Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):873-881. doi:10.7150/ijbs.8868 This issue Cite
Research Paper
Intermittent Hypothermia Is Neuroprotective in an in vitro Model of Ischemic Stroke
Sui-yi Xu1,2, Ya-fang Hu1, Wei-pin Li2, Yong-ming Wu1, Zhong Ji1, Sheng-nan Wang1, Ke Li3, Su-yue Pan1 ![]()
1. Department of Neurology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China;
2. Department of Neurosurgery, Shenzhen Key Laboratory of Neurosurgery, Shenzhen Second People's Hospital, Shenzhen University 1st Affiliated Hospital, Shenzhen 518035, China;
3. Research Center of Clinical Medicine, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China.
Received 2014-2-18; Accepted 2014-7-14; Published 2014-7-29
Abstract
Objective: To investigate whether the intermittent hypothermia (IH) protects neurons against ischemic insult and the potential molecular targets using an in vitro ischemic model of oxygen glucose deprivation (OGD).
Methods: Fetal rat cortical neurons isolated from Day E18 rat embryos were subjected to 90-min OGD and hypothermia treatments during reoxygenation before examining the changes in microscopic morphology, cell viability, microtubule- associated protein 2 (MAP-2) release, intracellular pH value and calcium, reactive oxygen species (ROS) generation, mitochondrial membrane potential (△Ψm) and neuronal death using cell counting kit (CCK-8), enzyme-linked immunosorbent assay (ELISA), BCECF AM, Fluo-3 AM, DCFH-DA and dihydroethidium (DHE), JC-1 staining and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL), respectively.
Results: 90-min OGD induced morphologic abnormalities, cell viability decline, MAP-2 release, intracellular acidosis, calcium overload, increased ROS generation, △Ψm decrease and cell death in primary neurons, which was partially inhibited by continuous hypothermia (CH) and intermittent hypothermia (IH). Interestingly, 6-h CH was insufficient to reduce intracellular calcium overload and stabilize mitochondrial membrane potential (△Ψm), while 12-h CH was effective in reversing the above changes. All IH treatments (6×1 h, 4×1.5 h or 3×2 h) effectively attenuated intracellular free calcium overload, inhibited ROS production, stabilized mitochondrial membrane potential (△Ψm) and reduced delayed cell death in OGD-treated cells. However, only IH intervals longer than 1.5 h appeared to be effective in preventing cell viability loss and intracellular pH decline.
Conclusion: Both CH and IH were neuroprotective in an in vitro model of ischemic stroke, and in spite of shorter hypothermia duration, IH could provide a comparable neuroprotection to CH.
Keywords: Ischemic stroke, Hypothermia, Neuroprotection, Primary neuronal culture.
Introduction
Numerous clinical trials have been conducted to test potential neuroprotective compounds for ischemic stroke. Miserably, all these attempts have clinically failed. The failure can be partially explained by the fact that cerebral ischemia initiates a complex series of signaling cascades contributing to neuronal cell death in the affected brain, while all the tested agents only target a single event of injury. Apparently, these agents are not effective enough in protecting neural cells against ischemic injury [1]. Neuroprotection is now focusing on the development of multi-target nonpharmacological strategies, such as therapeutic hypothermia [2].
Among all tested hypothermia strategies, intracarotid cold saline infusion (ICSI) appears to be most promising [3]. In a recent study, we showed a synergic effect of ICSI and the neuroprotective agent following ischemic stroke [4]. ICSI protects the brain from ischemic injury from two facets: ICSI-induced hypothermia per se that slows the metabolic rate of affected tissue and the accumulation of lactate acid while improving glucose utilization and intra-artery flushing which is believed to help flush out the toxic chemicals accumulated in the ischemic region [5]. Currently, there are two ICSI approaches under testing in the treatment of ischemic stroke, continuous ICSI and intermittent ICSI. Continuous ICSI requires a large volume of cold saline to achieve a therapeutic hypothermia and thus generates significant side effects such as hematocrit dilution. Intermittent ICSI can significantly reduce the volume of cold saline; using a rat model of focal cerebral ischemia, intermittent ICSI could lower down brain temperature effectively, resulting in prolonged continuous hypothermia and greater neuroprotection, while minimizing its impact on hematocrit comparing to continuous ICSI [6]. Of note, intermittent ICSI could solve the problem of infusion volume; however it may produce re-warming complications to stroke patients. To date, it remains unknown whether the fluctuation of brain temperature would compromise the neuroprotective effect of hypothermia in ischemic stroke.
In this study, we applied an in vitro oxygen glucose deprivation (OGD) model of primary cortical neuronal cultures to investigate whether the intermittent hypothermia (IH) protected neurons against ischemic insult and the molecular changes underlying IH-afforded neuroprotection.
Materials and methods
Primary cortical neuronal culture
The animal experiments were approved by the Ethics Committee of Southern Medical University. Pregnant Sprague Dawley rats were purchased from the Experimental Animal Center of Southern Medical University. Day E18 embryos were microdissected under a stereoscope (Olympus, Japan). Primary neuronal cultures were performed as previously described [7]. The purity of neuronal culture was found to be over 95% by flow cytometry analysis (Thermo, USA) or microscopic examination after immunostaining with β-tubulin III.
