Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results and Discussion
Material and methods
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):895-908. doi:10.7150/ijbs.9454 This issue Cite
Research Paper
The First Mitochondrial Genomes of Antlion (Neuroptera: Myrmeleontidae) and Split-footed Lacewing (Neuroptera: Nymphidae), with Phylogenetic Implications of Myrmeleontiformia
Yan Yan1, Yuyu Wang1, Xingyue Liu1 ![]() , Shaun L. Winterton2, Ding Yang1
, Shaun L. Winterton2, Ding Yang1 ![]()
1. Department of Entomology, China Agricultural University, Beijing 100193, China.
2. California State Collection of Arthropods, California Department of Food & Agriculture, Sacramento, California 95832, United States of America.
Received 2014-4-21; Accepted 2014-7-21; Published 2014-8-15
Abstract
In the holometabolous insect order Neuroptera (lacewings), the cosmopolitan Myrmeleontidae (antlions) are the most species-rich family, while the closely related Nymphidae (split-footed lacewings) are a small endemic family from the Australian-Malesian region. Both families belong to the suborder Myrmeleontiformia, within which controversial hypotheses on the interfamilial phylogenetic relationships exist. Herein, we describe the complete mitochondrial (mt) genomes of an antlion (Myrmeleon immanis Walker, 1853) and a split-footed lacewing (Nymphes myrmeleonoides Leach, 1814), representing the first mt genomes for both families. These mt genomes are relatively small (respectively composed of 15,799 and 15,713 bp) compared to other lacewing mt genomes, and comprise 37 genes (13 protein coding genes, 22 tRNA genes and two rRNA genes). The arrangement of these two mt genomes is the same as in most derived Neuroptera mt genomes previously sequenced, specifically with a translocation of trnC. The start codons of all PCGs are started by ATN, with an exception of cox1, which is ACG in the M. immanis mt genome and TCG in N. myrmeleonoides. All tRNA genes have a typical clover-leaf structure of mitochondrial tRNA, with the exception of trnS1(AGN). The secondary structures of rrnL and rrnS are similar with those proposed insects and the domain I contains nine helices rather than eight helices, which is common within Neuroptera. A phylogenetic analysis based on the mt genomic data for all Neuropterida sequenced thus far, supports the monophyly of Myrmeleontiformia and the sister relationship between Ascalaphidae and Myrmeleontidae.
Keywords: mitochondrial genome, Neuroptera, Myrmeleontiformia, phylogeny.
Introduction
Neuroptera (lacewings) are a holometabolous insect order, belonging to the superorder Neuropterida, with ca. 6000 extant species sorted in 16 families. The order consists of elegant insects, and notable for their transparent wings laced with intricate venation, and is characterized by the association of the male ninth gonocoxites with the gonarcus and in the larvae by the peculiar composite sucking mouthparts, discontinuous gut and Malphigian tubules modified for silk production [1, 2]. The earliest definitive neuropteran is hitherto known from the Late Permian [2], indicating a long evolutionary history of this order. The ongoing discoveries of a species rich and taxonomically diverse extinct fossil neuropteran fauna around the world suggest that this order may be less diverse today than in the past (provide some recently published references of fossil neuropteran described).
The phylogenetic relationships among the various neuropteran families are still unresolved with competing hypotheses concerning the number and composition of suborders as well as the higher-level relationships. The morphology based three-suborder-concept [3-5] divides Neuroptera into Nevrorthiformia, Myrmeleontiformia and Hemerobiiformia with the latter two being sister groups [4]. On the other hand, several other studies based on morphological as well as molecular data resulted in phylogenies differing in multiple aspects of interfamilial relationships except for the almost universal support for a paraphyletic Hemerobiiformia and a monophyletic Myrmeleontiformia [3-8].
Myrmeleontiformia is always recovered as a well-supported monophyletic group in almost all studies. This suborder consists of five extant families, i.e. Ascalaphidae, Myrmeleontidae, Nemopteridae, Nymphidae and Psychopsidae [1, 4, 8, 9]. Ascalaphidae (owlflies) and Myrmeleontidae (antlions) are cosmopolitan in distribution, and Ascalaphidae contains ca. 430 species whereas Myrmeleontidae represents the most species-rich lacewing family with ca. 1630 species [10]. Nemopteridae (spoon- and thread-winged lacewings) is a distinctive group characterized by highly specialized (e.g. elongated, thread-like or spoon-shaped) hind wings and distributed in the Afrotropical, Palaearctic, Australian and Neotropical regions [11]. Nymphidae (split-footed lacewings) is a small family endemic to the Australian-Malesian region with ca. 35 species [12]. Psychopsidae (silky lacewings) is also a spectacular group with butterfly-like wing shape and narrowly distributed in Australia, Asia and Africa with ca. 26 extant species [13].
Despite the undisputed monophyly of Myrmeleontiformia, the interfamilial relationships within this suborder are still controversial. MacLeod identified a series of characteristics that define the group based on the larval head capsule, all associated with increase size of the head and wider articulation of the jaws resulting in more generalized prey selection [9]. Relationships among the various families of Myrmeleontiformia vary among authors. Almost all recent studies agree that Psychopsidae are sister to remaining Myrmeleontiformia, that Ascalaphidae and Myrmeleontidae are sister families and that Nymphidae are an intermediate clade between the two [4, 5, 8, 9, 14-17]. Yet, the phylogenetic position of Nemopteridae remains problematic with some authors proposing, based solely on morphological data, that Nemopteridae are sister to Psychopsidae in a clade sister to all other Myrmeleontiformia [4, 7]; more synthetic analyses of combined molecular and morphological data find Nemopteridae as sister to Ascalaphidae+Myrmeleontidae [8].
The mitochondrial genome analysis is becoming increasingly common in phylogenetic studies, population genetics and phylogeography [18], providing more phylogenetic information and multiple genome-level characteristics than shorter sequences of individual genes [19-22]. More than 600 insect mt genomes of arthropods have been sequenced, most of them from the mega-diverse orders such as Hemiptera, Hymenoptera, Coleoptera, Diptera, and Lepidoptera [23, 24]. However, there are only nine mt genomes of Neuroptera fully sequenced and published to date, representing five families, i.e. Ascalaphidae, Chrysopidae, Ithonidae, Mantispidae and Osmylidae [23, 25]. Within Myrmeleontiformia, the mt genomes of only two species of Ascalaphidae (Libelloides macaronius Scopoli, 1763 and Ascaloptynx appendiculatus (Fabricius, 1793)) have been sequenced [30, 31].
Herein, we present two complete mt genomes of Myrmeleon immanis Walker, 1853 and Nymphes myrmeleonoides Leach, 1814, as the first representatives of Myrmeleontidae and Nymphidae, respectively. We compare the genomic structure and composition, such as gene order, nucleotide composition, codon usage, secondary structure of tRNAs, compositional biases, and investigate the phylogenetic status of Myrmeleontidae and Nymphidae within Neuroptera based on a phylogenomic analysis.
