Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):940-947. doi:10.7150/ijbs.9241 This issue Cite
Research Paper
Abnormality in Wnt Signaling is Causatively Associated with Oxidative Stress-Induced Intestinal Tumorigenesis in MUTYH-Null Mice
Takuro Isoda1, 2, Yoshimichi Nakatsu1, Kazumi Yamauchi1, Jingshu Piao1, Takashi Yao3, Hiroshi Honda2, Yusaku Nakabeppu4, Teruhisa Tsuzuki1 ![]()
1. Department of Medical Biophysics and Radiation Biology, Faculty of Medical Sciences, Kyushu University, Fukuoka, Japan,
2. Department of Clinical Radiology, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan,
3. Department of Human Pathology, School of Medicine, Juntendo University, Tokyo, Japan,
4. Division of Neurofunctional Genomics, Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan,
Received 2014-3-28; Accepted 2014-8-4; Published 2014-8-23
Abstract
MUTYH is a DNA glycosylase that excises adenine paired with 8-oxoguanine to prevent mutagenesis in mammals. Biallelic germline mutations of MUTYH have been found in patients predisposed to a recessive form of familial adenomatous polyposis (MAP: MUTYH-associated polyposis). We previously reported that Mutyh-deficient mice showed a high susceptibility to spontaneous and oxidative stress-induced intestinal adenoma/carcinoma. Here, we performed mutation analysis of the tumor-associated genes including Apc, Ctnnb1, Kras and Trp53 in the intestinal tumors of Mutyh-deficient mice. In the 62 tumors, we identified 25 mutations in Apc of 18 tumors and 36 mutations in Ctnnb1 of 36 tumors. Altogether, 54 out of the 62 tumors (87.1%) had a mutation in either Apc or Ctnnb1; no tumor displayed mutations simultaneously in the both genes. Similar to MAP, 60 out of 61 mutations (98.3%) were identified as G:C to T:A transversions of which 85% occurred at either AGAA or TGAA sequences. Immunohistochemical analyses revealed the accumulation of β-catenin in the nuclei of tumors. No mutation was found in either Kras or Trp53 in the tumors. These results indicate that the uncontrolled activation of Wnt signaling pathway is causatively associated with oxidative stress-induced intestinal tumorigenesis in the Mutyh-deficient mice.
Keywords: MAP, DNA repair, oxidative DNA damage, Wnt signaling pathway, mutagenesis
Introduction
Oxidative DNA damage appears to cause either mutagenesis or cell death, thereby resulting in various age-related diseases such as cancer and multi-organ dysfunction (1). 8-oxoguanine (8-oxoG), which is the major modified base found in the oxidized DNA, is highly mutagenic DNA lesion since DNA polymerases incorporate adenine as well as cytosine opposite 8-oxoG with almost equal frequency, thereby causing G:C to T:A transversion (2). In mammalian cells, 8-oxoG-related mutagenesis is prevented by MTH1, OGG1 and MUTYH. MTH1 has an 8-oxo-dGTPase activity hydrolyzing 8-oxo-dGTP to monophosphate form; 8-oxo-dGMP, thus preventing incorporation of 8-oxo-dGTP into DNA (3)(4), and found that MTH1 protein plays a crucial role in suppressing spontaneous mutagenesis as well as carcinogenesis (5)(6). OGG1 is a DNA glycosylase that excises 8-oxoG paired with cytosine, and suppress spontaneous and UV-induced tumorigenesis in mammal (7)(8)(9)(10)(11)(12). MUTYH removes adenine misincorporated opposite 8-oxoG and also removes 2-hydroxyadenine (generated by oxidation of adenine) misincorporated opposite guanine (13)(14). We previously established Mutyh-deficient mice (15)(16), and showed that Mutyh-deficient mice had susceptibility to tumorigenesis, especially adenoma and adenocarcinoma in the intestinal tracts (17). We also showed that oral administration of KBrO3, an oxidizing agent known to induce the 8-oxoG in the genome, dramatically enhanced the tumor-formations in small intestines of Mutyh-deficient mice (17).
