver hematopoiesis at mid-gestation [12, 13]. To circumvent embryonic lethality, CbfβGFP/GFP knock-in mice were generated and Cbfβ-/- embryos were rescued by Tek-GFP/Cbfβ [Cbfβ-/-Tg(Tek-GFP/Cbfβ)] or Gata1-Cbfβ [Cbfβ-/-Tg(Gata1-Cbfβ)] transgene [14-16]. These mice died around birth and exhibited numerous skeletal defects, indicating that a fully functional Cbfβ was essential for embryonic skeletal development [14-16]. Cbfβ/Runx2 double transgenic mice exhibited severe osteopenia and revealed that the Runx2/Cbfβ complex inhibits osteoblastogenesis at a late stage of differentiation [17].
Indian hedgehog (Ihh) is a master regulator of both chondrocyte and osteoblast differentiation during endochondral bone formation [18, 19]. Studies have shown that Runx2 binds to the Ihh promoter and enhances its expression. Ihh, secreted by prehypertrophic/hypertrophic chondrocytes, promotes chondrocyte proliferation [18, 19]. In addition, Ihh inhibits chondrocyte hypertrophy and keeps chondrocytes in the proliferative pool by inducing expression of parathyroid hormone-related protein (PTHrP), which in turn inhibits Ihh expression via the so-called “Ihh-PTHrP negative-feedback loop” [18, 19].
Nonetheless, the impact of Cbfβ deficiency in chondrocytes during postnatal skeletal development is largely unknown. Thus, we generated chondrocyte-specific Cbfβ-deficient mice (Cbfβf/fCol2α1-Cre mice) by crossing Col2α1-Cre mice [20] with Cbfβf/f [21] mice. Generally, the Cbfβf/fCol2α1-Cre mice survived to adulthood and displayed severe skeletal malformation (i.e. CCD-like features). Notably, the Cbfβ deficiency in chondrocytes impaired not only growth plate formation but also trabeculae morphogenesis by regulating chondrocyte-regulating genes including those involved in Ihh/PTHrP negative-feedback loop.
Results
Postnatal 7-day-old (P7) Cbfβf/fCol2α1-Cre mice had dwarfism with shortened limbs and hypoplastic skeletons from defective bone mineralization
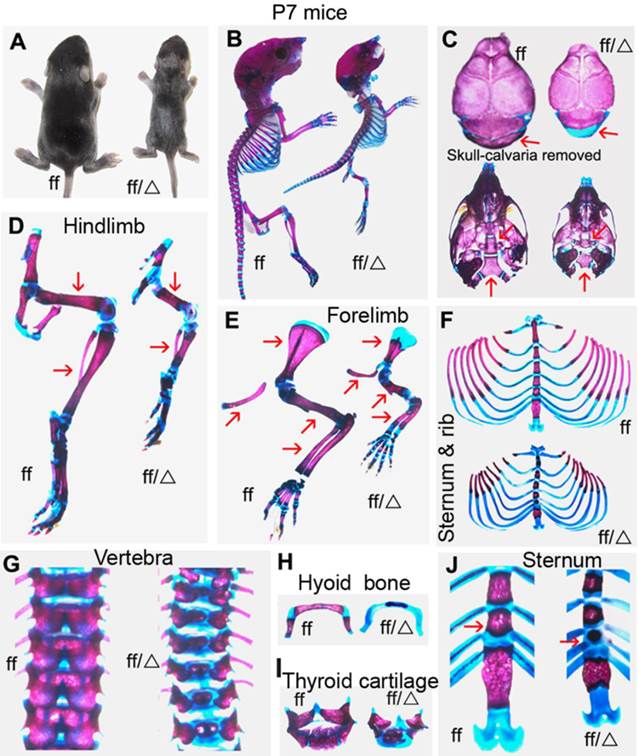
To investigate the role of Cbfβ in chondrocyte development during postnatal skeletogenesis, we generated Cbfβf/fCol2α1-Cre mice, deleting the Cbfβ gene specifically in the chondrocyte lineage. Unlike CbfβGFP/GFP knock-in mice, Cbfβ-/-Tg(Tek-GFP/Cbfβ) mice and Cbfβ-/-Tg(Gata1-Cbfβ) mice, Cbfβf/fCol2α1-Cre mice survived to adulthood. Postnatal seven-day-old (P7) Cbfβf/f Col2α1-Cre mice exhibited a dwarfed stature with shortened limbs (Fig. 1A). Alizarin red & Alcian blue staining revealed decreased bone mineralization and delayed ossification in several skeletal elements of the Cbfβf/f Col2α1-Cre mice (Fig. 1B). The skulls of the mutant mice appeared basically normal compared to wild-type (WT) mice, except for the non-mineralized occipital bone (Fig. 1C). Several defects could be observed in the basicranium of the Cbfβf/f Col2α1-Cre mice. The basisphenoid bone and the basioccipital bone were connected by a cartilaginous bone in the WT mice, which was missing in the mutant mice (Fig. 1C). The basioccipital bone of the mutant mice was also smaller than that of WT mice (Fig. 1C). Hindlimbs (Fig. 1D) and forelimbs (Fig. 1E) were shortened in the mutant mice, while the epiphyseal growth plates were elongated. Delayed ossification was observed in ribs (Fig. 1F), vertebrae (Fig. 1G), hyoid bone (Fig. 1H) and thyroid cartilage (Fig. 1I). Additionally, the sternal body was shortened greatly while sternal angle remained a similar length (Fig. 1J).