Results and Discussion
The mt genome of Myrmeleon immanis is a typical circular DNA with 15,799 bp (GenBank accession number KJ461323), which comprises 37 genes with a major A+T-rich region of 971 bp (Figure 1, Supplementary Material: Table S1), while the mt genome of Nymphes myrmeleonoides is a similar circular DNA 15,713 bp in length (GenBank accession number KJ461322) with a noncoding region of 812 bp (Figure 1, Supplementary Material: Table S2). The mt genomes of the two species are slightly smaller than those of other sequenced Neuroptera, whose size range from 15,877 bp (A. appendiculatus) to 16,723 bp (Chrysopa pallens). Both mt genomes consist of 22 transfer RNA genes, 13 protein-coding genes and two ribosomal RNA genes. Twenty-three genes are located on the J strand, while the remaining fourteen genes are located on the N strand. In N. myrmeleonoides the gene overlaps are 19 bp distributed in seven gene junctions, while in M. immanis the gene overlaps are 29 bp located in 12 gene junctions.
Mitochondrial map of Myrmeleon immanis and Nymphes myrmeleonoides. The tRNAs are denoted by the color blocks and are labeled according to the IUPACIUB single-letter amino acid codes. Gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left.
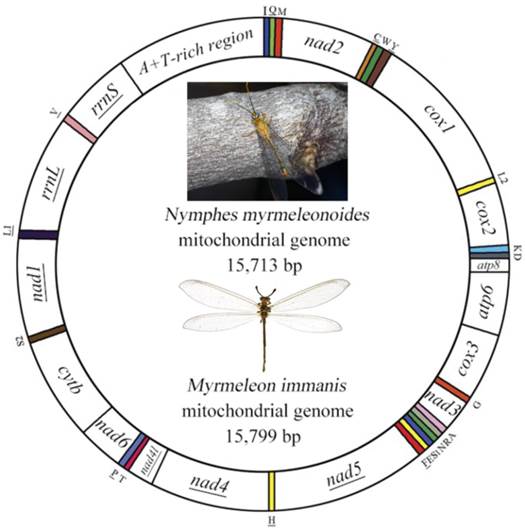
Genome organization
Gene arrangement is identical in both M. immanis and N. myrmeleonoides mt genomes. Compared with Drosophila yakuba (Burla) (Diptera: Drosophilidae), whose genome exhibits the typical ground pattern for insect mt genomes [26], the gene arrangement is relatively conserved except for one rearrangement in M. immanis and N. myrmeleonoides (Figure 1, Supplementary Material: Tables S1-S2). In most insect mt genomes, the assumed ancestral arrangement for these three tRNA genes is trnW-trnC-trnY, but in M. immanis and N. myrmeleonoides the arrangement is trnC-trnW-trnY. Among the known mt genomes of Neuropterida, the arrangement of W-C-Y is clearly plesiomorphic and is found in Raphidioptera, Megaloptera and a single family of Neuroptera (Osmylidae: Thyridosmylus) [23]; all other Neuroptera have the rearrangement C-W-Y, which is a derived characteristic for the bulk of the family [23, 25, 27-31].
Protein-coding genes
Among the 13 PCGs in the M. immanis and N. myrmeleonoides mt genomes, nine PCGs are located in the J strand, while the other four are located in the N strand (Figure 1). The length of all PCGs for M. immanis is 11,189 bp and 11,182 bp in N. myrmeleonoides. The overall AT content of the 13 PCGs is 74.3% in the M. immanis mt genome, ranging from 67.5% (cox1) to 83.5% (nad6), while in the N. myrmeleonoides mt genome, the AT content of the 13 PCGs is 79.0%, ranging from 72.9% (cox1) to 86.4% (nad6).
Generally, the start codon is mostly ATT and ATG for the PCGs, but ATA represents an alternative start codon for a few PCGs in the mt genomes of M. immanis and N. myrmeleonoides. In the M. immanis mt genome, ATA is a start codon for nad1, nad3, and nad5, while in N. myrmeleonoides, ATA as the start codon is only found in nad1 (Tables S1-S2). In cox1 the start codon is ACG in the M. immanis and TCG in N. myrmeleonoides.
Most PCGs have a start codon as ATG in nad4, nad4l, cox2, cox3, atp6, and cytb. However, the other PCGs in Neuropterida families contain some unusual start codons, like TTG, ACG, TCG, TTA, CTG. Comparing with the codon usage of the PCGs in the Neuropterida species, it is obvious that the start codon of cox1 is highly variable (Supplementary Material: Table S3). The start codons vary mostly in cox1 with many types of uncommon start codons like CTG in Mongoloraphidia harmandi, TTA in A. appendiculatus and TCG in several other species. The nad1 genes in Neuropterida also show the various start codons, such as TTG, ATG and ATA.
Stop codons are far less variable than start codons in the Neuropterida. Most stop codons of PCGs are complete or incomplete TAA, except in nad1 and nad3 which have TAG as the end codon in several species. The end codon of the 13 PCGs in M. immanis and N. myrmeleonoides are exactly the same. The end codon of nad1 is TAG. The cox1 and cox2 end with a thymine followed by a tRNA gene instead of an end codon. The other PCGs end with a conserved triplet of TAA. The triplet stop codon (TAA, TAG) always has a one or two bp overlap with the down-stream tRNA gene, and these overlaps could be the 'backup' to prevent translation read through if the transcripts are not cleaved correctly [32].
In the mt genomes of both M. immanis and N. myrmeleonoides, an 'ATGATAA' overlap is present between atp8 and atp6. This feature is shared by other species with known mt genomes in Neuropterida, in which the atp8 and atp6 are located on the J strand with an overlap of 7 bp. On the N strand, an overlap,of 'ATGTTAA' between nad4 and nad4l was also observed in the M. immanis and N. myrmeleonoides mt genomes. However, within Myrmeleontiformia, the overlap of nad4 and nad4l is not conserved because the mt genomes of two owlfly species (Ascaloptynx appendiculatus [30] and Libelloides macaronius [31]) show a contiguous pattern between nad4 and nad4l.
