Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Acknowledgements
References
Author Biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(9):990-996. doi:10.7150/ijbs.9832 This issue Cite
Short Research Communication
Implication of Transient Receptor Potential Vanilloid Type 1 in 14,15-Epoxyeicosatrienoic Acid-induced Angiogenesis
Kuo-Hui Su1,*, Kuan-I Lee1,*, Song-Kun Shyue2, Hsiang-Ying Chen1, Jeng Wei3, ![]() , Tzong-Shyuan Lee 1,
, Tzong-Shyuan Lee 1, ![]()
1. Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, 11221 Taiwan;
2. Institute of Biomedical Sciences, Academia Sinica, Taipei, 11529 Taiwan;
3. Heart Center, Cheng-Hsin General Hospital, Taipei, 11221 Taiwan.
*These authors equally contributed to this work.
Received 2014-6-6; Accepted 2014-8-13; Published 2014-9-6
Abstract
14,15-epoxyeicosatrienoic acid (14,15-EET) is implicated in regulating physiological functions of endothelial cells (ECs), yet the potential molecular mechanisms underlying the beneficial effects in ECs are not fully understood. In this study, we investigated whether transient receptor potential vanilloid receptor type 1 (TRPV1) is involved in 14,15-EET-mediated Ca2+ influx, nitric oxide (NO) production and angiogenesis. In human microvascular endothelial cells (HMECs), 14,15-EET time-dependently increased the intracellular level of Ca2+. Removal of extracellular Ca2+, pharmacological inhibition or genetic disruption of TRPV1 abrogated 14,15-EET-mediated increase of intracellular Ca2+ level in HMECs or TRPV1-transfected HEK293 cells. Furthermore, removal of extracellular Ca2+ or pharmacological inhibition of TRPV1 decreased 14,15-EET-induced NO production. 14,15-EET-mediated tube formation was abolished by TRPV1 pharmacological inhibition. In an animal experiment, 14,15-EET-induced angiogenesis was diminished by inhibition of TRPV1 and in TRPV1-deficient mice. TRPV1 may play a crucial role in 14,15-EET-induced Ca2+ influx, NO production and angiogenesis.
Keywords: transient receptor potential vanilloid 1, epoxyeicosatrienoic acid, angiogenesis, calcium, endothelial cell.
Introduction
Epoxyeicosatrienoic acids (EETs), the cytochrome P450 epoxygenase (CYP)-derived metabolites of arachidonic acid, play an important role in vascular functions by regulating the membrane hyperpolarization of smooth muscle cells by activating calcium (Ca2+)-activated potassium channels [1-3]. EETs also have beneficial effects on the pathophysiological functions of endothelial cells (ECs) [1-3]. For example, 14,15-EET increases the activity of endothelial nitric oxide synthase (eNOS) and NO bioavailability [2, 3]. Additionally, 14,15-EET induces the expression of vascular endothelial cell growth factor and promotes angiogenesis in ECs [1, 2]. However, much still remains to be learned about the molecular mechanism underlying EET-mediated protection in ECs.
The transient receptor potential vanilloid type 1 (TRPV1), a non-selective cation channel, is a crucial integrator of multiple chemical and physical stimuli in primary sensory neurons [4]. Activation of TRPV1 in neurons increases Ca2+ entry, which elevates intracellular Ca2+ level, thus excitating sensory neurons [4, 5]. Recently, growing evidence suggests that TRPV1 is expressed in ECs and plays an important role in regulating vascular tone and EC functions [6-8]. This TRPV1 function is particularly important because ECs may be directly exposed to various endogenous TRPV1 ligands under pathophysiological conditions such as ischemia [4, 6-9]. However, whether TRPV1 is involved in the 14,15-EET-conferred benefits in ECs remains to be investigated.
In this study, we examined the role of TRPV1 in 14,15-EET-promoted angiogenesis in ECs and mice. We found that administration of 14,15-EET may trigger TRPV1-dependent Ca2+ influx and NO production, which leads to increased EC tube formation and angiogenesis.