OGD and hypothermia treatments
Neurons at 3 DIV were used for OGD experiments. In brief, the original phenol red-free neurobasal medium (Gibco, USA) was removed and the cells were washed three times with phosphate-buffered saline (PBS) to ensure a glucose-free culture condition. A small volume of PBS was added to create a thin layer that was sufficient to cover the neurons in the cell culture plates [8]. Cells were then incubated in an anaerobic glovebox (Thermo, USA) that was flushed with 95% N2/5% CO2 for 15 min. The chamber was sealed and kept at 37℃ for 90min.
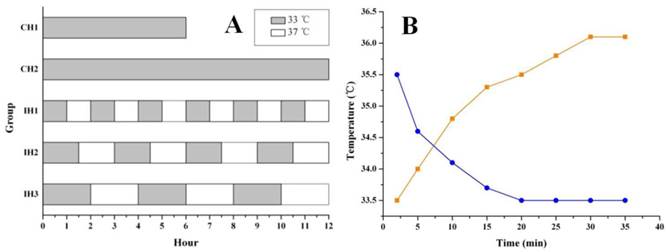
After 90 min of OGD treatment, the neurons were returned to normal culture conditions and subjected to hypothermia treatment, as shown in Figure 1A. Hypothermia intervention included both continuous hypothermia (CH) and intermittent hypothermia (IH). Hypothermia was achieved by transferring the cells into a 33℃ culture incubator (Thermo, USA) and normothermia was achieved by returning the cells back to the 37℃ culture incubator (Both cell incubators were supplied with 5% CO2). Two hypothermia durations 6 and 12 h were tested for both CH and IH groups. The intervals between hypothermia and normothermia were 1 h, 1.5 h and 2 h for IH1, IH2 and IH3 groups, respectively. A temperature probe (Physitemp, USA) was applied to monitor the temperature changes of the culture medium when moving the cells back and forth between 33℃ and 37℃ culture incubators (Figure 1B). TUNEL assay was performed 48 h after OGD treatment to assess cell death, while other parameters were measured at 12 h after OGD treatment.
Graph A showing the hypothermia administration after OGD. The gray stripe represented the hypothermic culture time at 33℃. The white gaps were normothermic intervals at 37℃. The real temperature change in culture vessel was monitored at different time points until the platform (graph B). The blue/orange curves showed the cooling/rewarming cycles in hypothermic/normothermic incubator.
Cell-viability testing
Cell viability was assessed using the CCK-8 kit (Beyotime, China) according to manufacturer's instruction. In brief, 20 μL of CCK-8 solution and 200 μL of cell culture supernatants (105 cells) were added to each well of the microtiter plates (Corning, USA). The reaction system was incubated at 37℃ for 1 hour. The absorbance was measured in a microplate reader (Biotek, USA) at 450 nm.
Enzymatic assessment of MAP-2 release in OGD-treated cells
An enzyme linked immunosorbent assay (ELISA) kit (Blue Gene, China) was used to assess microtubule associated protein 2 (MAP-2) release in OGD-treated cells. In brief, 100 μL cell culture supernatants were added to antibody pre-coated microtiter plate and incubated with 50 μL conjugate at 37℃ for 1 h before adding MAP-2's substrate. After 15-min incubation at 25℃, a 50 μL stop solution was added and absorbance was assessed at 450 nm.
Intracellular acidosis detection
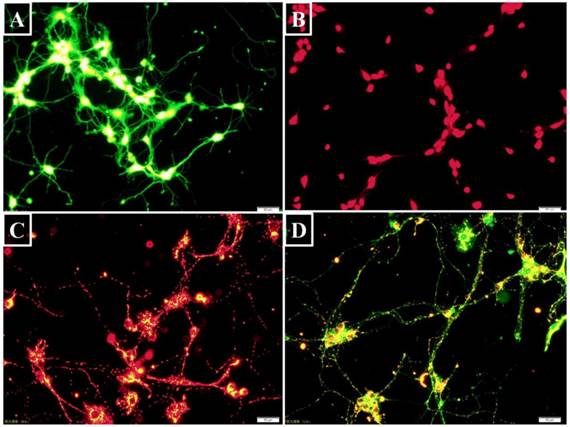
Intracellular pH value was assessed using fluorescent probe 2',7'-bis- (2-carboxyethyl)-5-(and-6)-carboxyfluorescein-acetoxymethyl ester (BCECF AM) (Beyotime, China). After entering the cell, it is hydrolyzed to BCECF that is trapped inside the cells. BCECF can then be excited to emit green fluorescence (Figure 2A). The neurons were digested by a 0.25% trypsin-EDTA solution (Sigma, USA) and harvested by centrifuging at 800 rpm (Beckman, USA). BCECF AM was diluted with anhydrous dimethyl sulfoxide (DMSO) to a final concentration of 5 μM [9]. BCECF AM and the neurons were co-incubated in dark for 30 minutes at 37℃ before flow cytometry analysis with an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Intracellular targets labeling with fluorescent probes (Bar=50 μm). (A) BCECF was a pH probe to detect intracellular acidosis. The green fluorescence intensity was dependent on intracellular pH level. (B) The hydrolyzed ethidium showing red fluorescence was correlated with intracellular superoxide- anion level. (C) J-aggregates showed red fluorescence when △Ψm was high. (D) On the contrary, JC-1 monomer appeared green fluorescence when △Ψm was low.