P7 Cbfβf/fCol2α1-Cre mice exhibited dwarfism with hypoplastic skeletons and decreased mineralization. (A) Photographic image of postnatal seven-day-old (P7) Cbfβf/fCol2α1-Cre (ff/Δ) and wild-type (WT, ff) mice. (B) Whole-mount Alizarin red S/Alcian blue staining of P7 Cbfβf/fCol2α1-Cre and WT mice to analyze bone/cartilage development. (C) Alizarin red S/Alcian blue staining of skull bones (cranium and cranial base) of P7 Col2α1-Cre and WT mice. Arrowheads indicate the defective occipital bone, basisphenoid bone and basioccipital bone in the mutant mice. (D, E) Alizarin red S/Alcian blue staining of hindlimbs (D) and forelimbs (E) of P7 Cbfβf/fCol2α1-Cre and WT mice. Arrowhead indicates the diaphysis was shortened in the mutant mice compared to WT. (F-J) Alizarin red S/Alcian blue staining of sternums/ribs (F), vertebrae (G), hyoid bones (H), thyroid cartilages (I) and sternum (J) of P7 Cbfβf/fCol2α1-Cre and WT mice. Arrowhead indicates the shortened sternum body in the mutant mice. Data are representative for 6 mice per group.
P7 and Postnatal 18-day-old (P18) Cbfβf/fCol2α1-Cre mice had dwarfism with shortened limbs, lower bone density and malocclusion
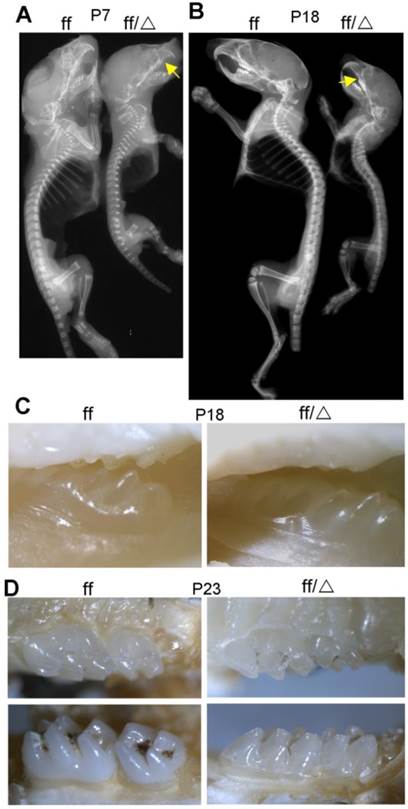
X-ray analysis was performed to assess bone density of WT and Cbfβf/f Col2α1-Cre mice (Fig. 2). The mutant mice displayed dwarfism with shortened limbs at both P7 and P18. The whole-body bone density was lower in P7 (Fig. 2A) and P18 (Fig. 2B) Cbfβf/fCol2α1-Cre mice compared to their WT cohorts. The reduced bone density was most obvious as observed in ribs, vetebrae, sternum, forelimbs and hindlimbs. Consistently, the ossification of ribs, vertebrae, sternum, forelimbs and hindlimbs was delayed as shown by Alizarin red & Alcian blue staining (Fig. 1). Interestingly, the X-ray image also showed malocclusion in the Cbfβf/fCol2α1-Cre mouse teeth (arrow, Fig. 2A, B). To confirm the tooth defect, photographic images of the P18 (Fig. 2C) and P23 (Fig. 2D) WT and Cbfβf/fCol2α1-Cre mice were taken using a stereomicroscope. The photographic images indicated that the eruption of the maxillary second molar was delayed in the Cbfβf/fCol2α1-Cre mice.
P7 and P18 mice had reduced skeletal mineralization. (A) X-ray analysis of P7 Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice shows decreased bone density in the mutant mice. (B) X-ray analysis of P18 Cbfβf/fCol2α1-Cre and WT mice shows decreased bone density in the mutant mice. Arrows indicated malocclusion in the mouse teeth. (C-D) Photographic images of (C) P18 and (D) P23 mice teeth, indicating eruption of the maxillary second molar was delayed in the Cbfβf/fCol2α1-Cre mice. Data are representative for 5 mice per group.