Materials and Methods
Reagents
14,15-EET and 14,15-EE5ZE were from Cayman (Ann Arbor, MI). Quest Fluo-8 NW Calcium Assay Kit was from AAT Bioquest (AAT Bioquest Inc., Sunnyvale, CA, USA). Ca2+ free medium, capsaicin, capsazepine (CPZ), SB366791, HC030031 (HC), 1,2-Bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid/acetoxymethyl ester (BAPTA/AM), ethylene glycol tetraacetic acid (EGTA), Griess's reagent, and Drabkin's reagent kit 525 were from Sigma-Aldrich (St. Louis, MO, USA). TurboFect was from Fermentas (Glen Burnie, MD, USA). Matrigel was from BD Biosciences (San Jose, CA, USA).
Cell culture
Human microvascular endothelial cells (HMECs) were obtained from the Centers for Disease Control (Atlanta, GA, USA) and grown in Medium 200 (Cascade Biologics Inc., Portland, OR, USA) supplemented with low serum growth supplement (Cascade Biologics, Portland, OR, USA) with 95% air and 5% CO2 at 37°C. The final concentrations of the components in Medium 200 contained 2% fetal bovine serum (FBS) (GibcoBRL, Grand Island, NY, USA), 1 μg/ml hydrocortisone, 10 ng/ml human epidermal growth factor, 3 ng/ml human fibroblast growth factor, 10 μg/ml heparin, and 1% antibiotic-antimycotic mixture (GibcoBRL, Grand Island, NY, USA). Cells at confluence were subcultured at a 1:3 ratio and used at passages 3 through 8. Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 100 unit/ml penicillin, and 100 μg/ml streptomycin (HyClone, Logan, UT) at 37ºC.
Detection of Ca2+ influx
HMECs or HEK293 cells were seeded in 24-well plates and grown to confluence, washed, and Fluo-8 NW-loading solution was added for 1 h at 37ºC. Medium was replaced with fresh medium containing test compound. Fluorescence was measured by fluorometry (Molecular Devices, Sunnyvale, CA, USA) with 490-nm excitation and 525-nm emission. Images were photomicrographed under a Nikon TE2000-U microscope with QCapture Pro 6.0 software (QImaging, BC, Canada).
Plasmid construction and transient transfection
The coding regions for TRPV1 wild type (WT), TRPV1-Y671K or TRPV1-Y671D with the pcDNA3.1 (+) plasmid were kindly provided by Dr. Carla Nau (Department of Anesthesiology, Friedrich-Alexander-University, Erlangen-Nuremberg, Erlangen, Germany) [10]. Each TRPV1 DNA fragment was digested with EcoRI and XbaI, then cloned into the pCMV5N-Flag vector. DNA sequences were verified by sequencing analysis. Transient transfection involved use of TurboFect according to the manufacturer's instructions. Briefly, 1 μg vector, TRPV1 WT, or TRPV1 mutants was transfected into HEK293 cells by use of TurboFect for 24 h.
Measurement of NO production
The method for detecting NO production was as described [11]. Briefly, accumulated nitrite (NO2-), the stable breakdown product of NO, in culture media, was measured by mixing an equal volume of Griess's reagent and then incubating at room temperature for 15 min. Azo dye production was analyzed by use of an SP-8001 UV/VIS spectrophotometer (Metertech, Taipei, Taiwan) with absorbance at 540 nm. Sodium nitrite was used as a standard.
Tube formation assay
Tube formation assay was as described previously [12]. Briefly, Matrigel was coated onto 24-well plates and polymerized for 30 min at 37ºC. Cells were seeded onto the Matrigel layer and incubated with the indicated treatments for 12 h. The morphology of cells was observed by microscopy and quantified by counting the number of branch points.
Animals
All animal experiments were approved by the Animal Care and Utilization Committee of National Yang-Ming University. Wild type (WT) C57BL/6 mice were purchased from the National Laboratory Animal Center, National Science Council (Taipei, Taiwan); TRPV1-/- mice (B6.129X1-Trpv1tm1Jul) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and backcrossed to C57BL/6 mice for at least 6 generations. Mice were housed in barrier facilities with a 12-h light/dark cycle and fed normal chow diet. PCR of genomic DNA was used to confirm TRPV1-/- and genotypes.