Intracellular calcium measurement
Fluo-3 AM (Beyotime, China) is the most commonly used fluorescent probe for the detection of intracellular calcium ion concentration. It is cleaved into Fluo-3 by esterase and trapped inside the cells. Fluo-3 AM was diluted to a final concentration of 5 μM with anhydrous DMSO and co-incubated in dark with the harvested neurons for 1 h at 37℃ to load the probe [10]. Calcium ion fluorescence was measured by flow cytomety with an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Reactive oxygen species detection
A reactive oxygen species (ROS) assay kit (Beyotime, China) was used to detect ROS using the fluorescent probe DCFH-DA, which can freely penetrate the cell membrane. DCFH-DA is not fluorescent by itself. After entering the cell, it is hydrolyzed to DCFH that is trapped inside the cells. Intracellular ROS oxidize the DCFH to generate fluorescent DCF, which is correlated with the quantity of ROS in the cell [11]. DCFH-DA was diluted to a final concentration of 10 μM with neurobasal medium. Adherent neurons were digested to form a cell suspension and incubated with DCFH-DA at 37℃ for 20 minutes [12]. The fluorescence in each group was assessed by flow cytometry with an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Dihydroethidium (DHE) is a fluorescent superoxide-anion probe (Beyotime, China). Following uptake by living cells, intracellular superoxide anions act on DHE to dehydrogenate it to ethidium that combines with DNA or RNA to generate red fluorescence (Figure 2B). DHE was diluted to a final concentration of 5 μM with DMSO. The cells were harvested as above and incubated with DHE at 37℃ for 30 minutes before flow cytometry analysis of DHE fluorescence with an excitation wavelength of 488 nm and an emission wavelength of 535 nm [13].
Mitochondrial depolarization assessment
A mitochondrial membrane potential (△Ψm) assay kit (Beyotime, China) was used to assess early apoptosis. When △Ψm is high, the fluorescence probe JC-1 accumulates in the mitochondrial matrix to form red fluorescent J-aggregates (Figure 2C). Conversely, when △Ψm was low, JC-1 does not accumulate and exist as a green fluorescent monomer in the mitochondrial matrix (Figure 2D) [14]. This change in JC-1 fluorescence reflects mitochondrial membrane functional change and is used as an early indicator of apoptosis [15]. J-aggregates and monomers were assessed by flow cytometry with an excitation wavelength of 525 nm or 490 nm and emission wavelength of 590 nm or 530 nm, respectively.
Cell death determination
TdT-mediated dUTP Nick-End Labeling (TUNEL) was performed using a one-step assay kit (Beyotime, China). The cell suspension was harvested and washed with PBS. The neurons were fixed with 4% paraformaldehyde and penetrated with 0.1% Triton X-100, as previously described [16]. Then, the cells were incubated in dark with 50 μL TUNEL solutions for 60 minutes at 37℃ before flow cytometry analysis with an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Data analysis
Data were expressed as mean ± standard error (SE). Statistical analyses were performed using IBM SPSS (Version 19.0, USA). One-way analysis of variance (ANOVA) was used to assess the differences in all observed indicators between the different groups. The least significant difference (LSD) or Dunnett test was used for multiple comparisons. Statistical significance was set at p<0.05.
Results and Discussion
IH ameliorates OGD-induced morphological abnormalities in neurons
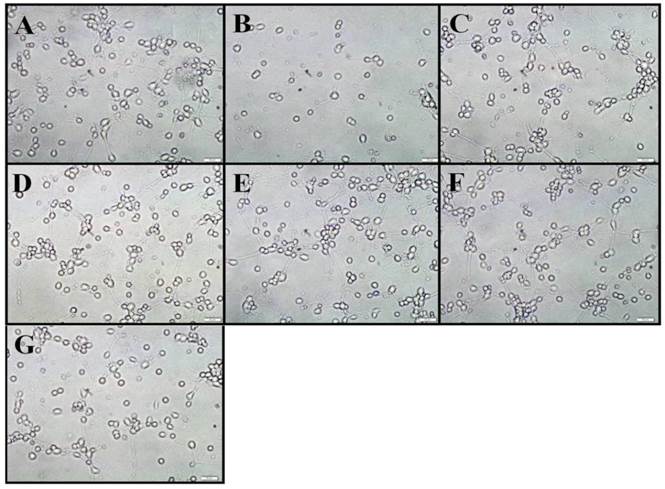
We first examined the morphological changes in OGD-treated neurons and whether intermittent hypothermia treatment during reoxygenation could prevent this change. Three bright fields were randomly chosen for microscopic observation. In the control group, the cells showed strong body refraction, vigorous axonal growth and network-like connection with surrounding neurons. Exposure the neurons to 90-min OGD with 12 h reoxygenation resulted in a significant reduction in cell density and axonal disintegration and beaded degeneration. Of note, both CH and IH treatments almost completely abolished the morphological changes induced by OGD treatment. As shown in Figure 4, hypothermia-treated cells showed similar body morphology and only a few residual axonal fractures when compared to the control group.