Transfer RNAs
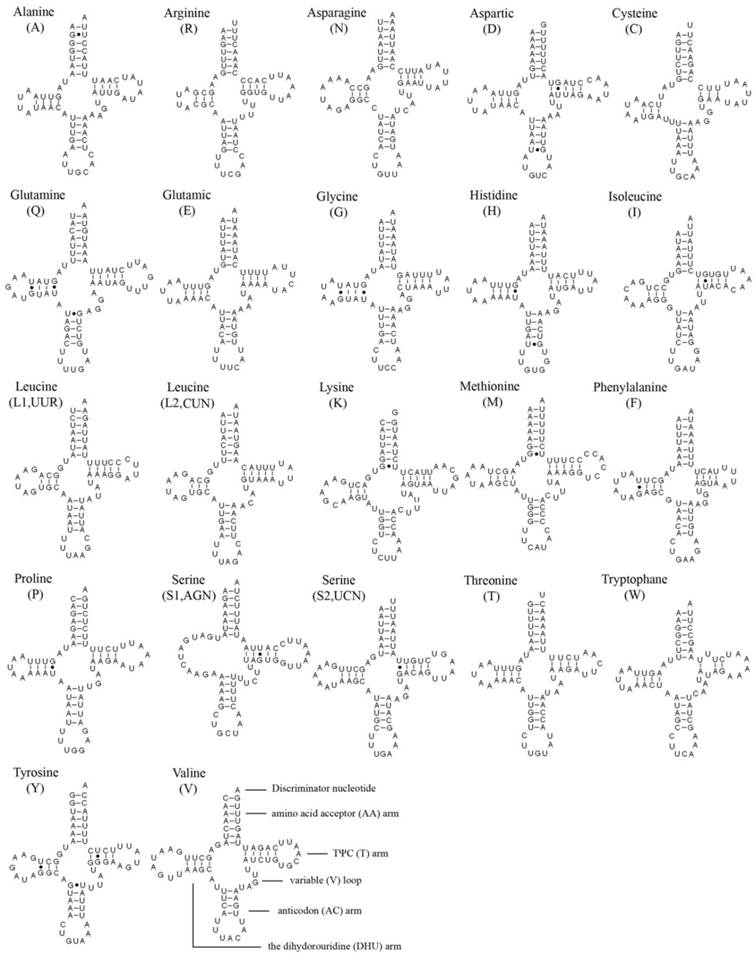
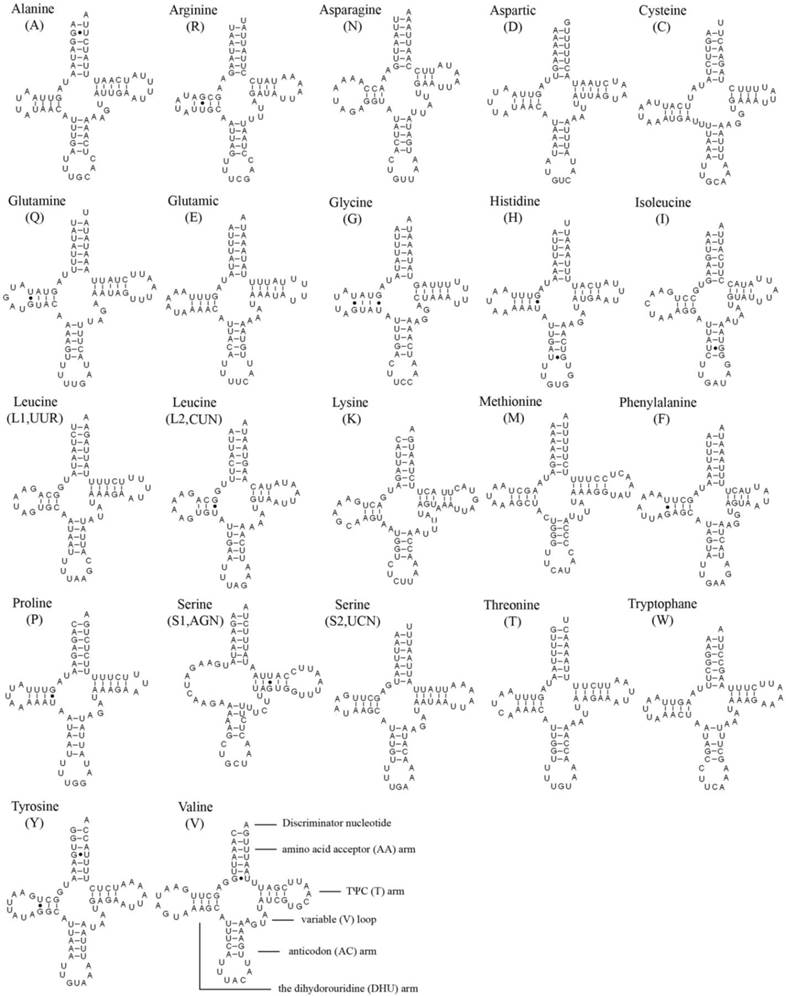
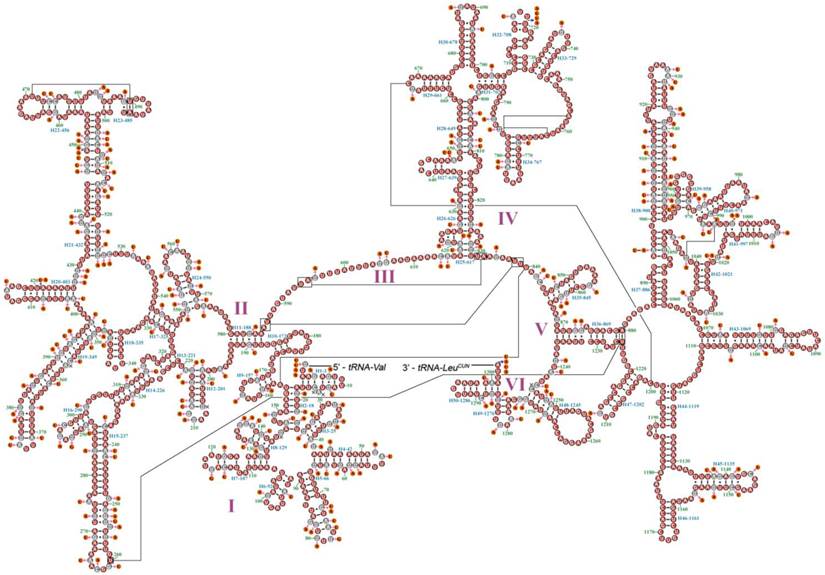
In the 22 tRNA genes, 14 genes are located on the J strand, while the others are encoded on the N strand. The length of a single tRNA ranges from 64 bp to 72 bp in both M. immanis and N. myrmeleonoides. The aminoacyl (AA) stem and the AC loop are conserved with 7 bp, but the AA arm of trnI in the M. immanis mt genome has just 6 bp. The DHU arm and TΨC arm are more variable, ranging from 3-5 bp. The AC arm is always 5 bp with some exceptions like 4 bp of trnK. Except for trnS1(AGN), all the other tRNAs form into the typical cloverleaf structure, and the DHU arm of trnS1(AGN) in the two species is a large loop instead of the conserved stem-and-loop structure (Figures 2-3). Some base pairs are not the classic A-U and C-G based on the secondary structure. In tRNAs of M. immanis and N. myrmeleonoides mt genomes, there are some mismatched base pairs such as G-U and U-U (Supplementary Material: Tables S4-S5). In Myrmeleon immanis, there are 20 G-U mismatched base pairs, and these mismatched base pairs are located in the AA stem (3 bp), the AC arm (4), the TΨC arm (5), and the DHU arm (8). However, in N. myrmeleonoides, there are 15 G-U mismatched base pairs located in the AA arm (3), AC arm (2), TΨC arm (1), and DHU arm (9). As for the U-U mismatch, the trnW gene has this type of mismatch in both mt genomes, and was observed in the anticodon (AC) arm in N. myrmeleonoides and the TΨC arm in M. immanis.