In vivo matrigel plug assay
Matrigel plug assay was described previously [11]. Briefly, Matrigel containing 50 U/ml heparin with or without 10 nM 14,15-EET or 10 μM CPZ was injected subcutaneously into the left and right flanks of mice. Mice were killed after 7 days with use of CO2. The plugs were carefully dissected from the host tissue. Matrigel plugs were photographed, and the hemoglobin content was measured by use of Drabkin's reagent kit 525 to quantify blood vessel formation.
Statistical analysis
The experiments were performed at least 5 times. Results are presented as mean ± SEM. Mann-Whitney U test was used to compare 2 independent groups. Kruskal-Wallis test followed by Bonferroni posthoc analysis was used to account for multiple testing. SPSS v18.0 (SPSS Inc, Chicago, IL) was used for analysis. Differences were considered statistically significant at P<0.05.
Results and Discussion
TRPV1 activation is required for 14,15-EET-mediated Ca2+ influx
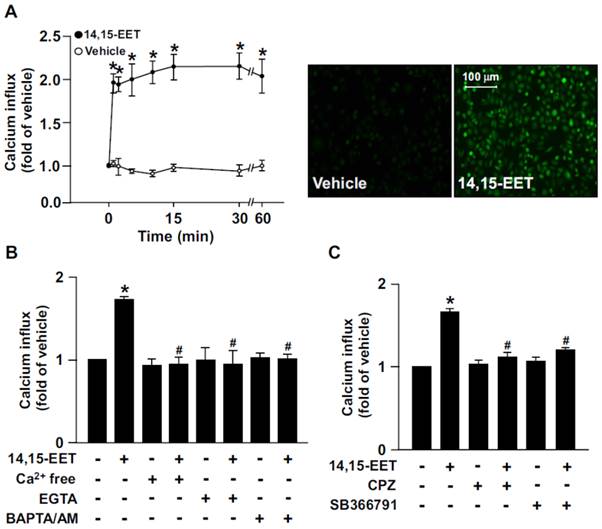
Growing evidence has suggested that 14,15-EET is implicated in regulating EC physiological functions such as angiogenesis [13, 14]. However, the potential molecular mechanisms underlying the beneficial effects in ECs are not fully understood. In HMECs, treatment with 14,15-EET (10 nM) time-dependently increased Ca2+ influx within 60 min; it increased intracellular Ca2+ as early as 1 min after treatment, which was sustained for up to 60 min (Fig. 1A). Additionally, the 14,15-EET-increased Ca2+ level was diminished after treating with the Ca2+ chelators EGTA (500 μM) or BAPTA/AM (10 μM) or removal of extracellular Ca2+ (Fig. 1B). To address the importance of TRPV1 in 14,15-EET-induced Ca2+ influx, HMECs were pre-treated with the TRPV1 inhibitor CPZ (10 μM) or SB366791 (10 μM) to inhibit the Ca2+ channel activity of TRPV1. Treatment with CPZ or SB366791 nearly abrogated the 14,15-EET-triggered Ca2+ influx (Fig. 1C). These results suggest that TRPV1 plays an important role in regulating 14,15-EET-induced Ca2+ influx in ECs.
14,15-EET elicits transient receptor potential vanilloid type 1 (TRPV1)-dependent Ca2+ influx in human microvascular endothelial cells (HMECs). (A) HMECs were treated with 14,15-EET (10 nM) for the indicated times. Fluorescence images were photographed by fluorescence microscopy at 10 min after 14,15 EET treatment. (B) HMECs were pre-treated with Ca2+ free medium, EGTA (500 μM), an extracellular Ca2+ chelator, or BAPTA/AM (10 μM), an intracellular Ca2+ chelator, for 2 h then with 14,15-EET (10 nM) for 10 min. (C) HMECs were pre-treated with the TRPV1 pharmacological antagonist capsazepine (CPZ, 10 μM) or SB366791 (10 μM), for 2 h and then incubated with 14,15-EET (10 nM) for 10 min. The intracellular level of Ca2+ was analyzed by Fluo-8 calcium assay. Data are mean ± SEM from 5 independent experiments. *, P<0.05 versus vehicle; #, P<0.05 versus 14,15-EET.