MUTYH-associated polyposis (MAP) is a human hereditary colorectal polyposis caused by biallelic-inherited mutations in MUTYH (18)(19). The clinical features of MAP resemble to attenuated familial adenomatous polyposis (AFAP) characterized with less polyps and later onset than familial adenomatous polyposis (FAP) (20)(21)(22). A significant number of patients clinically diagnosed as adenomatous polyposis without germline mutations in APC are considered to carry MUTYH mutations. In most cases, polyps developed in MAP patients had mutations in APC, almost all of those were G:C to T:A transversions with the frequent occurrence in AGAA or TGAA sequence (18)(23)(24).
APC is an important factor of the canonical Wnt-signaling pathway, and APC mutations cause a failure in phosphorylation of β-catenin by glycogen synthase kinase 3 beta (GSK3β), resulting in the stabilization of β-catenin and its accumulation in nucleus without Wnt signal. High level of β-catenin in nucleus leads to up-regulation of various genes for such as c-Myc and Cyclin D1 with T-cell factor/lymphoid enhancing factor (TCF/LEF) (25)(26). APC is also known as the tumor suppressor gene, germline mutation of which is responsible for FAP (27)(28). Somatic APC mutations were found in more than 80% of sporadic colorectal cancers (CRCs) and CTNNB1 (β-catenin) mutations were also found in about a half of CRCs lacking APC mutation (25)(29)(30). Because APC and CTNNB1 mutations are mutually exclusive, it is considered that each mutation has nearly equal effect on β-catenin stability and TCF/LEF transactivation, albeit some differences such as the effect on invasiveness (31). Accordingly, unrestrained activation of the Wnt signaling pathway, resulted from the mutations in APC or in CTNNB1, is considered to be associated with early premalignant lesion, such as aberrant crypt foci and small polyps (32).
In addition to APC or CTNNB1, mutations in KRAS and TP53 are frequently found in colorectal cancer in human. KRAS mutations, mainly at codon 12, were found in approximately a half of colorectal cancers (26)(33)(34). TP53 (official symbol: TP53 in human, and Trp53 in mouse) is a tumor suppressor gene involved in various cellular functions, such as cell-cycle control, apoptosis and maintenance of genetic stability. Defects of TP53, resulting from mutations or loss of heterozygosity, were found in many of colorectal cancers (35)(36). KRAS mutations, but not TP53 mutations, are commonly observed in tumors from MAP patients (37)(38).
In this study, we performed pathological evaluation among the small intestinal tumors developed in wild-type and Mutyh-deficient mice treated with KBrO3 according to the Vienna classification of gastrointestinal epithelial neoplasia (39). To gain an insight into the process of the oxidative stress-induced tumor-development in MUTYH-deficient genetic background, we analyzed mutations in the tumor-associated genes, such as Apc, Ctnnb1, Kras and Trp53 in the tumors developed in wild-type and Mutyh-deficient mice treated with KBrO3.
Materials and methods
Animals and KBrO3 treatment
Mutyh-deficient mice used in this study were previously generated, and backcrossed to C57BL/6J for more than 10 generations (17). Genomic DNA samples and histological slides of the small intestinal tumors were prepared from the mice treated with KBrO3 as previously reported (17). Briefly, wild-type C57BL/6J mice or congenic Mutyh-deficient mice were treated with 0.2% KBrO3 in drinking water for 16 weeks. Body weight and amount of water consumption were measured once a week. At the end of the KBrO3-treatment, mice were sacrificed and the tissues were fixed with 4% buffered-formaldehyde and then stored in 70% ethanol. All animals were kept in specific pathogen free (SPF) conditions. All animal care and handling procedures were approved by the Institutional Animal Care and Use Committee of Kyushu University, and followed the Guideline for Proper Conduct of Animal Experiments, Science Council of Japan.
Preparation of DNA from small intestinal tumor
Inspections of the small intestines were carefully carried out and the small intestinal tumors were removed under dissecting microscope. Preparations of DNA from the removed tumors were performed using DNeasy Tissue Kit (QIAGEN) according to the manufacturer's protocol.