Cbfβf/fCol2α1-Cre mice displayed elongated but deformed growth plates
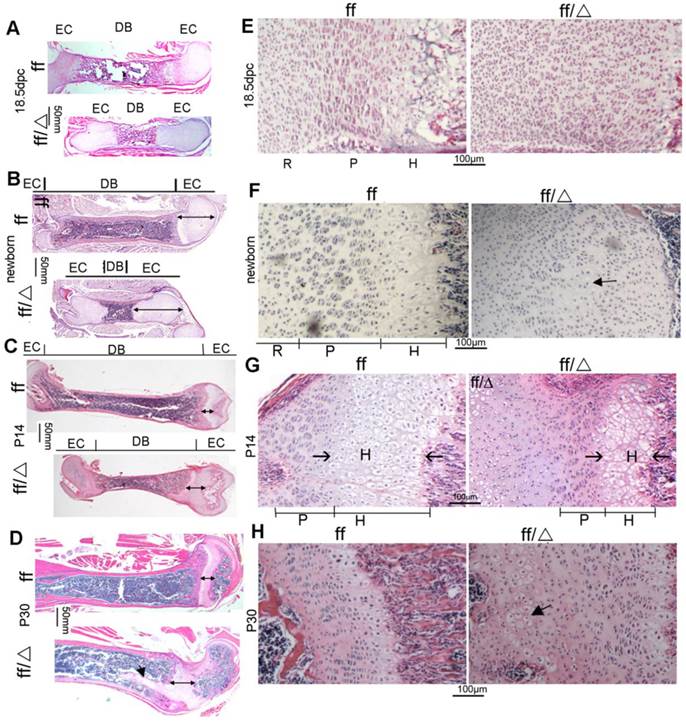
H&E staining was performed on 18.5 dpc WT and Cbfβf/fCol2α1-Cre embryonic femoral frozen sections, as well as newborn, postnatal 14-day-old (P14) and postnatal 30-day-old (P30) WT and Cbfβf/fCol2α1-Cre mice femoral paraffin slides (Fig. 3). Consistent with the short-limb deformity, the femurs, and their diaphyses were shortened in the mutant mice compared to their WT cohorts (Fig. 3A, B, C, D). However, the epiphyseal growth plates were elongated in the mutant mice at all ages (Fig. 3A, B, C, D, double ended arrow). The ratio of femoral diaphysis to bone length was three-fold less in 18.5dpc and newborn mutant mice compared to the newborn WT mice (Fig. 3A, B). This ratio decreased to 1.5-fold lower in the P14 mutant mice compared to P14 WT mice (Fig. 3C). Nonetheless, the elongation and deformity of the growth plates became more severe as the mice aged (Fig. 3A-D). Notably in the P30 mice, the growth plates protruded deep into the bone marrow abnormally (Fig. 3D). In the WT mice, growth plates were all organized into three layers, the resting zone, the proliferation zone and the hypertrophic zone at the 18.5dpc and newborn stages of development (Fig. 3E, F, left panels). The three-layer structure could hardly be observed in the mutant 18.5dpc embryo and newborn mouse growth plates (Fig. 3E, F, right panels). In the mutant 18.5dpc embryo and newborn mice, most chondrocytes appear like immature cells in the resting zone and the column-shaped proliferative chondrocytes were absent (Fig. 3E, F, right panels). Quite a few hypertrophic chondrocytes could be found near the primary spongiosa of the newborn mutant mice (Fig. 3F, right panel). The three-layer structure didn't form in the mutant mouse growth plates until P14, with a drastically shortened hypertrophic zone and a slightly shortened proliferative zone (Fig. 3G). In the P30 mutant mouse growth plates, the arrangement of proliferative columns was not as neat as that in WT mice, and several hypertrophic chondrocyte-like cells were presented in the resting zone near the secondary spongiosa (Fig. 3H). Collectively, data showed that the mutant mice had severe defects in the growth plate development, which became more severe as the mice aged. The mutant growth plates were elongated and comprised of massive immature chondrocytes, the proliferative cells were disrupted, and hypertrophy was delayed.
Cbfβf/fCol2α1-Cre mice had abnormal growth plates and delayed chondrocyte development. (A-D) H&E staining of femoral sections from (A) 18.5dpc, (B) newborn, (C) P14, and (D) P30 Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice. Double ended arrow indicates the length of the epiphyseal growth plates. Arrow in (D) indicates mutant growth plates protruded into the diaphysis. (E-H) H&E staining showed growth plates from (E) 18.5dpc, (F) newborn, (G) P14, and (H) P30 Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice. Arrow in (F) indicates the hypertrophic chondrocytes in the mutant mice. Arrow in (G) indicates the hypertrophic zone. Arrow in (H) indicates the hypertrophic chondrocytes presented in the resting zone of the mutant growth plates. EC, epiphyseal cartilage; DB, diaphysis bone; R, resting zone; P, proliferative zone; H, hypertrophic zone. Data are representative for 5 mice per group.
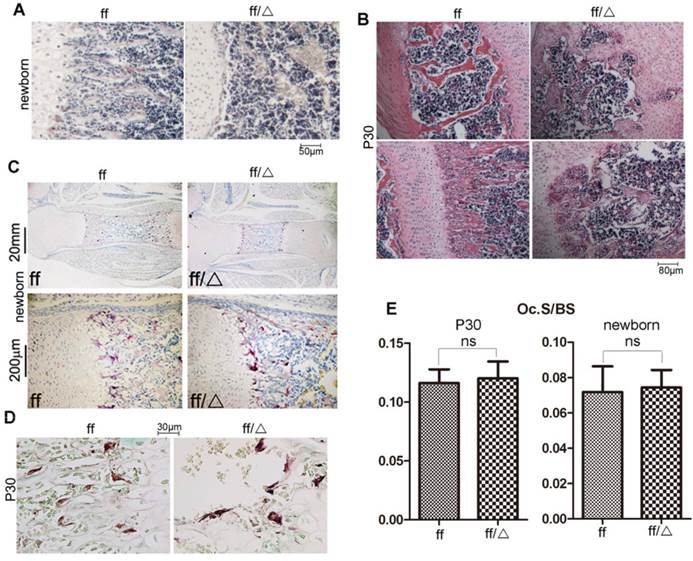
Endochondral bone formation was delayed in the Cbfβf/fCol2α1-Cre mice with unaffected osteoclastogenesis
Trabecular bone formation could be observed in the primary spongiosa of newborn WT mice, but was absent in the newborn mutant mice (Fig. 4A). Trabecular bone (Fig. 4B, lower panel) and lamellar bone (Fig. 4B, upper panel) were well formed in the P30 WT mice, but were severely reduced in the P30 Cbfβf/fCol2α1-Cre mice. Additionally, growth plates protrude deep into the diaphysis (Fig. 3D), and the secondary spongiosa was also occupied by cartilaginous bones (Fig. 4B, upper panel) in the P30 mutant mice. Since trabecular bone formation was impaired in the mutant mice, we performed TRAP staining to detect the formation of osteoclasts. Osteoclastogenesis was not affected at either the newborn stage or P30 in the Cbfβf/fCol2α1-Cre mice compared to WT (Fig. 4C-E). To summarize, deletion of Cbfβ in chondrocytes impaired bone formation in both primary and secondary spongiosa but didn't affect osteoclast formation.