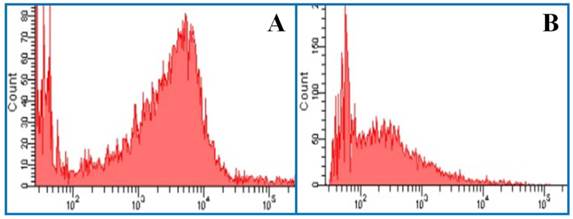
Histogram of flow cytometry. (A) The mean fluorescence intensity of J-aggregates. Excitation wavelength = 525 nm. Emission wavelength = 590 nm. (B) The mean fluorescence intensity of JC-1 monomer. Excitation wavelength = 490 nm. Emission wavelength = 530 nm.
Cell morphology observation under an inverted microscope. Graph A-G showed normal, OGD, CH1, CH2, IH1, IH2, IH3 group, respectively. It demonstrated that OGD led to neuronal loss and axonal disintegration. IH could provide a comparable neuroprotection to CH. Bar=50 μm.
Lack of oxygen and glucose supply is centered in the process of ischemia- or OGD-induced injury, resulting in energy deficiency, cell depolarization, and cytotoxic edema resulting from excessive intracellular accumulation of salt and water, and eventually cell death. Hypothermia slows the metabolic rate of ischemic tissue and improves glucose utilization and is thus believed to be able to alleviate ischemic injury. Indeed, hypothermia has been shown to reduce cytotoxic edema [17]. In another study, cooling the cells to 33℃ led to a nearly 50% decrease in neuronal death under a microscope [18]. These results are consistent with our morphological observations. However, neuronal morphological abnormalities alone may be inadequate to reflect the functional status of ischemic neurons because injured cells may still look “normal”. As thus, the efficacy of hypothermia treatment may be over- or under-estimated if simply using morphology as the outcome parameter. Therefore, we investigated several other intracellular molecular changes closely associated with ischemic injury.
The effect of IH on cell viability
The cell viability was assessed using the CCK-8 kit. As shown in Figure 5A. 90-min OGD induced a significant decrease in cell viability when compared with normal control group. Both 6-h CH (CH1) and 12-h CH (CH2) treatments significantly inhibited cell viability loss in OGD-treated cells. However in IH groups, neuroprotection was only observed for IH2 (4×1.5 h) and IH3 (3×2 h) treatments, but not for IH1 (6×1 h).
The overall data suggested that different hypothermia strategies provided different levels of neuroprotection. (A) CCK-8 = Cell viability (n=8). (B) MAP-2 = Microtubule associated protein 2 (n=10). (C) BCECF-AM = pH (n=4). (D) Fluo-3 AM = Free calcium ion (n=5). (E) DCFH-DA = ROS (n=3). (F) Dihydroethidium = Superoxide anion (n=5). (G) JC-1 = Monomer/J-aggregates (n=3). (H) TUNEL (n=5). *p<0.05, compared with OGD group.
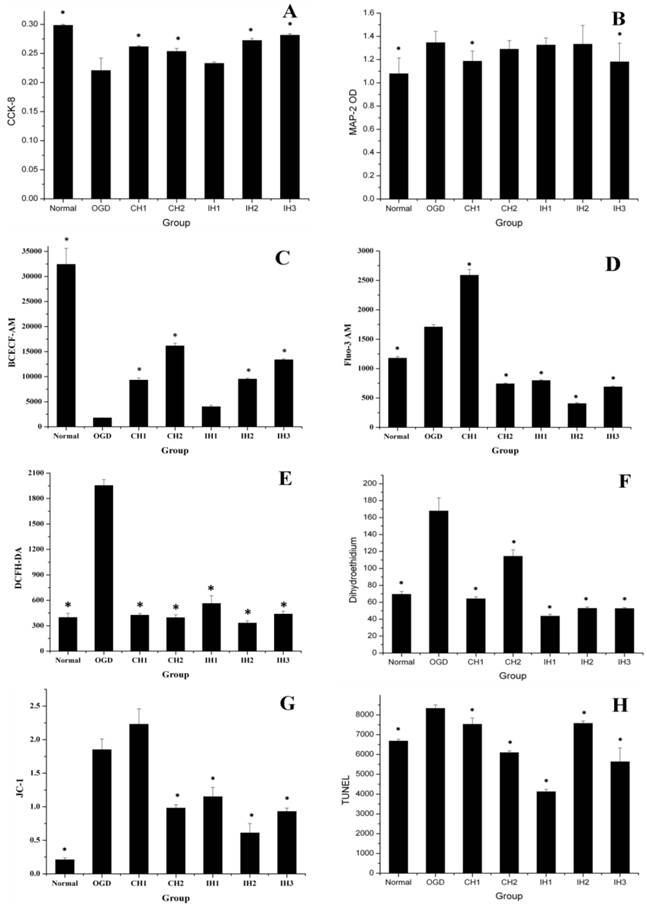
Hypothermia has been shown to protect microglia and oligodendrocyte cells against ischemic injury in vitro [19, 20]. In our experiment, our results that IH1 group failed to show a protection suggest that 1-h of hypothermia duration with 1-h interval may not be adequate to reverse ischemic injury induced by 90-min OGD. This result suggests that a minimum duration of initial cooling may be required for IH to produce a neuroprotection. However, future studies are needed to clarify whether the subsequent cycles are actually needed or not.