Secondary structures of 22 tRNA genes of Myrmeleon immanis.
Secondary structures of 22 tRNA genes of Nymphes myrmeleonoides.
Ribosomal RNAs
The length of rrnL and rrnS of the M. immanis mt genome is 1,304 bp and 777 bp, respectively, and the length of the rrnL and rrnS in N. myrmeleonoides is 1,320 bp and 786 bp. The overall structures of the two rRNAs largely resemble the previously published secondary structures for L. macaronius [31].
A proposed secondary structure model for rrnS of M. immanis and N. myrmeleonoides is shown in Figure 4. The secondary structure of rrnS contains three domains and 34 helices, which is similar with that of some other endopterygote orders (i.e. Coleoptera, Diptera, Hymenoptera, and Lepidoptera) [33-36]. There are a limited number of non-canonical pairings in the rrnS of the two mt genomes. The rrnS has 189 canonical pairings (A-U and G-C) and 42 non-canonical pairings (G-U: 31, U-U: 5, C-U: 2, G-A: 3 and C-A: 1).
Secondary structures of rrnS of Myrmeleon immanis and Nymphes myrmeleonoides. Red circles indicate the conserved nucleotides in the rrnS of M. immanis and N. myrmeleonoides. Gray circles indicate the variable nucleotides in the rrnS of M. immanis. Orange circles indicate the variable nucleotides in the rrnS of N. myrmeleonoides. Pink arrows indicate the replacement of different nucleotide in N. myrmeleonoides. Blue arrows indicate insertion of additional nucleotides in N. myrmeleonoides. Each helix is numbered progressively from the 5' to the 3' end together with the first nucleotide belonging to the helix itself. Domains are labeled with Roman numerals. Tertiary structures are denoted by boxed bases joined by solid lines.
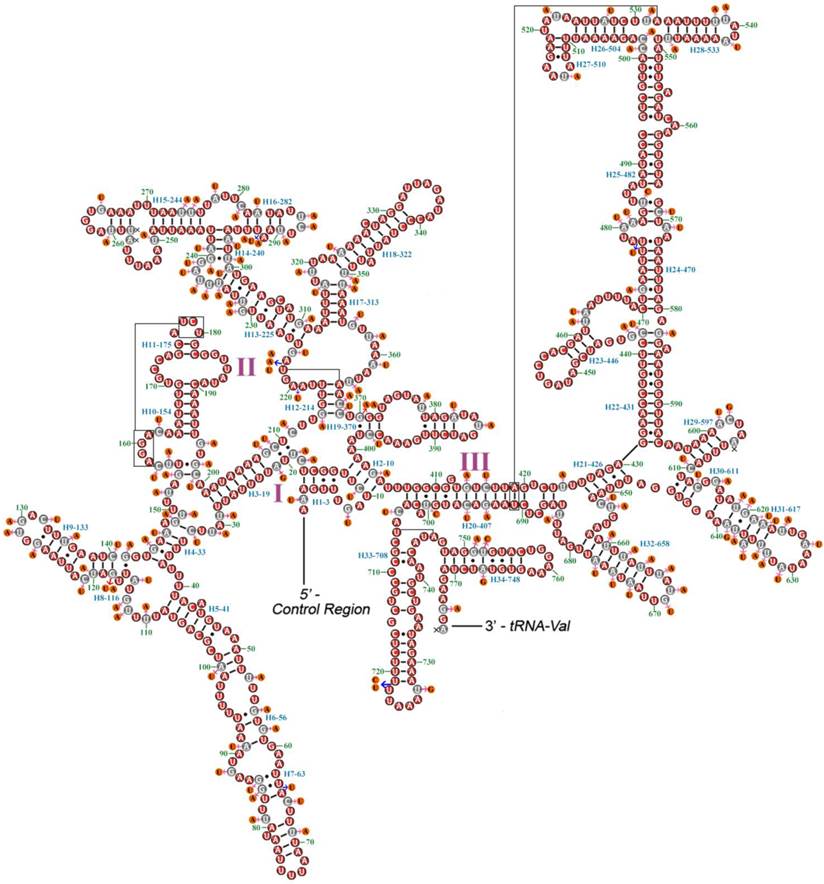
A proposed secondary structure model for rrnL of M. immanis and N. myrmeleonoides is shown in Figure 5. The secondary structure of rrnL is also generally congruent with the secondary structure models of previously published endopterygote insects, and have 5 canonical domains (I-II, IV-VI) with an absence of domain III. The five domains and lack of domain III is also a typical trait in arthropods [37]. The secondary structure of rrnL has 50 helices. Domain I contains nine helices rather than eight helices which is typical for neuropteran rrnL [23]. The rrnL has 321 canonical pairings (A-U and G-C) and 65 non-canonical pairings (G-U: 50, U-U: 11, C-U: 1, G-A: 1 and C-A: 2). The distribution of the conserved nucleotides is uneven in the secondary structure of rrnL, with lowest level of invariable positions on domains I-II and highest level on domain IV.
Secondary structures of rrnL of Myrmeleon immanis and Nymphes myrmeleonoides. Red circles indicate the conserved nucleotides in the rrnL of M. immanis and N. myrmeleonoides. Gray circles indicate the variable nucleotides in the rrnL of M. immanis. Orange circles indicate the variable nucleotides in the rrnL of N. myrmeleonoides. Pink arrows indicate the replacement of different nucleotide in N. myrmeleonoides. Blue arrows indicate insertion of additional nucleotides in N. myrmeleonoides. Each helix is numbered progressively from the 5' to the 3' end together with the first nucleotide belonging to the helix itself. Domains are labeled with Roman numerals. Tertiary structures are denoted by boxed bases joined by solid lines.
Non-coding region
The major A+T rich-region, i.e. the control region, of the M. immanis and N. myrmeleonoides mt genomes is located between rrnS and trnI, and the A+T base composition of this region is 86.8% and 93.1%, respectively. The high rate of A+T base composition is caused by the repeated motifs containing almost exclusively A/T at different lengths. In the control region of the M. immanis mt genome, the most abundant TA motif occurs 194 times, AT occurs 187 times, TAA occurs 78 times, TTAA occurs 27 times, AATTT occurs 12 times, and TTATATAT occurs 3 times. In the control region of N. myrmeleonoides, the TA motif occurs 198 times, AT occurs 201 times, TAA occurs 80 times, AATT occurs 38 times, TTTAA occurs 13 times, and ATATAA occurs 10 times. Additionally, some short tandem repeat sequences exist in the control region of M. immanis, e.g. (AT)3, (AT)4, (AT)5, (TA)3, (TA)4, (TA)5, (TA)6, (TAT)3, and (ATT)3. Whereas, in the control region of N. myrmeleonoides, the short tandem repeat sequences are (TA)3, (TA)4, (TA)5, (TA)6, (TA)9, (AT)3, (AT)4, (AT)5, (AT)6, (AT)9, (TAA)3, and (TTTAA)2. These tandem repeat elements can be considered as microsatellites and are useful in the study of geographical population structure [38].