Cbfβf/fCol2α1-Cre mice had impaired endochondral bone formation. (A) H&E staining showed the femoral primary spongiosa in the newborn Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice. (B) H&E staining showed the femoral primary and secondary spongiosa in the P30 Cbfβf/fCol2α1-Cre and WT mice. (C) TRAP staining of the femurs of newborn Cbfβf/fCol2-Cre and WT mice to analyze osteoclast formation. (D) TRAP staining of the femurs of P30. (E) Quantification of Osteoclast surface per bone surface of WT and Cbfβf/fCol2-Cre newborn and P30 mice, and data were presented as mean SEM (ns, not significant). Oc.S/BS, osteoclast surface per bone surface. Data are representative for 6 mice per group.
SEM (ns, not significant). Oc.S/BS, osteoclast surface per bone surface. Data are representative for 6 mice per group.
Cbfβ deficiency impaired chondrocyte proliferation and patched (Ptc1) expression
Efficient ablation of Cbfβ in mutant mouse growth plates was confirmed by immunochemistry (IHC) staining using the anti-Cbfβ antibody (Fig. 5A, E). Consistently, Alcian blue staining of primary-cultured chondrocytes in a micro-mass culture model showed a drastic decrease of chondrogenesis of Cbfβf/fCol2α1-Cre cells (Fig. 5B). Femoral PCNA staining also revealed that proliferative chondrocytes were decreased in the mutant growth plates (Fig. 5C, E). Indian hedgehog (Ihh), binding to membrane protein patched 1 (Ptc1), is a major regulator of bone development, coordinating chondrocyte proliferation, chondrocyte differentiation and osteoblast differentiation [18, 19, 22, 23]. Our IHC staining showed that Ptc1 was expressed in the proliferative and the pre-hypertrophic chondrocytes in postnatal 3-day-old (P3) WT mouse femurs (Fig. 5D, E). Ptc1 expression was also drastically decreased in the P3 mutant mice compared to WT cohorts (Fig 5D, E). We also performed TUNEL staining to detect apoptotic chondrocytes in the growth plate of P14 mice. The apoptotic cells appeared in the extremity of hypertrophic zone adjacent to the calcified trabecular bones. Loss of Cbfβ increased hypertrophic chondrocyte apoptosis (Fig. 5F, G), which may contribute to the shortening of the hypertrophic zone of the growth plates in postnatal Cbfβf/fCol2α1-Cre mice.
Cbfβf/fCol2α1-Cre deficiency resulted in reduced proliferation and decreased Ptc1 expression. (A) Immunohistochemistry (IHC) staining to detect expression of Cbfβ in the growth plates of newborn Cbfβf/fCol2-Cre (ff/Δ) and WT (ff) mouse femurs. (B) Chondrocytes from newborn Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice were cultured and then stained with Alcian blue. (C) PCNA staining of the growth plates of tibial sections from P7 Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice. (D) IHC to detect Ptc1 expression in P3 Cbfβf/fCol2α1-Cre and WT mice growth plates. The left two panels show lower magnification of the femurs. The other four panels show higher magnification of the femoral growth plates. H, hypertrophic zone (E) quantification of Cbfβ-positive cell ratio in hypertrophic zone, PCNA-positive cell ratio in proliferation zone and Ptc1-positive cell ratio in pre-hypertrophic zone, and data were presented as mean SEM (**, p<0.01; ***, p<0.005). (F) TUNEL staining to detect apoptotic cells in P14 Cbfβf/fCol2α1-Cre and WT mice growth plates. (G) Quantification of TUNEL positive cell ratio in hypertrophic zone, and data were presented as mean
SEM (**, p<0.01; ***, p<0.005). (F) TUNEL staining to detect apoptotic cells in P14 Cbfβf/fCol2α1-Cre and WT mice growth plates. (G) Quantification of TUNEL positive cell ratio in hypertrophic zone, and data were presented as mean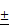 SEM (**, p<0.01; ***, p<0.005). Data are representative for 4 mice per group.
SEM (**, p<0.01; ***, p<0.005). Data are representative for 4 mice per group.
Cbfβ deficiency leads to downregulation of Ihh, Cyclin D1, and PTHrP-R in the chondrocytes
The cell cycle is regulated by a group of Cyclin/CDK protein complexes, among which is Cyclin D1 that plays an important role in chondrocyte proliferation as a target of Ihh [24]. To address the mechanism underlying the effects of Cbfβ on chondrocytes, we examined the expression levels of Cyclin D1, Ihh and PTHrP by immunofluorescent staining of tibial sections (Fig. 6A-C). Interestingly, Cyclin D1 was down-regulated in the mutant growth plates (Fig. 6A). This finding showed that Cbfβ might regulate, at least in part, chondrocyte proliferation by regulating Ihh-induced Cyclin D1 expression. Endochondral ossification is controlled by Indian hedgehog (Ihh) through induction of parathyroid hormone-related peptide (PTHrP) which negatively regulates chondrocyte maturation [18, 19]. Because Cbfβ functions in concert with Runx proteins, we reasoned that Cbfβ might regulate chondrocyte development in the same way as the Runx proteins. Ihh expression was shown to be suppressed in Runx2-/- mice [11]. Consistently, Ihh was also drastically downregulated in the growth plate of Cbfβ-deficient tibiae (Fig. 6B). Ihh has been shown to be regulated by a negative feedback loop mediated by PTHrP [18, 19]. Our results showed that the PTHrP receptor (PPR) was up-regulated in the mutant mice (Fig. 6C), indicating the Ihh-PTHrP negative feedback loop was disrupted in the mutant mice.