PCR and DNA sequencing
Sixty-two small intestinal tumors obtained from 4 Mutyh-deficient mice were used for mutation analysis of Apc, Ctnnb1, Kras and Trp53, while 11 tumors developed in wild-type mice were analyzed for mutations only in Ctnnb1. Thirty- to fifty-nanogram of genomic DNA from each small intestinal tumor was used as the template for PCR. Amplified DNA was purified with QIAquick PCR Purification Kit (QIAGEN) according to the manufacturer's protocol. The purified DNA fragments were used as the template for direct sequencing with BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) and the sequences were determined with ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems). In the case of detecting two mutations in Apc from one tumor, we performed PCR with new primer sets designed for amplifying the fragment encompassing both mutation sites. The DNA fragments amplified with PrimeSTAR HS DNA polymerase (TAKARA) were inserted into the EcoRV site in pBluescript II SK(-) (Stratagene) and the cloned fragments were used as templates for sequencing. In the case of deletion found by direct sequencing, we confirmed the deleted region by sequencing using cloned amplified fragment as template. The information of the primers used and PCR conditions are available upon request.
Histological analysis
The small intestinal tumors were removed, embedded in paraffin and sectioned (3 µm). After being deparaffinized and re-hydrated, the sections were stained with hematoxylene and eosin. The evaluation of the tumors was performed according to the Vienna classification (39).
Immunohistochemistry
Immunohistochemistry were performed on 3 µm thick paraffin-embedded sections of the small intestinal tumors with anti-β-catenin antibody. The sections were deparaffinized in xylen and re-hydrated through graduated ethanol at room temperature. Tissue sections were soaked in 10 mM citrate buffer (pH6.0) and then subjected to antigen retrieval by microwaving for 20 minutes before the primary antibodey reaction. After treatment with 3% hydrogen peroxide for 5 minutes and 50 mM Tris-HCl (pH7.5) containing 1% BSA for 15 minutes, a 1:4000 diluted rabbit polyclonal anti-β-catenin antibody (Sigma) was applied to the sections, and incubated either at room temperature for 1 hr or at 4°C overnight. The detections were carried out with avidin-biotin-enzyme complex (ABC) method using LSAB+ System-HRP (DAKO) according to the manufacturer's protocol. The tumors with more than 5% of nuclear stained cells were counted for positive.
Results
Pathological analysis of small intestinal tumors
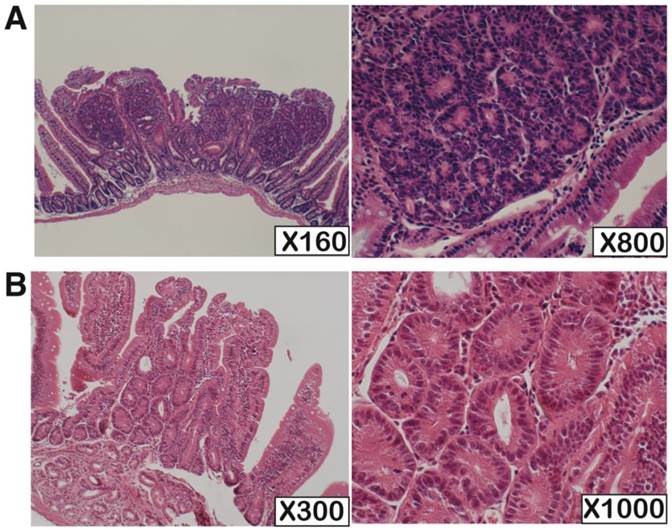
We performed pathological analysis of 73 and 10 small intestinal tumors derived from Mutyh-deficient (n=4) and wild-type mice (n=10), respectively (Table 1, Figure 1). The tumors had predominantly developed in the oral side of the small intestine. The sizes of the tumors developed in wild-type mice were apparently smaller than those in Mutyh-deficient mice. All tumors showed non-polypoid growth and their height was almost equal to normal villi. All the tumors showed cytological changes such as variably sized and enlarged nuclei, and also showed architectural changes with keeping the fundamental structure of glands, while no tumors showed evident invasion. Based on the Vienna classification of gastrointestinal epithelial neoplasia, all of these tumors were classified as category 4 (non-invasive high grade neoplasia), except for one case in a wild-type mouse that was classified as category 3 (non-invasive low grade neoplasia) because of milder cytological and architectural changes.