The effect of hypothermia treatment on MAP-2 release in OGD-treated cells
MAP-2 plays an important role in neuronal damage [21]. Therefore, we attempted to detect MAP-2 release in the supernatant that could reflect hypothermic neuroprotection. As shown in Figure 5B, MAP-2 levels in the supernatant significantly increased after an OGD. Unexpectedly, only CH1 and IH3 treatments reduced MAP-2 release induced by OGD treatment, while all other hypothermia treatments were not effective.
MAP-2 exists primarily in the neurons and dendrites of the central nervous system and is involved in neuronal development, structural stability and synaptic plasticity. MAP-2 within a neuron is gradually lost when the cell is damaged [22]. One-h OGD treatment has been shown to induce MAP-2 release, and 4 h of CH at 33℃ inhibits this damage [23]. In this study, we failed to observe a consistent effect of hypothermia treatment on MAP-2 release in OGD-treated neurons among different groups, indicating that MAP-2 might not be a sensitive parameter to assessing hypothermia's neuroprotection.
IH attenuates the fall of intracellular pH level
Intracellular pH value is a sensitive indicator of hypoxic metabolism. We assessed intracellular levels using the fluorescent probe BCECF AM. As shown in Figure 5C, the neuronal pH level significantly decreased after OGD. All of the hypothermia- treated groups attenuated the fall in intracellular pH levels, and CH and IH showed similar efficacy. In consistent with our result, Bao reported in an early study that hypothermia has been shown to reduce oxygen consumption and attenuate the fall in intracellular pH levels [24].
IH reduces intracellular free calcium overload
To evaluate the effect of CH and IH on reducing calcium overload, we used fluorescent probe FLUO-3 AM to measure intracellular free calcium. As shown in Figure 5D, OGD increased intracellular calcium compared with the normal group. The CH1 group could not inhibit the intracellular calcium overload, but CH2 and all of the IH groups were effective at inhibiting intracellular calcium overload.
Calcium overload is an important factor contributing to ischemic neuronal injury through promoting the generation of free radicals, endoplasmic reticulum injury and mitochondrial damage [25, 26]. Mitochondrial damage is another important pathway that leads to ischemic injury [27]. Mitochondrial calcium overload could initiate delayed neuronal death [28]. It has been shown that mild hypothermia at 35℃ effectively reduces intracellular free calcium levels in neurons 48 h after OGD treatment [29]. Our study further confirmed that a 6-h regimen of CH at 33℃ was not sufficient to reduce calcium overload. However, exposure of neurons to 12-h CH and IH were effective in reducing calcium overload.
IH inhibits ROS production in OGD-treated neurons
The levels of ROS detected by DCFH DA showed a significant increase after OGD treatment. As shown in Figure 5E, all CH and IH treatments inhibited ROS production. We also measured the generation of intracellular superoxide anions using the fluorescence probe DHE. As shown in Figure 5F, OGD significantly increased intracellular superoxide anion radicals compared with the normal control group. We found that all CH and IH treatments could effectively inhibit superoxide anion radical production induced by OGD and this result was true for both the CH and IH groups. Our results are consistent with previous findings that OGD elevated intracellular ROS levels in cultured neurons [30-32] and mild hypothermia inhibited ischemia-mediated ROS generation [33, 34].
IH stabilizes mitochondrial membrane potential (△Ψm)
When the mitochondrion is damaged, J-aggregates (red fluorescence) decline and accumulation of the JC-1 monomer (green fluorescence) increases. Therefore, the mean fluorescence intensity ratio (green/red fluorescence) is positively correlated with mitochondrial damage. As shown in Figure 5G, OGD caused significant damage to the mitochondria compared with the normal control group. All hypothermia treatments except CH1 were effective in stabilizing △Ψm.
The decline in △Ψm is an early indicator of apoptosis, which can occur after a transient OGD [35, 36]. The inner and outer membranes of neuronal mitochondria are deformed or ruptured after exposure to 30-90 min of OGD [28]. The neuronal body becomes blunt and the mitochondria are significantly depolarized [37]. These results are consistent with our observations. Moreover, hypothermia has been shown to protect the mitochondria against ischemia-induced apoptosis via inhibiting ROS production [38], which is further supported by our results here.