Analysis of gene expression in the cartilage of Cbfβf/f Col2α1-Cre mice by IF. (A-C) Immunofluorescence (IF) staining of the growth plates from tibial frozen sections of P7 Cbfβf/f Cbfβf/fCol2α1-Cre (ff/Δ) and WT (ff) mice with anti-CyclinD1 (A), anti-Ihh (B), and anti-PPR (C). (A-C) High-magnification figures were co-presented on the right panels. (D) Western blot analysis of the expression of Cbfβ, PPR, Ihh, PCNA, Cyclin D1, Runx2, and Runx3 in the limb epiphyseal cartilages of newborn Cbfβf/fCol2α1-Cre and WT mice. β-Tubulin was used as a loading control. (E) Quantification of protein expression level normalized to β-Tubulin in “D”. P, proliferative zone; R, resting zone; H, hypertrophic zone; pre-H, pre-hypertrophic zone. Data in A-C are representative for 3 mice per group.
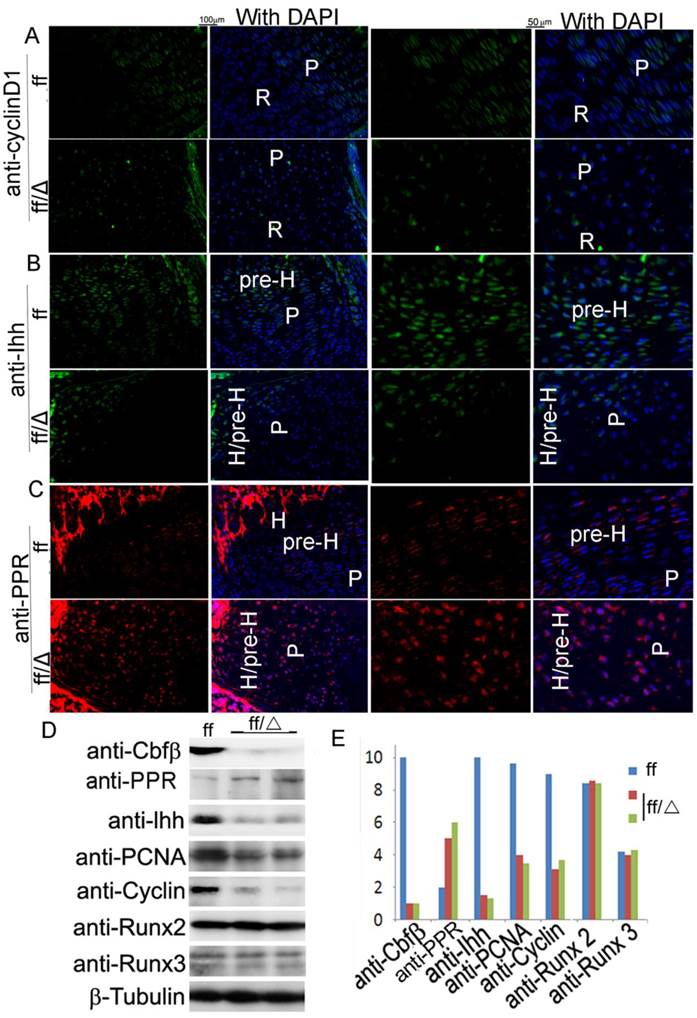
Furthermore, Western blot analysis showed that the levels of Cyclin D1 and PCNA were reduced by three-fold in the mutant mice (Fig. 6D, E). Strikingly, while the level of Ihh was reduced by ten-fold in the mutant mice, the level of PPR was increased by 2.5-fold, further confirming that the Ihh-PTHrP negative feedback loop was disrupted in the mutant mice (Fig. 6D, E). Our Western blot analysis also confirmed that the deletion of Cbfβ in the chondrocytes did not affect the expression levels of Runx2 and Runx3 (Fig. 6D, E). Increased PPR expression in the mutant mice inhibited chondrocyte maturation which contributes to elongation of the bone epiphysis and thereby affects endochondral ossification. Collectively, these results indicate that the Col2α1-Cre-mediated Cbfβ deletion affects chondrocytes by impacting the expression of Ihh and its targeted gene Cyclin D1 and disrupting the Ihh-PTHrP negative regulatory loop, which then affects chondrocyte maturation and endochondral ossification.
Discussion
We excised Cbfβ specifically in the chondrocyte lineage cells using the Col2α1-Cre mice (Fig. 1) to generate a novel mice model to study the role of Cbfβ in postnatal cartilage and endochondral bone development. Interestingly, the Cbfβf/fCol2α1-Cre mice displayed dwarfism, shortened limbs, delayed ossification, deformed growth plates and defective endochondral bone formation during postnatal development (Figs 1-3). The epiphyseal cartilages were unexpectedly elongated in the mutant mice, with a drastic decrease in proliferative and hypertrophic chondrocytes (Fig. 3E-H). Primary spongiosa formation was delayed in these mutant mice at birth, and P30 mutant mice exhibited reduced endochondral bone formation (Fig. 4A,B).