Pathological evaluation of KBrO3-induced small intestinal tumors
| Category 3 | Category 4 | Total | |||
|---|---|---|---|---|---|
| 4.1 | 4.2 | 4.3 | |||
| Wild type mice | 1 | 3 | 6 | 0 | 10 |
| Mutyh-deficient mice | 0 | 2 | 71 | 0 | 73 |
NOTE: The numbers in the table indicates the number of the tumors classified as each category. This evaluation was performed according to the Vienna classification of gastrointestinal neoplasia. Category 3: non-invasive low grade neoplasia, Category 4: non-invasive high grade neoplasia (4.1: high grade adenoma/displasia, 4.2: non-invasive carcinoma, 4.3: Suspicion of invasive carcinoma).
H.E. staining of the KBrO3-induced small intestinal tumors developed in Mutyh-deficient and wild type mouse. (A) One of the typical tumors from Mutyh-deficient mouse; classified as category 4. (B) An exceptional case classified as category 3 developed in wild-type mouse. Magnifications are indicated in lower right of the photos.
Somatic Apc mutations in the small intestinal tumors
To analyze Apc mutation, we amplified and sequenced the 5'-region of exon 15 of Apc, because many somatic mutations in this region were identified in the tumors from MAP patients (18)(23). Using 10 overlapping primer sets, the targeting region of Apc was amplified and used as templates for direct sequencings. We analyzed 62 tumors developed in 4 Mutyh-deficient mice treated with KBrO3. Twenty-five somatic mutations from 18 tumors were detected, and all the mutations were identified as G:C to T:A transversions, leading to the non-sense mutations (Table 2). Seventeen out of 25 mutations (68.0%) were found in the AGAA or TGAA sequences. Because 7 tumors displayed two mutations in the region analyzed, we examined whether these two mutations occurred in the same allele or not. After PCR amplification using specific primer sets for amplifying the fragment simultaneously covering the region of the two mutations, the amplified fragments were cloned into plasmid vectors. The sequence analyses of these plasmid clones revealed that no clones simultaneously carried both two mutations, thus demonstrating that these tumors were compound-heterozygotes in Apc alleles.
Somatic Apc mutations in the small intestinal tumors
| Mouse ID1) | Tumor ID2) | Nucleic acid change3) | Amino acid change4) | Secuence context5) |
|---|---|---|---|---|
| 44f | 1 | 93828 G→T | E1396X | TGAG |
| 44f | 5 | 92805 G→T | E1055X | AGAA |
| 44f | 6 | 93226 C→A | S1195X | TGAA |
| 44f | 6 | 93405 G→T | E1255X | AGAA |
| 44f | 9 | 92607 G→T | E989X | TGAA |
| 44f | 9 | 93481 C→A | S1280X | TGAT |
| 44f | 10 | 93141 G→T | E1167X | TGAA |
| 44f | 13 | 93343 C→A | S1234X | TGAA |
| 50m | 22 | 93220 C→A | S1193X | TGAT |
| 50m | 24 | 92694 G→T | E1018X | AGAA |
| 50m | 24 | 93708 G→T | G1356X | AGGA |
| 50m | 25 | 92550 G→T | G970X | TGGA |
| 50m | 28 | 92805 G→T | E1055X | AGAA |
| 50m | 28 | 93667 C→A | S1342X | TGAA |
| 50m | 29 | 92805 G→T | E1055X | AGAA |
| 50m | 29 | 92238 G→T | E853X | AGAG |
| 50m | 30 | 93042 G→T | E1134X | TGAA |
| 50m | 30 | 93099 G→T | E1153X | AGAA |
| 56f | 33 | 93099 G→T | E1153X | AGAA |
| 56f | 46 | 93492 G→T | E1284X | TGAA |
| 56f | 48 | 92826 G→T | E1062X | CGAG |
| 56f | 48 | 93102 G→T | E1154X | AGAA |
| 124m | 49 | 92811 G→T | E1057X | TGAA |
| 124m | 54 | 93498 G→T | G1286X | AGGA |
| 124m | 60 | 92199 G→T | E853X | AGAG |
NOTE: 62 tumors from 4 Mutyh-deficient mice were analyzed for Apc mutation.
1) The 4 Mutyh-deficient mice consisting of two males and two females were analyzed.
2) ID number of tumors carrying mutations in Apc. Sixteen tumors from each mouse were analyzed, except for 124m mouse from which 14 tumors were analyzed.
3) The number indicates a position of a mutation site. The numbers correspond to nucleotide sequence of APC deposited in GenBank (Accession: NC_000084).