The secondary structure of control region is simpler in the mt genomes of M. immanis and N. myrmeleonoides than that in Hymenopteran [39], Hemipteran [40] or Lepidopteran [41] insects with less conserved blocks or long tandem repeats. There is a poly-T stretche with 12 bp in the control region of the M. immanis and N. myrmeleonoides mt genomes, and an area with 24 bp identical sequences upstream from poly-T is present M. immanis and N. myrmeleonoides, i.e. ATGGTTCAATAAAATAATTCTCTCTTTTTTTTTTTT. This region is highly conserved within Neuroptera.
There is another long non-coding region in the mt genome of M. immanis between trnI and trnQ with 70 bp, and a similar non-coding region in the same position of the mt genome of N. myrmeleonoides is present with 58 bp. Additionally, a non-coding region is located between trnS2(UCN) and nad1, with 16 bp in the N. myrmeleonoides mt genome and 21 bp in the M. immanis mt genome. The other non-coding regions of the M. immanis mt genome are rather short, ranging from 1 to 9 bp, while N. myrmeleonoides has another long non-coding region of 29 bp between trnQ and trnM. The other short non-coding regions range from 1 to 14 bp in the N. myrmeleonoides mt genome.
Codon usage
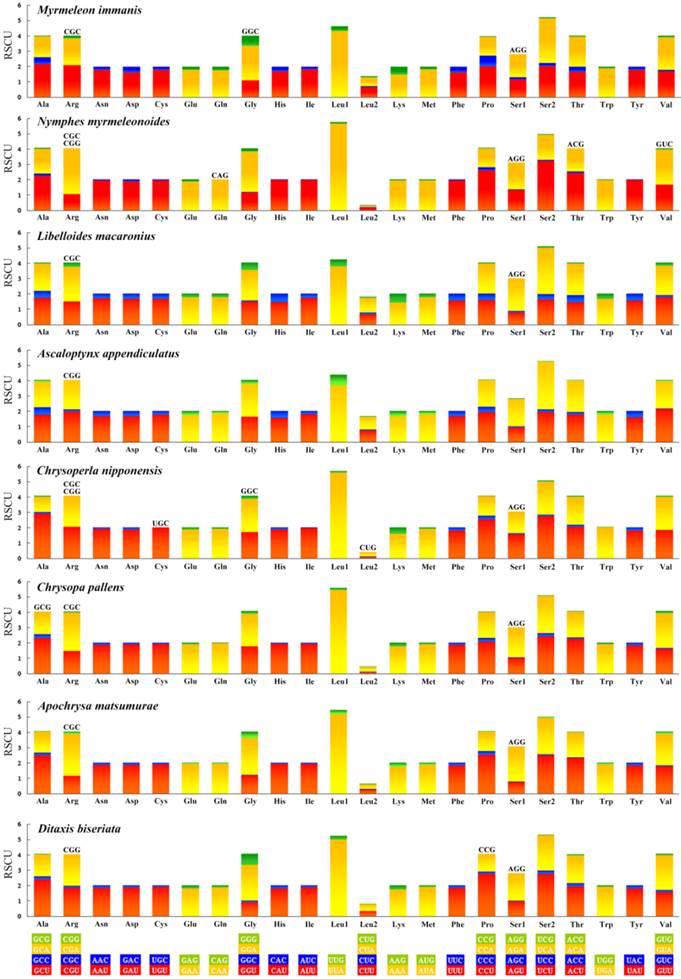
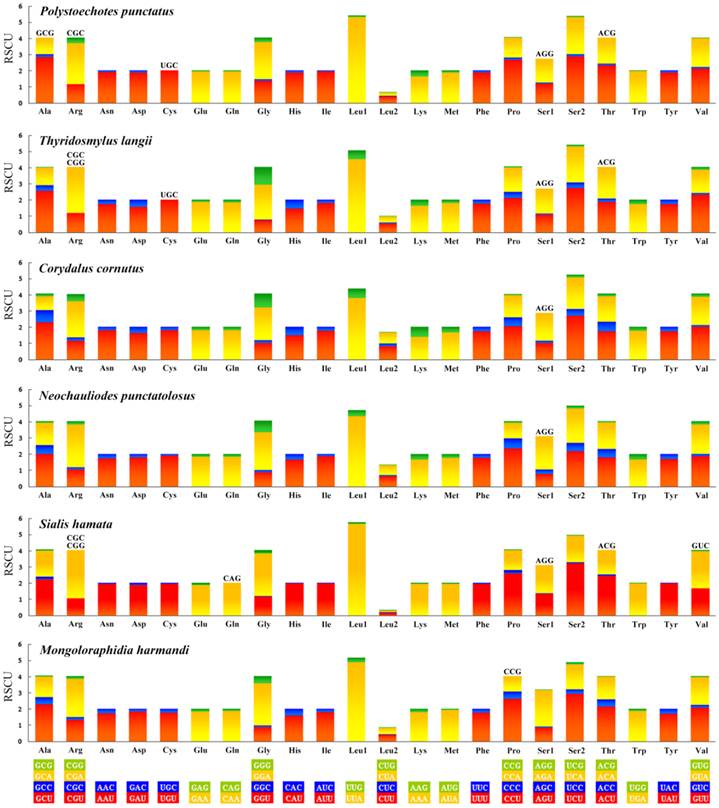
All the genetic codons in the Neuropterida mtDNAs use standard invertebrate mitochondrial genetic code. We exclude the stop codons since some of them are incomplete. This result (Figure 6) shows the relative synonymous codon usage (RSCU). It is obvious that the preferred codon usage is A/T in their third position rather than G/C. As for the four-fold degenerate codon usage, the third position prefers T than A in most codon families except Arg, Gly, Ser1, and Ser2 in several species. The result also shows the missing part of the codon in Neuropterida families, and these missing codons all have high content of GC with G or C in the third position. In the mt genome of M. immanis, the missing codons are Arg (CGC), Gly (GGC) and Ser1 (AGG), while in the mt genome of N. myrmeleonoides, the missing parts are Arg (CGC, CGG), Gln (CAG), Ser1 (AGG), Thr (ACG) and Val (GUC). In the Neuropterida mtDNAs, the missing codon mostly occurs in Arg and Ser1 with absence of CGC and AGG. The mt genomes of M. immanis and N. myrmeleonoides also show a significant bias to the A/T. The top six most frequently used triplets in these two mt genomes are the same, i.e. UUU (Phe), UUA (Leu), AUU (Ile), AUA (Met), UAU (Tyr) and AAU (Asn), and all these triplets consist of A and T, which may be due to the high AT composition in the mt genomes and PCGs of the M. immanis and N. myrmeleonoides.