To provide further evidence that TRPV1 activation is essential for 14,15-EET-promoted Ca2+ influx in ECs, we used a gain- or loss-of-function strategy to deplete the Ca2+ permeability of TRPV1 channel by overexpressing a full-length TRPV1 WT, TRPV1-Y671K (a TRPV1 mutant with defective Ca2+ permeability) or TRPV1-Y671D (a TRPV1 mutant with normal Ca2+ permeability) in HEK293 cells, a cell line lacking TRPV1 channels. Treatment with 14,15-EET or capsaicin (a specific TRPV1 agonist) significantly increased the intracellular level of Ca2+ in TRPV1-overexpressed HEK293 cells but not vector-transfected HEK293 cells (Fig. 2A). Blockage of TRPV1 by TRPV1 antagonist CPZ or SB366791 abrogated 14,15-EET-induced Ca2+ influx in TRPV1-overexpressed HEK293 cells (Fig. 2B).
The essential role of TRPV1 in 14,15-EET-induced Ca2+ influx in human embryonic kidney 293 (HEK293) cells. (A) HEK293 cells were transfected with vector or TRPV1 WT plasmid, then treated with 14,15-EET (10 nM) or capsaicin (10 μM) for 10 min. (B) After transfection with TRPV1 WT plasmid, HEK293 cells were pretreated with the TRPV1 antagonist CPZ (10 μM) or SB366791 (10 μM) for 2 h, then 14,15-EET (10 nM) for 10 min. (C) HEK293 cells were transfected with TRPV1 WT, TRPV1-Y671K or TRPV1-Y671D plasmid and then treated with 14,15-EET (10 nM) for 10 min. The intracellular level of Ca2+ was measured by Fluo-8 calcium assay. Fluorescence images were photographed by fluorescence microscopy. Data are mean ± SEM. *, P<0.05 versus vector or TRPV1 WT transfection alone; #, P<0.05 versus TRPV1 WT + 14,15-EET.
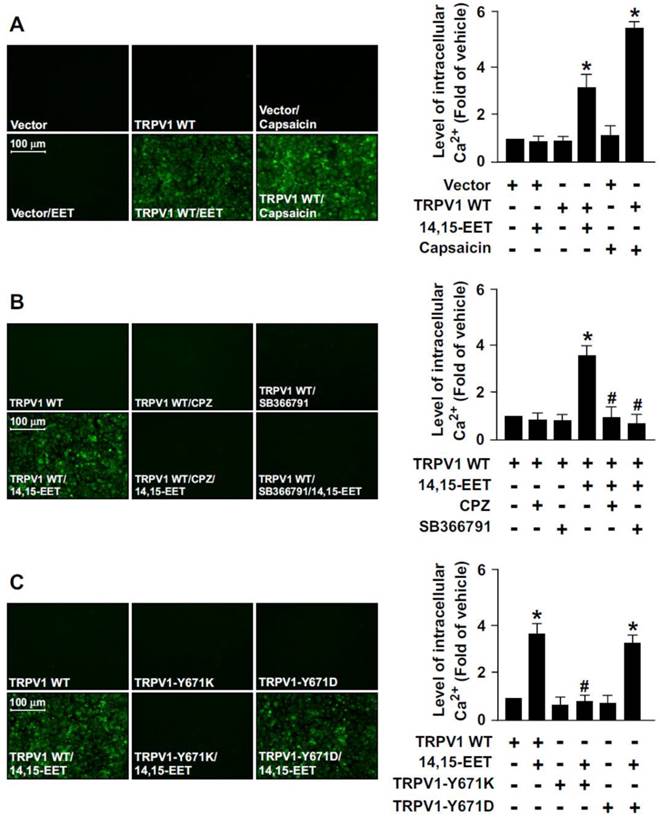
Moreover, 14,15-EET increased intracellular Ca2+ level in TRPV1 WT- or TRPV1 Y671D-overexpressed HEK293 cells but not TRPV1 Y671K-overexpressed HEK293 cells (Fig. 2C). However, such loss-of-function of TRPV1 experiments by transfection with non-viral vectors in HMECs were limited due to the poor transfection efficiency in primary ECs. Therefore, the effect of loss of TRPV1 function by TRPV1-Y671K mutant in HMECs was unclear. Collectively, these data suggest that TRPV1-mediated Ca2+ influx might be an important event in the 14,15-EET-conferred beneficial effects in ECs.