4) The number indicates a position of a changed amino acid. X represents a nonsense codon. The numbers correspond to amino acid numbers of APC deposited in GenBank (Accession: NP_031488).
5) Sequence context surrounding G:C to T:A mutations. G shown in bold indicates the mutation site. GAA sequence is underlined. Sequence of non-transcribed strand is italicized.
Somatic Ctnnb1 mutations in the small intestinal tumors
β-catenin is reported to have 4 putative GSK3β-phosphorylation sites. Phosphorylation of β-catenin by GSK3β in a complex with Axin and Apc is required to target β-catenin for degradation by the proteasome. Therefore, the mutations at the phosphorylation sites of β-catenin are considered to be related to its stability that would lead to the accumulation of β-catenin in nuclei, thereby up-regulating the expression of the target genes. In addition, the region of mouse (but not human) Ctnnb1 encoding GSK3 β-phospharylation sites of β-catenin contains AGAA sequences. Therefore, we analyzed mutations in exon 3 of Ctnnb1 encoding GSK3β-phospharylation sites of β-catenin. Among 62 tumors from 4 Mutyh-deficient mice and 11 tumors from 10 wild-type mice, 36 out of 62 tumors (58.1%) from Mutyh-deficient mice and 3 out of 11 tumors (27.3%) from wild-type mice showed a mutation in the region corresponding to the GSK3β-phosphorylation sites (Table 3). All the mutations in Mutyh-deficient mice were base substitution mutations, in which 35 mutations (97.2%) were identified as G:C to T:A transversions and 1 mutation (2.8%) as A:T to T:A transversion. Remarkably, 34 G:C to T:A transversions occurred at the AGAA sequences associated with either Ser33 or Ser37, the putative phosphorylation sites, changing them to tyrosine. Two mutations found on either side of codon for Ser33, which converted the original amino acids (Asp32 or Gly34) to Valine. No tumor carried mutations in both Apc and Ctnnb1 genes, simultaneously. In wild-type mice, 2 base substitution mutations and a 24-bp-deletion were identified. Both base substitution mutations were G:C to A:T transitions, not related to 8-oxoG-induced transversions. One of them occurred at Ser41 changing it to Isoleucine, where no mutation was found in Mutyh-deficient mice.
Mutation analysis for Ctnnb1
| Genotype | Amino acid change1) | Nucleic acid change2) | Sequence context3) | Number of mutations |
|---|---|---|---|---|
| Wild type | 37S→F | 17219C→T | AGAA | 1 |
| 41T→I | 17231C→T | ACCA | 1 | |
| del (31-39) 4) | 24-bp deletion | - | 1 | |
| Mutyh-deficient | 32D→V | 17204A→T | GATT | 1 |
| 33S→Y | 17207C→A | AGAA | 26 | |
| 34G→V | 17210G→T | GGAA | 1 | |
| 37S→Y | 17219C→A | AGAA | 8 |
1) The number indicates a position of a changed amino acid. The numbers correspond to amino acid numbers of β-catenin deposited in GenBank (Accession: NP_001159374).
2) The number indicates a position of a mutation site. The numbers correspond to nucleotide sequence of Ctnnb1 deposited in GenBank (Accession: NC_000075).
3) Sequence context surrounding G:C to T:A mutations. Letters shown in bold indicate the mutation site. GAA sequence is underlined. Sequence of non-transcribed strand is italicized.
4) Nine amino acids from no.31 to 39 were replaced by newly arisen one serine resulted from the 24-bp deletion.
No tumors had mutations in either Kras or Trp53
It has been shown that KRAS mutations were common in tumors developed in MAP patients, and all the mutations were G:C to T:A transversions at first G of codon12 (37)(38). In addition, TP53 muations were also detected in tumors from MAP patients, albeit at relatively low frequency (37). Thus, we further searched for the mutations in exon 2 of Kras (containing codon 12 and 13) and in exon 5 to 8 of Trp53 (conserved region) in tumors from KBrO3-treated Mutyh mice. Neither Kras nor Trp53 mutations was found in the regions analyzed.