IH reduces cell death induced by OGD
In this study, TUNEL assay was used to assess cell death at 48 h after OGD [18]. The mean fluorescence intensity of TUNEL staining (Figure 5H) was significantly enhanced after OGD treatment, which was significantly reduced in all hypothermia- treated groups. The similar effect of CH has been previously reported in OGD-induced neurons [39]. Exposure to 30-90 min of OGD has been shown to cause varying degrees of neuronal injury [40]. The 90-min OGD was relatively short and cannot be compared to dense adult stroke in clinic. Future studies should explore different durations of OGD exposure to determine the effective time window.
Of note, 6-h CH is not sufficient to reduce OGD-induced intracellular calcium overload and stabilize mitochondrial membrane potential (△Ψm), while 12-h CH was effective in inhibiting these changes. For IH treatment, the intermittent intervals appeared to be a key factor determining the efficacy of neuroprotection. Interestingly, when the 6-h CH was divided into IH patterns of 6×1 h, 4×1.5 h or 3×2 h, intracellular calcium overload and △Ψm decrease were abolished in OGD-treated neurons. However, too short or long hypothermia intervals may be invalid or even harmful. Several key questions remain to be addressed to optimize IH therapy in future studies, such as hypothermia multitude, hypothermia intervals, and therapeutic time window.
There are several limitations of this study and cautions should be taken into data interpretation. First, fluorescent probe is only capable of provide semi-quantitative results and specificity is always an issue with this technique. Second, in this study, there was no delay between 90-min OGD and hypothermia. However, in the real world, most stroke patients cannot arrive at the hospital within 90 min after symptom onset, and when hypothermia treatment is delayed; its efficacy may decrease greatly. Lastly, using primary fetal neuronal cultures as our experimental model may be caveat because fetal neurons have a much greater anaerobic capacity and are more resistant to hypoxic injury than adult neurons.
Conclusion
In summary, we showed that different intermittent hypothermia strategies provided different levels of protection against ischemic neuronal injury (Table 1) via partially restoring cell viability, inhibiting intracellular acidosis, ROS generation, calcium overload and cell death. In addition, the neuroprotective effects were comparable between intermittent hypothermia and continuous hypothermia.
Comparison of experimental indicators.
| Group | Time | Morphology | CCK-8 | MAP-2 | BCECF | Fluo-3 | DCFH | DHE | JC-1 | TUNEL |
|---|---|---|---|---|---|---|---|---|---|---|
| CH1 | 6 h | + | + | + | + | - | + | + | - | + |
| CH2 | 12 h | + | + | - | + | + | + | + | + | + |
| IH1 | 6×1 h | + | - | - | - | + | + | + | + | + |
| IH2 | 4×1.5 h | + | + | - | + | + | + | + | + | + |
| IH3 | 3×2 h | + | + | + | + | + | + | + | + | + |
Notes:'—'represents invalid, '+' means effective.
Abbreviations
BCECF AM: 2',7'-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein-acetoxy methyl ester. CH: continuous hypothermia. CCK-8: cell counting kit 8. DIV: days in vitro. DHE: dihydroethidium. DMSO: dimethyl sulfoxide. IH: intermittent hypothermia. ICSI: intracarotid cold saline infusion. MAP-2: microtubule- associated protein 2. OD: optical density. OGD: oxygen glucose deprivation. PBS: phosphate-buffered saline. ROS: reactive oxygen species. TdT: terminal deoxynucleotidyl transferase. TUNEL: TdT-mediated dUTP Nick- End Labeling.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NO. 81271521), the Specialized Research Fund for the Doctoral Program of Higher Education (NO. 20124433110017), Guangdong Provincial Science & Technology Plan (NO. 20120309), the Scientific Research Item of Guangdong Provincial Education Department (NO. 2012KJCX0028). This work was also supported by Shenzhen Development and Reform Commission's Stroke Screening and Prevention Public Service Platform (www.szpb.gov.cn).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xu SY, Pan SY. The failure of animal models of neuroprotection in acute ischemic stroke to translate to clinical efficacy. Med Sci Monit Basic Res. 2013;19:37-45 doi:883750 [pii]
2. Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011;10:372-82 doi:10.1016/S1474-4422(11)70016-3S1474-4422(11)70016-3 [pii]