TRPV1 signaling is essential for 14,15-EET-activated production of NO
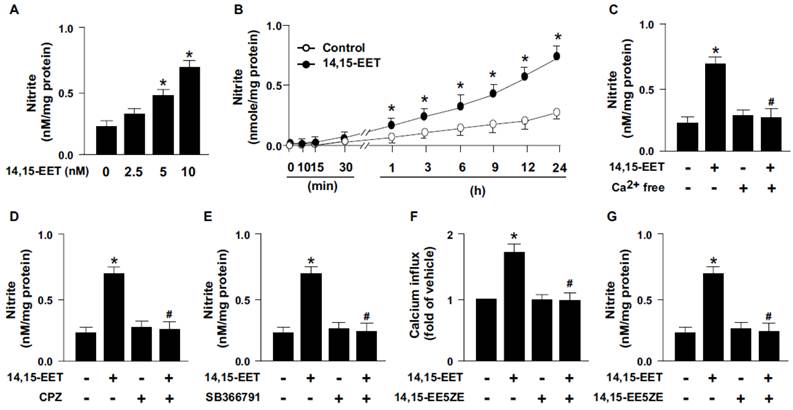
14,15-EET has been reported to regulate the activation of eNOS and NO production [15, 16]. We next examined whether TRPV1 contributes to 14,15-EET-induced NO production. Treatment with 14,15-EET dose- and time-dependently increased NO production from HMECs (Fig. 3A and B). As well, pretreatment with Ca2+ free medium or pharmacological inhibition of TRPV1 with CPZ or SB366791 diminished the 14,15-EET-induced NO production (Fig. 3C-E). Thus, TRPV1 may play an important role in 14,15-EET-increased NO production. In addition to the direct binding with ligands, the activity of TRPV1 channels can be regulated by second messenger pathways induced by receptors/cognate ligands [5]. Therefore, we used 14,15-EET antagonist 14,15-EE5ZE to investigate the possible involvement of EET receptor in 14,15-EET-elicited TRPV1 activation and NO bioavailbility in ECs. Our data demonstrated that blockage of 14,15-EET effect by its antagonist 14,15-EE5ZE abolished 14,15-EET-mediated Ca2+ influx (Fig. 3F) and consequent NO production (Fig. 3G), suggesting 14,15-EET may not directly bind with TRPV1 and EET receptor might be the upstream molecule for 14,15-EET-mediated TRPV1 activation. Moreover, EETs have biological actions involved in regulating vascular functions such as regulation of vascular tone, inflammation and angiogenesis [1-3]. All these actions are related to the endothelial-derived NO bioavailability, a crucial molecule for maintaining cardiovascular function under physiological and pathological conditions [17, 18]. Production of NO is mediated by eNOS activity, which is regulated by kinase-dependent signaling including Ca2+-dependent signaling. TRPV1, a non-selective cation channel, is a crucial regulator of Ca2+ influx in neural cells and in non-neuronal cells such as myocardiocytes and ECs [19, 20]. TRPV1 also contributes to the function of ECs, including NO production and angiogenesis by its ligands capsaicine and evodiamine [6-8, 21]. However, whether TRPV1-Ca2+ signaling is involved in the 14,15-EET-mediated pleiotropic effect in ECs such as angiogenesis is still not fully understood.