Cbfβ may control skeletal development by regulating the Ihh-PTHrP negative feedback loop
Induction of Ihh by Runx2 through the heterodimeric complexes with Cbfβ is significantly repressed in the cartilage of the Cbfβf/fCol2α1-Cre mice (Fig. 6B, D, E). Consistently, the expression of Cyclin D1 was also repressed in the mutant growth plates (Fig. 6A, D, E). PTHrP, induced by Ihh, in turn inhibits chondrocyte maturation and Ihh expression [18]. This Ihh-PTHrP negative regulatory loop maintains the chondrocyte proliferation pool and keeps pace of chondrocyte maturation [18]. Interestingly, the PTHrP receptor was upregulated in the mutant mice (Fig. 6C, D, E). While chondrocytes in Ihh-null mice display inhibited proliferation and pre-maturation [19], both were impeded in Cbfβf/f Col2α1-Cre mice (Fig. 3). Enhanced chondrocyte hypertrophy, which was supposed to be observed with a low level of Ihh, may be compensated for by high expression of PTHrP receptor in the mutant mice. In addition, Ihh is also critical to endochondral osteoblastogenesis [23]. Consistently, decreased trabecular bone was observed in the mutant mice in endochondral bone.
Hedgehog signaling, including Ihh, in mature osteoblasts regulates bone resorption by controlling RANKL expression [25]. Interestingly, although Ihh expression was down-regulated in the Cbfβf/f Col2α1-Cre mice, osteoclast number was not changed. Thus, it is possible that Cbfβ deficiency in the growth plates may influence the secretion of other cytokines and cell signaling molecules (e.g. Wnt, IL6, TNFa), aside from Ihh, which may play compensatory functions in osteoclast formation.
Cbfβ acted as a doorkeeper for cartilage and bone development in endochondral skeletal development
The hypertrophic zone not only serves as a scaffold for bone formation, but also produces osteoblastogenesis-related signals (e.g. Ihh) [22, 23, 26, 27]. During the process of endochondral bone formation, hypertrophic chondrocytes undergo mineralization and apoptosis and are substituted gradually by osteoblasts, which lay down bone matrix proteins and minerals and develop into osteocytes [27]. Runx2 has been reported to be essential for chondrocyte hypertrophy [28, 29]. Deletion of Cbfβ resulted in retarded growth of trabecular bone and lamellar bone, and increased growth of epiphyseal cartilage, which protruded into both primary and secondary spongiosa (Fig. 4). The largely delayed chondrocyte hypertrophy keeps chondrocyte proliferation slow but constitutive, which contributes to the elongated growth plate. In addition, the signal to end cartilage growth and initiate bone formation was missing. This delayed ossification was not limited to long bones, but also occurs in vertebrae, ribs, and certain skull elements (Fig. 1).
Cbfβ deletion in the chondrocyte lineage causes some clinical features of CCD in mice
Cbfβf/fCol2α1-Cre mice show many clinical features of CCD such as hypoplastic clavicles, shortened limbs, and dwarfism [3]. Whereas CCD is known to stem mostly from Runx2 deficiency, there are reports of clinical features of CCD in some patients with Cbfβ haploinsufficiency [30, 31], indicating that lack of Cbfβ may also play a role in the pathogenesis of CCD. Our findings further support the idea that Cbfβ deficiency may be responsible for some cases of CCD which have not been linked with Runx2 deficiency [14]. Nevertheless, although Cbfβf/fCol2α1-Cre mice displayed malocclusion, supernumerary teeth and/or tooth defect, another critical feature of CCD, was not observed in Cbfβf/fCol2α1-Cre mice. This observation indicates that deletion of Cbfβ in the chondrocyte lineage cells does not fully recapitulate the clinical features of CCD in mice. Most importantly, this finding suggests that CCD's clinical features may result from a lack of Cbfβ in chondrocytes during postnatal development.
In summary, our study reveals the crucial role of Cbfβ in chondrocyte development and postnatal bone formation by regulating chondrocyte development for endochondral ossification. We show a working model by which Cbfβ may inhibit PPR and promote Ihh to keep the proper regions of non-hypertrophic chondrocytes and the proper regions of hypertrophic chondrocytes, and to preserve endochonral bone formation. Taken together, this work provides important insights into the role of Cbfβ during postnatal skeletal development and also supports a role for Cbfβ in the pathogenesis of CCD. Further understanding of the role of Cbfβ in postnatal bone development may provide a greater understanding of the role of Cbfβ in skeletal development and many bone diseases stemming from defective bone development.
Materials and methods
Generation of Cbfβ CKO mice
The Col2α1-Cre mouse line was kindly provided by Dr. Rosa Serra from The University of Alabama at Birmingham (UAB) (Birmingham, AL, USA). Cbfβf/f mice (Jackson Laboratory, B6.129P2-Cbfβtm1Itan/J) were crossed with Col2α1-Cre mice to generate Cbfβf/+Col2α1-Cre mice, which were intercrossed to obtain homozygous CKO (Cbfβf/f Col2α1-Cre) mice. The genotypes of the mice were determined by polymerase chain reaction (PCR) using tail DNA and following primers, Cbfβ deletion & flox, 5'-TGTCTGAAGACAACTACAGTGTAC-3', 5'-CAACCCATTACCATTGGATTGGC-3', 5'-CTCTCTGAACACTATATCAGTTCC-3'; cre, 5'-CCTGGAAAATGCTTCTGTCCGTTTGCC-3', 5'-GGCGCGGCAACACCATTTTT-3'. All mice were maintained under a 12-hour light-dark cycle with ad libitum access to food and water at the UAB Animal Facility. The study was approved by the UAB Institutional Animal Care and Use Committee and conformed to NIH guidelines.