3. Ji Y, Wu Y, Ji Z, Song W, Xu S, Wang Y. et al. [Comparison of neuroprotective effects of hypothermia induced by different methods in rats with early cerebral ischemia]. Nan fang yi ke da xue xue bao = Journal of Southern Medical University. 2012;32:89-92
4. Song W, Wu YM, Ji Z, Ji YB, Wang SN, Pan SY. Intra-carotid cold magnesium sulfate infusion induces selective cerebral hypothermia and neuroprotection in rats with transient middle cerebral artery occlusion. Neurol Sci. 2012 doi:10.1007/s10072-012-1064-3
5. Ji Y, Hu Y, Wu Y, Ji Z, Song W, Wang S. et al. Therapeutic time window of hypothermia is broader than cerebral artery flushing in carotid saline infusion after transient focal ischemic stroke in rats. Neurol Res. 2012;34:657-63 doi:10.1179/1743132812Y.0000000061ner2822 [pii]
6. Ji YB, Wu YM, Ji Z, Song W, Xu SY, Wang Y. et al. Interrupted intracarotid artery cold saline infusion as an alternative method for neuroprotection after ischemic stroke. Neurosurgical focus. 2012;33:E10. doi:10.3171/2012.5.FOCUS1215
7. Xu SY, Wu YM, Ji Z, Gao XY, Pan SY. A modified technique for culturing primary fetal rat cortical neurons. J Biomed Biotechnol. 2012;2012:803930. doi:10.1155/2012/803930
8. Wise-Faberowski L, Raizada MK, Sumners C. Oxygen and glucose deprivation-induced neuronal apoptosis is attenuated by halothane and isoflurane. Anesth Analg. 2001;93:1281-7
9. Chen M, Zou X, Luo H, Cao J, Zhang X, Zhang B. et al. Effects and mechanisms of proton pump inhibitors as a novel chemosensitizer on human gastric adenocarcinoma (SGC7901) cells. Cell Biol Int. 2009;33:1008-19 doi:10.1016/j.cellbi.2009.05.004S1065-6995(09)00132-2 [pii]
10. Kuang CY, Yu Y, Guo RW, Qian DH, Wang K, Den MY. et al. Silencing stromal interaction molecule 1 by RNA interference inhibits the proliferation and migration of endothelial progenitor cells. Biochem Biophys Res Commun. 2010;398:315-20 doi:10.1016/j.bbrc.2010.06.088S0006-291X(10)01214-3 [pii]
11. Xu S, Zhou Z, Zhang L, Yu Z, Zhang W, Wang Y. et al. Exposure to 1800 MHz radiofrequency radiation induces oxidative damage to mitochondrial DNA in primary cultured neurons. Brain Res. 2010;1311:189-96 doi:10.1016/j.brainres.2009.10.062S0006-8993(09)02299-9 [pii]
12. Li Y, Bao Y, Jiang B, Wang Z, Liu Y, Zhang C. et al. Catalpol protects primary cultured astrocytes from in vitro ischemia-induced damage. Int J Dev Neurosci. 2008;26:309-17 doi:10.1016/j.ijdevneu.2008.01.006S0736-5748(08)00016-6 [pii]
13. Li Z, Shi K, Guan L, Cao T, Jiang Q, Yang Y. et al. ROS leads to MnSOD upregulation through ERK2 translocation and p53 activation in selenite-induced apoptosis of NB4 cells. FEBS Lett. 2010;584:2291-7 doi:10.1016/j.febslet.2010.03.040S0014-5793(10)00260-7 [pii]
14. Ma J, Zhang L, Li S, Liu S, Ma C, Li W. et al. 8,9-Epoxyeicosatrienoic acid analog protects pulmonary artery smooth muscle cells from apoptosis via ROCK pathway. Exp Cell Res. 2010;316:2340-53 doi:10.1016/j.yexcr.2010.05.013S0014-4827(10)00274-0 [pii]
15. Zhu XJ, Shi Y, Peng J, Guo CS, Shan NN, Qin P. et al. The effects of BAFF and BAFF-R-Fc fusion protein in immune thrombocytopenia. Blood. 2009;114:5362-7 doi:10.1182/blood-2009-05-217513blood-2009-05-217513 [pii]
16. Ma Y, Wang B, Li W, Ying G, Fu L, Niu R. et al. Reduction of intersectin1-s induced apoptosis of human glioblastoma cells. Brain Res. 2010;1351:222-8 doi:10.1016/j.brainres.2010.05.028S0006-8993(10)01135-2 [pii]
17. Drury PP, Bennet L, Gunn AJ. Mechanisms of hypothermic neuroprotection. Seminars in fetal & neonatal medicine. 2010;15:287-92 doi:10.1016/j.siny.2010.05.005
18. Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002;22:21-8 doi:10.1097/00004647-200201000-00003
19. Diestel A, Troeller S, Billecke N, Sauer IM, Berger F, Schmitt KR. Mechanisms of hypothermia-induced cell protection mediated by microglial cells in vitro. The European journal of neuroscience. 2010;31:779-87 doi:10.1111/j.1460-9568.2010.07128.x
20. Xiong M, Li J, Ma SM, Yang Y, Zhou WH. Effects of hypothermia on oligodendrocyte precursor cell proliferation, differentiation and maturation following hypoxia ischemia in vivo and in vitro. Experimental neurology. 2013;247:720-9 doi:10.1016/j.expneurol.2013.03.015
21. Johnson GV, Jope RS. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. Journal of neuroscience research. 1992;33:505-12 doi:10.1002/jnr.490330402
22. Zhang SX, Underwood M, Landfield A, Huang FF, Gison S, Geddes JW. Cytoskeletal disruption following contusion injury to the rat spinal cord. Journal of neuropathology and experimental neurology. 2000;59:287-96