Relative synonymous codon usage (RSCU) of the mt genomes of Neuropterida. Stop codon is not given.
Base composition
The mt genomes of M. immanis and N. myrmeleonoides show a strong bias toward A and T (M. immanis: A=38.7%, T=36.9%, C=14.4%, G=10.1%; N. myrmeleonoides: A=39.8%, T=40.7%, C=11.1%, G=8.4%), which is very typical of invertebrate mt genomes (Figure 7). The three parameters: AT-skew, GC-skew, and A+T content are usually used in the investigation of the nucleotide-compositional behavior of mt genomes [42, 43]. Results of the comparative analysis of the A+T%, AT- and GC-Skew within the sequenced Neuropterida mt genomes is summarized in a three-dimensional scatter-plot chart (Figure 7). Considering the whole mt genome, the A+T% in N. myrmeleonoides is higher than any other species of sequenced mt genomes in Neuropterida, while the A+T% in M. immanis is lower than the average AT content. The AT- and GC-Skew indicate the strand bias in the base composition [44-46]. The average AT-Skew of sequenced Neuropterida mt genomes is 0.0138, ranging from -0.0367 in Apochrysa matsumurae to 0.0711 in L. macaronius, whereas the average GC-Skew is -0.1863, spanning from -0.2590 in Corydalus cornutus to -0.1374 in Chrysoperla nipponensis (Supplementary Material: Table S6). The AT- and GC-Skew are 0.0238 and -0.1755 in the M. immanis mt genome, while they are -0.0112 and -0.1385 in N. myrmeleonoides.
Three-dimensional scatter-plot of the AT-, GC-Skew and A+T% of the mt genomes of Neuropterida. L.m.: Libelloides macaronius; A.a.: Ascaloptynx appendiculatus; C.n.: Chrysoperla nipponensis; C.p.: Chrysopa pallens; A.m.: Apochrysa matsumurae; D.b.: Ditaxis biseriata; P.p.: Polystoechotes punctatus; T.l.: Thyridosmylus langii; C.c.: Corydalus cornutus; N.p.: Neochauliodes punctatolosus; S.h.: Sialis hamata; M.h.: Mongoloraphidia harmandi; N.m.: Nymphes myrmeleonoides; M.i.: Myrmeleon immanis. Red circles: Ascalaphidae; blue circles: Chrysopidae; yellow circles: Mantispidae; olive circles: Ithonidae; mazarine circles: Osmylidae; purple circles: Megaloptera; green circles: Raphidiidae; orange circles: Myrmeleontidae; pink circles: Nymphidae.
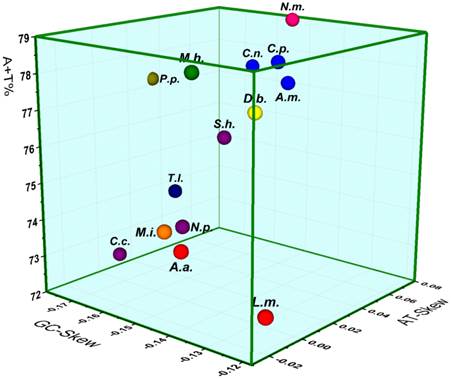
The three parameters of the Neuropterida genomes indicate some phylogenetic relationships of certain families (Figure 7). Three species of Chrysopidae, Chrysoperla nipponensis, Chrysopa pallens and Apochrysa matsumurae (blue balls), show the similar content of A+T and AT/GC-skew. Two species of Ascalaphidae, Libelloides macaronius and Ascaloptynx appendiculatus, also show the similar content of A+T and AT-skew with a little difference in GC-skew. Moreover, the genome of Myrmeleon immanis shows the similar content of those three parameters with that of A. appendiculatus and L. macaronius, while it is different from that of N. myrmeleonoides with a discrepancy of A+T% and AT-skew. This result confirms the close relationship between Ascalaphidae and Myrmeleontidae relative to Nymphidae.
Phylogeny
Among the four datasets used in the phylogenetic analyses, there are 13,257 sites in the PCG123R matrix (containing all three codon positions of PCGs, plus two rRNA genes), 11,076 sites in the PCG123 matrix (containing all three codon positions of PCGs), 9,565 sites in the PCR12R matrix (containing the first and the second codon positions of PCGs, plus the two rRNA genes) and 7,384 sites in the PCG12 matrix (containing the first and the second codon positions of PCGs).
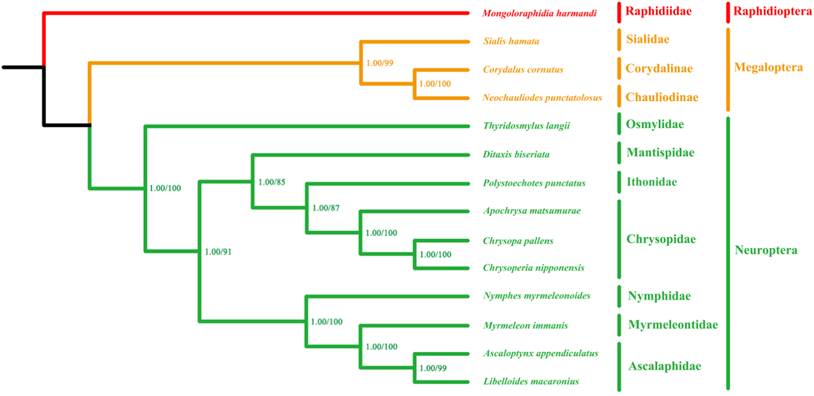
The phylogenetic trees inferred from both Bayesian and ML analyses have very similar topologies across on the four datasets. The branch support values of the PCG123R matrix are higher than the other matrices. Therefore, we show the phylogenetic trees of the PCG123R matrix in Figure 8. All species of Myrmeleontiformia in our analysis formed a monophylum, supporting the monophyly of this suborder. Second, the undoubted sister relationship between Ascalaphidae and Myrmeleontidae is also corroborated. Lastly, placement of Nymphidae as sister to to Ascalaphidae and Myrmeleontidae is corroborated [1, 4]. However, Nymphidae was alternatively considered to be the sister to Psychopsidae + Nemopteridae based on morphological data from the genital sclerites [7]. The recent comprehensive phylogenetic study on Neuropterida combining morphological and molecular data recovered that Nymphidae is the sister of the lineage comprising Nemopteridae + (Ascalaphidae + Myrmeleontidae) [8]. Our mt phylogenomic result is consistent with this hypothesis that Nymphidae together with Ascalaphidae and Myrmeleontidae form a monophyletic group. However, lack of Nemopteridae and Psychopsidae samples precludes any assessment of the placement of these families relative to the rest of Myrmeleontiformia. Future mt genomic study should be focused on Psychopsidae and Nemopteridae as well as some significant subfamilial taxa (e.g. Stilbopteryginae) in order to unravel the higher phylogeny within Myrmeleontiformia.