Immunohistochemistry
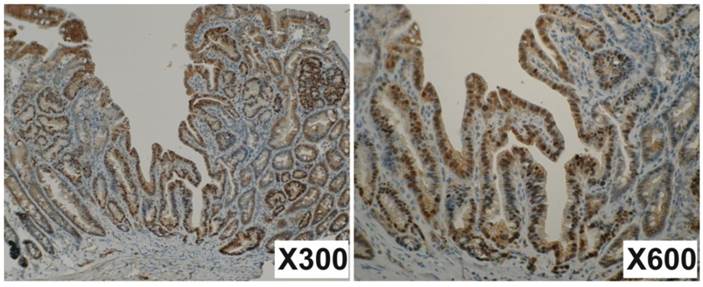
Mutations occurred both in Apc and in Ctnnb1 would result in the accumulation of β-catenin in the nucleus. To verify this possibility, immunohistochemistry for β-catenin was performed with 71 small intestinal tumors developed in the 4 Mutyh-deficient mice treated with KBrO3. The tumors with more than 5% of nuclear stained cells were counted for positive. Nuclear staining was identified in 41 tumors (57.7%), indicating the accumulation of β-catenin in the nuclei (Figure 2). These findings support the results of mutation analysis in Apc and Ctnnb1.
Immunohistochemistry for β-catenin. Many nuclear steins are observed in tumor cells but not in normal cells. The junctions of epithelial cells are also well stained. Magnifications are indicated in lower right of the photos.
Discussion
We previously reported that Mutyh-deficient mice showed the susceptibility to tumorigenesis, especially adenoma/carcinoma in the intestine, hemangioma in the liver and angiosarcoma in the spleen (17). The intestinal tumor susceptibility of Mutyh-deficient mice was further enhanced by treatment with KBrO3, a known oxidative renal carcinogen associated with 8-oxoG accumulations. The multiple tumor-formation in the mutant mice is consistent with the malignant capacity of multiple colorectal adenomatous polyposis in MAP patients, suggesting that Mutyh-deficient mouse is an animal model for investigating the pathogenesis of MAP. Thus, we performed the characterization of tumors induced in Mutyh-deficient mice treated with KBrO3.
The small intestinal tumors analyzed in this study were exclusively classified in category 4 according to the Vienna classification of gastrointestinal neoplasia (Table 1). Only one tumor developed in a wild-type mouse with KBrO3-treatment was classified in category 3. Even the tiny tumors were also classified in category 4. These results suggested that the KBrO3-induced small intestinal tumors show significantly dysplastic change in the early stage of tumor-development. This gives rise to a possibility that the chronic exposures of KBrO3 to intestinal mucosa in Mutyh-deficient mice might evoke concurrent mutations in multiple genes, although we could find any mutations neither in Kras nor in Trp53.
Tumors from MAP patients exhibit a significantly increased incidence of G:C to T:A somatic mutations in APC. These mutations exclusively occurred in AGAA or TGAA sequences. We examined mutations in the region of Apc in which many somatic mutations were identified in tumors from MAP patients, and found 25 somatic mutations in 18 out of 62 tumors. All the mutations were characterized as G:C to T:A transversions, and 68.0% (17 out of 25) of the mutations were found at either AGAA or TGAA sequence. These results are highly consistent with the findings in the tumors developed in MAP patients. We also analyzed Ctnnb1 for incidence of mutations because CTNNB1 mutations were found in about a half of colorectal cancers lacking APC mutation in humans (25)(29)(30). We identified 36 mutations (35 G:C to T:A transversions and 1 T:A to A:T transversion) in Ctnnb1 among 62 tumors. All the G:C to T:A transversions detected in Ctnnb1 occurred at the GAA sequence, 34 in AGAA and one in second G of GGAA sequences. Fifty-four out of 62 (87.1%) tumors developed in KBrO3-treated Mutyh-deficient mice carried mutations either in Apc or Ctnnb1. The immunohistochemistry confirmed β-catenin accumulation in the nuclei of the tumors harboring either Apc or Ctnnb1 mutations. These observations suggest the association between the defect in the Wnt-signaling pathway and multi-tumor formation in Mutyh-deficient mice treated with KBrO3. On the other hand, we could not detect any Kras mutations in the KBrO3-induced tumors, although KRAS mutations in codon12 were frequently observed in the cancers (64%) and adenomas (43%) of MAP patients (37, 38). These differences may be attributed to a short period (16 weeks) for tumor-formation under the consecutive and enhanced oxidative stress in Mutyh-deficient mice. It is also possible that Kras mutation may occur at a relatively late stage in multistep carcinogenesis, so that the mutation, if any, could not be detected with the methods we applied in this study.