23. Wang D, Zhao Y, Zhang Y, Zhang T, Shang X, Wang J. et al. Hypothermia protects against oxygen-glucose deprivation-induced neuronal injury by down-regulating the reverse transport of glutamate by astrocytes as mediated by neurons. Neuroscience. 2013;237:130-8 doi:10.1016/j.neuroscience.2013.01.056
24. Bao YH, Liang YM, Gao GY, Jiang JY. Lack of effect of moderate hypothermia on brain tissue oxygenation after acute intracranial hypertension in pigs. Journal of neurotrauma. 2010;27:433-8 doi:10.1089/neu.2007.0433
25. Paschen W, Frandsen A. Endoplasmic reticulum dysfunction--a common denominator for cell injury in acute and degenerative diseases of the brain? Journal of neurochemistry. 2001;79:719-25
26. Larsen GA, Skjellegrind HK, Moe MC, Vinje ML, Berg-Johnsen J. Endoplasmic reticulum dysfunction and Ca2+ deregulation in isolated CA1 neurons during oxygen and glucose deprivation. Neurochemical research. 2005;30:651-9
27. Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell calcium. 2010;47:122-9 doi:10.1016/j.ceca.2010.01.003
28. Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:5611-22 doi:10.1523/JNEUROSCI.0531-04.2004
29. Feiner JR, Bickler PE, Estrada S, Donohoe PH, Fahlman CS, Schuyler JA. Mild hypothermia, but not propofol, is neuroprotective in organotypic hippocampal cultures. Anesthesia and analgesia. 2005;100:215-25 doi:10.1213/01.ANE.0000142129.17005.73
30. Velly LJ, Canas PT, Guillet BA, Labrande CN, Masmejean FM, Nieoullon AL. et al. Early anesthetic preconditioning in mixed cortical neuronal-glial cell cultures subjected to oxygen-glucose deprivation: the role of adenosine triphosphate dependent potassium channels and reactive oxygen species in sevoflurane-induced neuroprotection. Anesthesia and analgesia. 2009;108:955-63 doi:10.1213/ane.0b013e318193fee7
31. Canas PT, Velly LJ, Labrande CN, Guillet BA, Sautou-Miranda V, Masmejean FM. et al. Sevoflurane protects rat mixed cerebrocortical neuronal-glial cell cultures against transient oxygen-glucose deprivation: involvement of glutamate uptake and reactive oxygen species. Anesthesiology. 2006;105:990-8
32. Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA. et al. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Annals of neurology. 2008;64:654-63 doi:10.1002/ana.21511
33. Tissier R, Chenoune M, Pons S, Zini R, Darbera L, Lidouren F. et al. Mild hypothermia reduces per-ischemic reactive oxygen species production and preserves mitochondrial respiratory complexes. Resuscitation. 2013;84:249-55 doi:10.1016/j.resuscitation.2012.06.030
34. McManus T, Sadgrove M, Pringle AK, Chad JE, Sundstrom LE. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. Journal of neurochemistry. 2004;91:327-36 doi:10.1111/j.1471-4159.2004.02711.x
35. Iijima T. Mitochondrial membrane potential and ischemic neuronal death. Neuroscience research. 2006;55:234-43 doi:10.1016/j.neures.2006.04.005
36. Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain research. 2003;993:140-5
37. Iijima T, Mishima T, Akagawa K, Iwao Y. Neuroprotective effect of propofol on necrosis and apoptosis following oxygen-glucose deprivation--relationship between mitochondrial membrane potential and mode of death. Brain research. 2006;1099:25-32 doi:10.1016/j.brainres.2006.04.117
38. Huang CH, Chen HW, Tsai MS, Hsu CY, Peng RH, Wang TD. et al. Antiapoptotic cardioprotective effect of hypothermia treatment against oxidative stress injuries. Academic emergency medicine: official journal of the Society for Academic Emergency Medicine. 2009;16:872-80 doi:10.1111/j.1553-2712.2009.00495.x
39. Zhou T, Jiang J, Zhang M, Fu Y, Yang Z, Jiang L. Protective effect of mild hypothermia on oxygen-glucose deprivation injury in rat hippocampal neurons after hypoxia. Molecular medicine reports. 2013;7:1859-64 doi:10.3892/mmr.2013.1410
40. Kaminuma T, Suzuki Y, Shirai K, Mizui T, Noda SE, Yoshida Y. et al. Effectiveness of carbon-ion beams for apoptosis induction in rat primary immature hippocampal neurons. Journal of radiation research. 2010;51:627-31
Author contact
![]() Corresponding author: Dr. Su-yue Pan, Department of Neurology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China. Email: pansuyue82com.
Corresponding author: Dr. Su-yue Pan, Department of Neurology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China. Email: pansuyue82com.