Phylogenetic relationships among Neuroptera families based on mt genomic data. Cladogram of relationships resulting from BI and ML analyses with Mongoloraphidia harmandi (Raphidioptera), Neochauliodes punctatolosus (Megaloptera), Sialis hamata (Megaloptera) and Corydalus cornutus (Megaloptera) as outgroups. Numbers at the nodes are Bayesian posterior probabilities (left) (10,000,000 generations) and ML bootstrap values (right) (100 replicates).
Material and methods
Sampling and DNA extraction
The specimen of M. immanis was collected on August 13, 2011 at Qianguo prairie, Songyuan, Jilin Province, China by Yuyu Wang using the light trap and N. myrmeleonoides was collected on November 13, 2007 by S. L. Winterton at the base of Mt. Coot-tha, Slaughter Falls, Brisbane, Queensland, Australia. After collection, specimens were preserved in 95% ethanol and deposited at -20℃ in the Entomological Museum of China Agricultural University (CAU) for long-term preservation. Total DNA of M. immanis and N. myrmeleonoides was extracted by using the TIANGEN (TIANamp Genomic DNA Kit) with the muscle of the thorax. The quality of DNAs was assessed through electrophoresis in a 1% agarose gel and staining with Gold View (nucleic acid stain replacing EB).
PCR amplification and sequencing
We amplified the mitochondrial genomes of M. immanis and N. myrmeleonoides by using overlap fragments which about 300-1200 bp. These fragments were amplified by PCR (Polymerase Chain Reaction) with 30 s at 95℃, 40 cycles of 10 s at 95℃, 50 s at 40℃, 2 min at 65℃, and 10 min at 65℃, then finishing with 10℃ indefinately. All the PCR used the NEB Long Taq DNA polymerase (New England BioLabs, Ipswich, MA) and the PCR products were detected by agarose gel electrophoresis to ensure the correctness of the result and the approximate size of the fragments. The primers of the fragments are listed in Supplementary Material: Table S8 for M. immanis mt genome and Table S9 for N. myrmeleonoides.
All fragments were sequenced in both directions by using the BigDye Terminator Sequencing Kit (Applied Bio Systems) and the ABI 3730XL Genetic Analyzer (PE Applied Biosystems, San Francisco, CA, USA) with two vector-specific primers and internal primers for primer walking.
Bioinformatic analysis
All sequenced fragments of the M. immanis and N. myrmeleonoides mt genomes were spliced manually and the uncertain site, which is located at the overlap of two fragments, is determined by comparing the original alignments. The 22 tRNA genes were identified by using the tRNAscan-SE Search Server v.1.21 [47] with a Cove cutoff score of 1 and the prediction of the genetic code followed the invertebrate mitochondrial DNA, while some tRNA genes were identified by the multiple sequence alignment of the tRNAs in Neuroptera. The secondary structures of tRNAs were also estimated by tRNAscan-SE Search Server v.1.21. The boundaries of 13 PCGs in the M. immanis and N. myrmeleonoides mt genomes were identified by comparing the boundaries of same PCGs in other Neuroptera mt genomes with MEGA 4.0 [48]. The rRNA genes and the control region were identified by the boundary of the tRNA genes and comparing with other insect mt genomes. The base composition and the codon usage were analyzed with MEGA 4.0 [48]. In addition, the AT-skew and GC-skew in the mt genomes were used for study on the feature of the base composition and calculated with the formulae: AT-skew = (A-T)/(A+T) and GC-skew = (G-C)/(G+C) [49]. We also used the OriginPro 9.0 [50] to generate the three-dimensional scatter plot of the mt genomes within the Neuropterida. The secondary structures of the rrnL and rrnS were inferred based on the model of Drosophila yakuba [26] and Libelloides macaronius [31]. Sequence motifs in the control region were identified using Spectral Repeat Finder program [51].
Phylogenetic analysis
To investigate the phylogenetic status of Myrmeleontidae and Nymphidae within Neuroptera, we selected all eight species of Neuroptera in which the complete mitochondrial genomes have been sequenced (see Supplementary Material: Table S7). Mongoloraphidia harmandi (Raphidioptera), Neochauliodes punctatolosus (Megaloptera), Sialis hamata (Megaloptera) and Corydalus cornutus (Megaloptera) were selected as outgroups because of the close relationship among Neuroptera, Megaloptera and Raphidioptera.
We used the Clustal X [52] for alignment of sequences of PCGs. Additionally, the two rRNAs alignments were conducted by the G-blocks Server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) based on more stringent selection. By using the MrBayes Version 3.1.2 [53] and a PHYML online web server [54, 55], we constructed the phylogenetic trees and the evolutionary model was estimated by Modeltest 3.7 [56], resulting the GTR+I+G model was optimal for analysis nucleotide alignments (PCGs and rRNAs). In the Bayesian inference, two simultaneous runs were conducted with 10,000,000 generations. Each set was sampled with a burnin of 25% of every 1000 generations. Trees inferred prior to stationarity were discarded as burnin, and the remaining trees were used to construct a 50% majority-rule consensus tree. In the ML analysis, the nodal supporting values were assessed by bootstrap re-sampling (BP) [57] calculated using 100 replicates. We used all the PCGs and delete any regions of ambiguous alignment, and subsequently concatenated all 13 PCGs (PCG123) (excluding the stop codon) to construct the phylogenetic tree initially, then included additionally the rrnL and rrnS (PCG123R) sequences to reconstruct the phylogenetic tree as a second result. We compared phylogenetic estimates to determine the most appropriate tree by considering the nodal supporting values.
Supplementary Material
Tables S1 - S9.
Abbreviations
mt: mitochondrial; PCG: protein-coding gene; atp6 and atp8: genes for the ATPase subunits 6 and 8; cox1-cox3: genes for cytochrome c oxidase subunits I-III; cytB: a gene for apocytochrome b; nad1-nad6 and nad4l: genes for NADH dehydrogenase subunits 1-6 and 4L; rrnL: large (16S) rRNA subunit (gene); rrnS: small (12S) rRNA subunit (gene); trnX (where X is replaced by one letter amino acid code of the corresponding amino acid), transfer RNA.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 31322501 and 31320103902), the National Key Basic Research Program of China (973 Program) (No. 2013CB127600), the Foundation for the Author of National Excellent Doctoral Dissertation of PR China (No. 201178) and the National Science Foundation of the United States of America (DEB-1144119).
Competing Interests
The authors have declared that no competing interest exists.