Skeletal preparation
For skeletal preparations, mice were skinned, eviscerated, fixed in 95% ethanol, stained with Alizarin red and/or Alcian blue and sequentially cleared in 1% KOH. Cartilage and mineralized bone were characterized by different colors (blue and red, respectively) after the stain, according to standard procedures [32].
Histology and tissue preparation
Histology and tissue preparation were performed as described previously [33]. Generally, femurs and tibiae of mice were harvested, skinned, and fixed in 4% paraformaldehyde overnight. Samples were then dehydrated in ethanol solution and decalcified in 10% EDTA for 1 to 4 weeks. For paraffin sections, samples were dehydrated in ethanol, cleared in xylene, embedded in paraffin, and sectioned at 6 μm with Leica microtome and mounted on Superfrost Plus slides (Fisher). For frozen sections, samples were infiltrated in 30% sucrose, embedded in OCT media, sectioned at 8 µm with a freezing microtome and affixed to Superfrost Plus Gold slides (Fisher). H&E staining was performed as described previously [34].
Immunofluorescence analysis
Osteoblast and chondrocyte genes were analyzed by immunofluorescence using the following primary antibodies: rabbit-anti-Ihh (Abcam, ab39634), rabbit-anti-Cyclin D1 (Santa Cruz, sc-753), mouse-anti-PTHrP-R (Santa Cruz, sc-12722); and these secondary antibodies: FITC-goat-anti-mouse IgG(H+L) and TR-goat-anti-rabbit IgG (H+L). Imaging was taken by Leica Confocal Microscope and Zeiss fluorescent microscope.
Preparation of protein samples from growth plate cartilage of newborn mice
Under a stereo microscope, hindlimbs from new-born mice were dissected from the body and soft tissues were removed. White parts of the limbs between the femora and tibiae were recognized as cartilage and were dissected from the bone tissues. Remaining soft tissues were carefully removed. The samples were then washed twice with 1X PBS, placed in a clean 2-ml EP tube and immersed with 100µl 2XSDS sample buffer (supplemented with DTT and protease inhibitors). Samples were lysed using an electronic homogenizer and boiled for 10 minutes.
Western blot analysis
Proteins were resolved on SDS-PAGE and electrotransferred onto nitrocellulose membranes. Immunoblotting was performed according to manufacturer instructions. Cbfβ, Runx3, Runx2, CyclinD1, PPR and Ihh protein levels were analyzed using the following primary antibodies: rabbit-anti-Cbfβ (Santa Cruz, sc-56751), rabbit-anti-Runx3 (Sigma, AV37263); rabbit-anti-Runx2 (Abcam, ab23981), and Horseradish peroxidase-linked anti-rabbit IgG (7074) and Horseradish peroxidase-linked anti-mouse IgG (7076) were from Cell Signaling. Other antibodies were the same as those used for immunofluorescence.
Proliferation assay
Proliferating cell nuclear antigen (PCNA) immunostaining was performed using a commercial kit (93-1143; Zymed Laboratories, Inc.) with horseradish peroxidase reaction and subsequent detection by DAB (Vector Laboratories SK-4100) as per the manufacturer's instructions.
Apoptosis assay
Fozen sections from P14 mouse femurs were used for apoptosis assay. Apoptotic chondrocytes were detected by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using a commercial kit (11684795910; Roche) following manufacturer's manual, counterstained by DAPI and subsequent detection by Zeiss fluorescent microscope.
Primary cell culture
Growth plate chondrocytes from newborn mice were derived as described [35] and micromass cultures were applied for 7 days [36]. Chondrocyte nodules were examined by Alcian blue staining [35].
Statistical analysis
Data were presented as mean ± SD (n=3 or more). Statistical significance was assessed using Student's t-test. Values were considered statistically significant at P<0.05. Results were representative of at least four individual experiments.