We also examined for Ctnnb1 mutations in the small intestinal tumors developed in wild-type mice. None of the mutations was G:C to T:A transversion; two were G:C to A:T transitions and one was a 24-bp deletion. Thus, the mutations found in the KBrO3-treated wild-type mice would not result from the mutagenic effects of 8-oxoG. These results indicates that Mutyh can sufficiently suppress the appearance of premalignant cells containing G:C to T:A transversions in wild-type mouse, even under the condition of enhanced oxidative stress.
The small intestinal tumors induced by KBrO3-treatment displayed more mutations in Ctnnb1 than in Apc, although APC mutation is mostly common in human colorectal cancer and MAP polyps. GSK3β-phosphoryration sites and their surroundings of β-catenin are highly conserved; there are no difference of amino acid sequence in the region between human and mouse. However, there are some differences in the DNA sequence corresponding to this region. In this study, Ctnnb1 mutations were frequently found in the codon for Ser33 and the corresponding DNA sequence is TCT in both human and mouse. There is a TTC sequence spanning codon 32 and 33 in mouse Ctnnb1 but not in human CTNNB1 because of the difference in codon-usage for Asp32; GAT in mouse but GAC in human. Because AGAA sequences (and its reverse-complementary sequence TTCT) appear to be the target sites for oxidative stress-induced mutagenesis in MUTYH-deficient genetic background, it is conceivable that this difference would be a main reason why Ctnnb1 mutations were predominantly observed in the tumors from Mutyh-deficient mice.
The observed high incidence of mutations either in Apc or Ctnnb1 imply that the initial event of tumor-formation would be a dysfunction of Wnt-signal pathway in intestinal epithelial cells of Mutyh-deficient mice treated with KBrO3. Our study further demonstrated that in agreement with the findings in MAP, G:C to T:A transversions were detected in the genes controlling Wnt-signal pathway with high frequency, and the mutations predominantly occurred at AGAA or TGAA sequences. Thus, in addition to the multiple tumor-formation in the mutant mice, specificity with respect to the genetic defects and its mutational spectra and specificity are also considerably match to the observations in the multiple colorectal adenomatous polyposis in MAP patients. We, therefore, consider that KBrO3-induced tumorigenesis using Mutyh-deficient mice is a useful experimental system for studying on molecular processes in the early development of adenoma and carcinoma in MAP, as well as for examining the preventive and therapeutic approaches for the oxidative stress-induced intestinal tumors (40).
Abbreviations
8-oxoG: 8-oxoguanine; MAP: MUTYH-associated polyposis; GSK3β: glycogen synthase kinase 3 beta
Acknowledgements
The authors thank Drs. Masumi Hidaka and Mizuki Ohno for discussion, and Ms. Fumiko Sasaki and Ms. Yasuko Togami for their technical assistance with the laboratory works.