References
1. New TR. Planipennia. Lacewings. Handbuch der Zoologie (Berlin). 1989;4:1-132
2. Grimaldi DA, Engel MS. Evolution of the Insects. New York: Cambridge University Press. 2005:337 p
3. Aspöck H, Liu X, Aspöck U. The family Inocelliidae (Neuropterida: Raphidioptera): a review of present knowledge. Mitt Dtsch Ges Allg Angew Ent. 2012;18:565-573
4. Aspöck U, Plant JD, Nemeschkal HL. Cladistic analysis of Neuroptera and their systematic position within Neuropterida (Insecta: Holometabola: Neuropterida: Neuroptera). Syst Entomol. 2001;26:73-86
5. Beutel RG, Friedrich F, Aspöck U. The larval head of Nevrorthidae and the phylogeny of Neuroptera (Insecta). Zool J Linn Soc. 2010;158:533-562
6. Haring E, Aspöck U. Phylogeny of the Neuropterida: a first molecular approach. Syst Entomol. 2004;29:415-430
7. Aspöck U, Aspöck H. Phylogenetic relevance of the genital sclerites of Neuropterida (Insecta: Holometabola). Syst Entomol. 2008;33:97-127
8. Winterton SL, Hardy NB, Wiegmann BM. On wings of lace: phylogeny and Bayesian divergence time estimates of Neuropterida (Insecta) based on morphological and molecular data. Syst Entomol. 2010;35:349-378
9. MacLeod EG. Comparative morphological studies on the head capsule and cervix of larval Neuroptera (Insecta). Cambridge, Massachusetts: Ph D Thesis, Harvard University. 1964
10. Stange LA. A systematic catalog, bibliography and classification of the world antlions (Insecta: Neuroptera: Myrmeleontidae). Memoirs of the American Entomological Institute. 2004;74:1-565
11. Tjeder B. Neuroptera-Planipennia. The lacewings of Southern Africa. 6. Family Nemopteridae. South African Animal Life (Lund, Sweden). 1967;13:290-501
12. New TR. Intergeneric relationships of recent Nymphidae. In: Gepp, J, Aspöck, H. & Hölzel, H. (Eds.), Progress in World's Neuropterology. Proceedings of the 1st International Symposium on Neuropterology. Privately printed, Graz (Austria). 1984:125-131
13. Oswald JD. Phylogeny, taxonomy, and biogeography of extant silky lacewings (Insecta: Neuroptera: Psychopsidae). Memoirs of the American Entomological Society. 1993;40:1-65
14. Beutel RG, Zimmermann D, Krauß M, Randolf S, Wipfler B. Head morphology of Osmylus fulvicephalus (Osmylidae, Neuroptera) and its phylogenetic implications. Org Divers Evol. 2010;10:311-329
15. Henry CS. An unusual ascalaphid larva (Neuroptera: Ascalaphidae) from southern Africa, with comments on larval evolution within the Myrmeleontoidea. Psyche. 1978;85:265-274
16. Withycombe CL. Some aspects of the biology and morphology of the Neuroptera with special reference to the immature stages and their possible phylogenetic significance. Trans Ent Soc London. 1925;73:303-411
17. Handlirsch A. Die fossilen Insekten und die Phylogenie der rezenten Formen. Ein Handbuch für Paläontologen und Zoologen. Part 7. Engelmann, Leipzig. 1906-1908.
18. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
19. Dowton M, Castro LR, Austin AD. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: the examination of genome 'morphology'. Invertebr Syst. 2002;16:345-356
20. Boore JL, Macey JR, Medina M. Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymol. 2005;395:311-348
21. Masta SE, Boore JL. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol Biol Evol. 2008;25:949-959
22. Boore JL. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol Evol. 2006;21:439-446
23. Zhao J, Li H, Winterton SL, Liu Z. Ancestral gene organization in the mitochondrial genome of Thyridosmylus langii (McLachlan, 1870) (Neuroptera: Osmylidae) and implications for lacewing evolution. PLoS ONE. 2013;8:e62943
24. Cameron SL. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu Rev Entomol. 2014;59:95-117
25. Wang Y, Liu X, Winterton SL, Yang D. Comparative mitogenomic analysis reveals sexual dimorphism in a rare montane lacewing (Insecta: Neuroptera: Ithonidae). PLoS ONE. 2013;8:e83986
26. Clary DO, Wolstenholme DR. The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985;22:252-271
27. Wang Y, Liu X, Winterton SL, Yang D. The first mitochondrial genome for the fishfly subfamily Chauliodinae and implications for the higher phylogeny of Megaloptera. PLoS ONE. 2012;7:e47302
28. Haruyama N, Mochizuki A, Sato Y, Naka H, Nomura M. Complete mitochondrial genomes of two green lacewings, Chrysoperla nipponensis (Okamoto, 1914) and Apochrysa matsumurae Okamoto, 1912 (Neuroptera: Chrysopidae). Mol Biol Rep. 2011;38:3367-3373
29. Cameron SL, Sullivan J, Song H, Miller KB, Whiting MF. A mitochondrial genome phylogeny of the Neuropterida (lace-wings, alderflies and snakeflies) and their relationship to the other holometabolous insect orders. Zool Scr. 2009;38:575-590
30. Beckenbach AT, Stewart JB. Insect mitochondrial genomics 3: the complete mitochondrial genome sequences of representatives from two neuropteroid orders: a dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome. 2009;52:31-38
31. Negrisolo E, Babbucci M, Patarnello T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics. 2011;12:221
32. Boore J. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda). BMC Genomics. 2006;7:182
33. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-123
34. Sheffield NC, Song H, Cameron SL, Whiting MF. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol. 2008;25:2499-2509
35. Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, Pande N, Shang Z, Yu N, Gutell RR. The comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3(2):1-31
36. Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol. 2006;15:657-686
37. Yuan M, Wei D, Wang B, Dou W, Wang J. The complete mitochondrial genome of the citrus red mite Panonychus citri (Acari: Tetranychidae): high genome rearrangement and extremely truncated tRNAs. BMC Genomics. 2010;11:597
38. Li H, Liu H, Shi A, Štys P, Zhou X, Cai W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE. 2012;7:e29419
39. Wei S, Shi M, Sharkey MJ, Van Achterberg C, Chen X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC genomics. 2010;11:371
40. Li H, Liu H, Cao L, Shi A, Yang H, Cai W. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int J Biol Sci. 2012;8:93
41. Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci. 2009;5(4):351-365
42. McMahon DP, Hayward A, Kathirithamby J. The mitochondrial genome of the 'twisted-wing parasite' Mengenilla australiensis (Insecta, Strepsiptera): a comparative study. BMC Genomics. 2009;10:1-15
43. Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics. 2008;9:1-15
44. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
45. Hassanin A, Léger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54:277-298
46. Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol. 2006;38:100-116
47. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955-0964
48. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
49. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
50. Mikrajuddin A, Khairurrijal. A simple method for determining surface porosity based on SEM images using OriginPro software. Indonesian J Phys. 2009;20:No.2
51. Sharma D, Issac B, Raghava G, Ramaswamy R. Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics. 2004;20:1405-1412
52. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acid Res. 1997;25:4876-4882
53. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
54. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
55. Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucl Acid Res. 2005;33:W557-W559
56. Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
57. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791
Author contact
![]() Corresponding authors: E-mail: xingyue_liucom, dyangcaucom.
Corresponding authors: E-mail: xingyue_liucom, dyangcaucom.