Abbreviations
Cbfβ: Core binding factor beta; Col2α1-Cre: Collagen 2α1 promoter driven Cre; Ptc1: patched; Ihh: Indian hedgehog; Runxs: runt-related transcription factors; CCD: cleidocranial dysplasia; P7: postnatal seven-day-old; WT: wild-type; PTHrP: parathyroid hormone-related peptide; PPR: PTHrP receptor; IHC: immunohistochemistry; IF: immunofluorescence; TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Acknowledgements
We thank Ms. Suzy Newton and Ms. Christie Paulson for their excellent assistance with the manuscript. We appreciate the assistance of the Center for Metabolic Bone Disease (P30 AR046031), Small Animal Phenotyping Core, Metabolism Core, and Neuroscience Molecular Detection Core Laboratory (P30 NS0474666) at UAB. This work was supported by the National Institutes of Health grants AR-44741 (Y.P.L.) and AR-055307 (Y.P.L.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. AML1, AML2, and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics. 1994;23:425-32
2. Westendorf JJ, Hiebert SW. Mammalian runt-domain proteins and their roles in hematopoiesis, osteogenesis, and leukemia. Journal of cellular biochemistry. 1999(Suppl 32-33):51-8
3. Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. Journal of medical genetics. 1999;36:177-82
4. Liakhovitskaia A, Lana-Elola E, Stamateris E, Rice DP, van 't Hof RJ, Medvinsky A. The essential requirement for Runx1 in the development of the sternum. Dev Biol. 2010;340:539-46 doi:10.1016/j.ydbio.2010.02.005
5. Kimura A, Inose H, Yano F, Fujita K, Ikeda T, Sato S. et al. Runx1 and Runx2 cooperate during sternal morphogenesis. Development (Cambridge, England). 2010;137:1159-67
6. Wang Y, Belflower RM, Dong YF, Schwarz EM, O'Keefe RJ, Drissi H. Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J Bone Miner Res. 2005;20:1624-36 doi:10.1359/JBMR.050516
7. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747-54
8. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K. et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755-64
9. Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR. et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765-71
10. Kobayashi H, Gao Y, Ueta C, Yamaguchi A, Komori T. Multilineage differentiation of Cbfa1-deficient calvarial cells in vitro. Biochem Biophys Res Commun. 2000;273:630-6 doi:10.1006/bbrc.2000.2981
11. Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K. et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952-63 doi:10.1101/gad.1174704
12. Sasaki K, Yagi H, Bronson RT, Tominaga K, Matsunashi T, Deguchi K. et al. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:12359-63
13. Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X. et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87:697-708
14. Kundu M, Javed A, Jeon JP, Horner A, Shum L, Eckhaus M. et al. Cbfbeta interacts with Runx2 and has a critical role in bone development. Nat Genet. 2002;32:639-44 doi:10.1038/ng1050
15. Miller J, Horner A, Stacy T, Lowrey C, Lian JB, Stein G. et al. The core-binding factor beta subunit is required for bone formation and hematopoietic maturation. Nat Genet. 2002;32:645-9
16. Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S. et al. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet. 2002;32:633-8 doi:10.1038/ng1015
17. Kanatani N, Fujita T, Fukuyama R, Liu W, Yoshida CA, Moriishi T. et al. Cbf beta regulates Runx2 function isoform-dependently in postnatal bone development. Dev Biol. 2006;296:48-61
18. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332-6 doi:10.1038/nature01657
19. Karp SJ, Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, McMahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development (Cambridge, England). 2000;127:543-8
20. Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145-6
21. Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F. et al. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med. 2007;204:1749-55 doi:10.1084/jem.20062456
22. Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS. et al. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6382-7 doi: 10.1073/pnas.0608449104
23. Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development (Cambridge, England). 2004;131:1309-18 doi:10.1242/dev.01006
24. Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development (Cambridge, England). 2001;128:5099-108
25. Mak KK, Bi Y, Wan C, Chuang PT, Clemens T, Young M. et al. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Developmental cell. 2008;14:674-88 doi:10.1016/j.devcel.2008.02.003
26. Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Developmental cell. 2002;2:389-406
27. Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. The international journal of biochemistry & cell biology. 2008;40:46-62 doi:10.1016/j.biocel.2007.06.009
28. Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y. et al. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275:8695-702
29. Drissi MH, Li X, Sheu TJ, Zuscik MJ, Schwarz EM, Puzas JE. et al. Runx2/Cbfa1 stimulation by retinoic acid is potentiated by BMP2 signaling through interaction with Smad1 on the collagen X promoter in chondrocytes. J Cell Biochem. 2003;90:1287-98 doi:10.1002/jcb.10677
30. Khan A, Hyde RK, Dutra A, Mohide P, Liu P. Core binding factor beta (CBFβ) haploinsufficiency due to an interstitial deletion at 16q21q22 resulting in delayed cranial ossification, cleft palate, congenital heart anomalies, and feeding difficulties but favorable outcome. AmJMedGenetA. 2006;140:2349-54
31. Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S. et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773-9
32. McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299-301 doi:10.1002/tera.1420220306
33. Yang S, Hao L, McConnell M, Xuedong Z, Wang M, Zhang Y. et al. Inhibition of Rgs10 Expression Prevents Immune Cell Infiltration in Bacteria-induced Inflammatory Lesions and Osteoclast-mediated Bone Destruction. Bone Research. 2013;1:267-81
34. Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP. Haploinsuffciency for Znf9 in Znf9+/- mice is associated with multiorgan abnormalities resembling myotonic dystrophy. Journal of molecular biology. 2007;368:8-17 doi:10.1016/j.jmb.2007.01.088
35. Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nature protocols. 2008;3:1253-60
36. DeLise AM, Stringa E, Woodward WA, Mello MA, Tuan RS. Embryonic limb mesenchyme micromass culture as an in vitro model for chondrogenesis and cartilage maturation. Methods in molecular biology (Clifton, NJ. 2000;137:359-75
Author contact
![]() Corresponding author: Wei Chen, MD: Department of Pathology, University of Alabama at Birmingham, SHEL 815, 1825 University Blvd, Birmingham, AL 35294, USA, Tel: 205.975.2605, Fax: 205.975.4919, E-mail: wechenedu or Yi-Ping Li, PhD: Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham, AL 35294, USA, Tel: 205.975.2606, Fax: 205 975.4919, E-mail: ypliedu.
Corresponding author: Wei Chen, MD: Department of Pathology, University of Alabama at Birmingham, SHEL 815, 1825 University Blvd, Birmingham, AL 35294, USA, Tel: 205.975.2605, Fax: 205.975.4919, E-mail: wechenedu or Yi-Ping Li, PhD: Department of Pathology, University of Alabama at Birmingham, SHEL 810, 1825 University Blvd, Birmingham, AL 35294, USA, Tel: 205.975.2606, Fax: 205 975.4919, E-mail: ypliedu.