Funding
This study was financially supported in part by Grant-in-aid for Scientific Research (20310031, 25241012) from the Japan Society for the promotion of Science and a Grant-in-aid for Cancer Research for the Third Term Comprehensive 10-Year Strategy for Cancer Control (13803744) from the Ministry of Health, Labour and Welfare, Japan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915-22
2. Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431-4
3. Mo JY, Maki H, Sekiguchi M. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc Natl Acad Sci USA. 1992;89:11021-5
4. Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H. et al. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem. 1993;268:23524-30
5. Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y. et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc Natl Acad Sci USA. 2001;98:11456-61
6. Egashira A, Yamauchi K, Yoshiyama K, Kawate H, Katsuki M, Sekiguchi M. et al. Mutational specificity of mice defective in the MTH1 and/or the MSH2 genes. DNA Repair (Amst). 2002;1:881-93
7. Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:8010-5
8. Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94:7429-34
9. Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T. et al. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene. 1997;14:2857-61
10. Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K. et al. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637-52
11. Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M. et al. Ogg1 Knockout-associated Lung Tumorigenesis and Its Suppression by Mth1 Gene Disruption. Cancer Res. 2003;63:902-5
12. Kunisada M, Sakumi K, Tominaga Y, Budiyanto A, Ueda M, Ichihashi M. et al. 8-Oxoguanine Formation Induced by Chronic UVB Exposure Makes Ogg1 Knockout Mice Susceptible to Skin Carcinogenesis. Cancer Res. 2005;65:6006-10
13. Ohtsubo T, Nishioka K, Imaiso Y, Iwai S, Shimokawa H, Oda H. et al. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000;28:1355-64
14. Ushijima Y, Tominaga Y, Miura T, Tsuchimoto D, Sakumi K, Nakabeppu Y. A functional analysis of the DNA glycosylase activity of mouse MUTYH protein excising 2-hydroxyadenine opposite guanine in DNA. Nucleic Acids Res. 2005;33:672-82
15. Hirano S, Tominaga Y, Ichinoe A, Ushijima Y, Tsuchimoto D, Honda-Ohnishi Y. et al. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J Biol Chem. 2003;278:38121-4
16. Tominaga Y, Ushijima Y, Tsuchimoto D, Mishima M, Shirakawa M, Hirano S. et al. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004;32:3198-211
17. Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K. et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599-604
18. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT. et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227-32
19. Mazzei F, Viel A, Bignami M. Role of MUTYH in human cancer. Mutat Res. 2013;743-744:33-43
20. Jo W-S, Chung DC. Genetics of hereditary colorectal cancer. Semin Oncol. 2005;32:11-23
21. Lipton L, Tomlinson I. The multiple colorectal adenoma phenotype and MYH, a base excision repair gene. Clin Gastroenterol Hepatol. 2004;2:633-8
22. Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RKS. et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791-9
23. Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT. et al. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum Mol Genet. 2002;11:2961-7
24. Sampson JR, Jones S, Dolwani S, Cheadle JP. MutYH (MYH) and colorectal cancer. Biochem Soc Trans. 2005;33:679-83
25. Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1-24
26. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55-67
27. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H. et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589-600
28. Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A. et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665-9
29. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S. et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229-33
30. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational Analysis of the APC / β -Catenin / Tcf Pathway in Colorectal Cancer Pathway in Colorectal Cancer1. Cancer Res. 1998;58:1130-4
31. Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML. β-Catenin Mutations Are More Frequent in Small Colorectal Adenomas Than in Larger Adenomas and Invasive Carcinomas. Cancer Res. 1999;59:1442-4
32. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN. et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235-7
33. Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ. et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293-7
34. Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327:298-303
35. Vogelstein B, Fearon EE, Hamilton SR, Kern SE, Preisinger AC, Leppert M. et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525-32
36. Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon J V. et al. p53 mutations in colorectal cancer. Proc Natl Acad Sci USA. 1990;87:7555-9
37. Lipton L, Halford SE, Johnson V, Novelli MR, Jones A, Cummings C. et al. Carcinogenesis in MYH-Associated Polyposis Follows a Distinct Genetic Pathway. Cancer Res. 2003;63:7595-9
38. Nielsen M, de Miranda NFCC, van Puijenbroek M, Jordanova ES, Middeldorp A, van Wezel T. et al. Colorectal carcinomas in MUTYH-associated polyposis display histopathological similarities to microsatellite unstable carcinomas. BMC Cancer. 2009;9:184
39. Schlemper RJ. The Vienna classification of gastrointestinal epithelial neoplasia. Gut. 2000;47:251-5
40. Takahashi-Yanaga F, Yoshihara T, Jingushi K, Igaw K, Tomooka K, Watanabe Y. et al. DIF-1 inhibits tumor growth in vivo reducing phosphorylation of GSK-3β and expressions of cyclin D1 and TCF7L2 in cancer model mice. Biochem Pharmacol. 2014;89:340-8
Author contact
![]() Corresponding author: Teruhisa Tsuzuki, Department of Medical Biophysics and Radiation Biology, Faculty of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan. Phone: +81-92-642-6141; fax: +81-92-642-6145; E-mail: tsuzukikyushu-u.ac.jp
Corresponding author: Teruhisa Tsuzuki, Department of Medical Biophysics and Radiation Biology, Faculty of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan. Phone: +81-92-642-6141; fax: +81-92-642-6145; E-mail: tsuzukikyushu-u.ac.jp