TRPV1 is required for 14,15-EET-induced nitric oxide (NO) production. (A) HMECs were treated with 0, 2.5, 5, 10 nM 14,15-EET for 24 h or (B) treated with 10 nM 14,15-EET for indicated times. HMECs were pre-treated with Ca2+ free medium (C), 10 μM CPZ (D), or 10 μM SB366791 (E) for 2 h, then incubated in 10 nM 14,15-EET for 24 h. (F and G) HMECs were pretreated with 14,15-EE5ZE for 2 h and then incubated with 10 nM 14,15-EET for 10 min or 24 h. The intracellular level of Ca2+ was measured by Fluo-8 calcium assay and the level of nitrite in culture medium was measured by Griess's reagent. Data are mean ± SEM from 5 independent experiments. *, P<0.05 versus 0 min or vehicle; #, P<0.05 versus 14,15-EET.
TRPV1 plays a crucial role in 14,15-EET-mediated promotion of angiogenesis
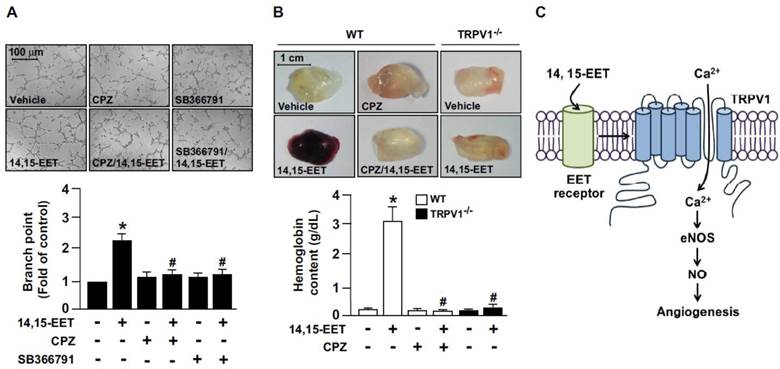
We next explored whether TRPV1 is involved in 14,15-EET-promoted angiogenesis. The combination of 14,15-EET and CPZ or SB366791 markedly attenuated 14,15-EET-induced tube formation (Fig. 4A). To confirm the in vitro studies, we used Matrigel plug assay to access the role of TRPV1 in angiogenesis in vivo. In WT mice, administration of 14,15-EET increased angiogenesis, which was prevented by treatment with CPZ (Fig. 4B). Moreover, genetic deletion of TRPV1 abolished the 14,15-EET-induced angiogenesis of Matrigel plugs (Fig. 4B). Taken together, our functional analysis demonstrated that 14,15-EET promoted EC tube formation, which is consistent with findings from previous studies. [13, 14] More importantly, we demonstrated that blocking TRPV1 inhibited the 14,15-EET-induced tube formation, which indicates the critical role of TRPV1 in the 14,15-EET-induced beneficial effects in ECs. Our in vivo experiments further confirmed that the function of TRPV1 is not limited to the cell model. Mixing CPZ, the TRPV1 inhibitor, with Matrigel plugs significantly reduced vascularization with or without 14,15-EET. In addition, TRPV1 deletion attenuated 14,15-EET-induced angiogenesis. In terms of EC function, our findings agree with previous studies that TRPV1 plays a crucial role in vascular functions in response to physiological and pathological insults [4, 6-9]. Thus, these results indicated the essential role of TRPV1 in 14,15-EET-mediated EC angiogenesis (Fig. 4C).
TRPV1 activation plays a critical role in 14,15-EET-increased angiogenesis. (A) HMECs were cultured in pre-coated Matrigel and pretreated with 10 μM CPZ or 10 μM SB366791 for 2 h, then incubated with 10 nM 14,15-EET for 12 h. Bar graphs indicate the total number of tube branch points in 5 randomly selected microscopy views. Data are mean ± SEM from 5 independent experiments. *, P<0.05 versus vehicle; #, P<0.05 versus 14,15-EET. (B) Eight week-old mice were subcutaneously injected with Matrigel plugs containing 50 U/ml heparin with or without 14,15-EET (10 nM) or CPZ (10 μM) and TRPV1-/- mice were subcutaneously injected with Matrigel with or without 14,15-EET (10 nM). After 7 days, Matrigel plugs were isolated and photographed. The haemoglobin content of the plugs was measured to indicate functional angiogenesis. Data are mean ± SEM from 9 mice. *, P<0.05 versus WT mice without 14,15-EET; #, P<0.05 versus WT mice with 14,15-EET. (C) The proposed molecular mechanism of TRPV1 activation by 14,15-EET for endothelial NO production and angiogenesis. TRPV1 is involved in 14,15-EET-mediated increase of intracellular Ca2+, NO production and induction of endothelial angiogenesis.
In addition to TRPV1, at least other 18 TRP channel isoforms have been found in ECs and implicated in the regulation of vascular functions [4, 22, 23]. For instance, TRPC1, TRPC3 and TRPC6 are involved in bradykinin-, ATP-, 11,12-EET- or acetylcholine-induced Ca2+ influx in ECs; all these factors are vasodilatation stimulators. Additionally, endothelial TRPV4 is a mechanosensitive ion channel and plays an important role in flow- or 5,6-EET-induced vasodilatation [24]. Moreover, genetic deletion of TRPV4 decreased acetylcholine-induced Ca2+ entry in ECs and impaired endothelium-dependent, NO-mediated vasorelaxation [22, 25]. Experimental evidence showed that TRPV subunits can form hetero- or homotetramers within vasculature [4, 23]. The N-terminus of TRPV1 channels contains several ankyrin repeats that can bind to intracellular proteins such as calmodulin and AMP-activated protein kinase to activate the downstream signaling pathways on stimulation [4, 6-8]. However, whether TRPV1 interacts with other TRP members or works in concert to promote 14,15-EET-mediated angiogenesis remains to be investigated.
Conclusion
In summary, this study illustrates that TRPV1 is engaged in 14,15-EET-elicited angiogenesis. The molecular mechanism revealed in this study may provide advanced information for better understanding the therapeutic value of EETs in treating eNOS-related cardiovascular diseases.
Acknowledgements
This work was supported by grants from Cheng-Hsin General Hospital (103F003C04) and the National Science Council (101-2628-B-010-001-MY2, 101-2811-B-010-039, 102-2628-B-010-001-MY3, 102-2811-B-010-035), Taiwan.
Competing Interests
The authors declare no competing interests.
References
1. Cheranov SY, Karpurapu M, Wang D. et al. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood. 2008;111:5581-91
2. Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010;459:881-95
3. Pfister SL, Gauthier KM, Campbell WB. Vascular pharmacology of epoxyeicosatrienoic acids. Adv Pharmacol. 2010;60:27-59
4. Baylie RL, Brayden JE. TRPV channels and vascular function. Acta Physiol (Oxf). 2011;203:99-116
5. Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357-72
6. Yang D, Luo Z, Ma S. et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010;12:130-41
7. Ching LC, Kou YR, Shyue SK. et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc Res. 2011;91:492-501
8. Ching LC, Chen CY, Su KH. et al. Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol Med. 2012;18:805-15
9. Wei J, Ching LC, Zhao JF. et al. Essential role of transient receptor potential vanilloid type 1 in evodiamine-mediated protection against atherosclerosis. Acta Physiol (Oxf). 2013;207:299-307
10. Mohapatra DP, Wang SY, Wang GK. et al. A tyrosine residue in TM6 of the vanilloid receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol Cell Neurosci. 2003;23:314-24
11. Su KH, Shyue SK, Kou YR. et al. β receptor integrates the erythropoietin signaling in activation of endothelial nitric oxide synthase. J Cell Physiol. 2011;226:3330-9
12. Su KH, Yu YB, Hou HH. et al. AMP-activated protein kinase mediates erythropoietin-induced activation of endothelial nitric oxide synthase. J Cell Physiol. 2012;227:3053-62
13. Zhang B, Cao H, Rao GN. Fibroblast growth factor-2 is a downstream mediator of phosphatidylinositol 3-kinase-Akt signaling in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. J Biol Chem. 2006;281:905-14
14. Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996-1012
15. Chen R, Jiang J, Xiao X. et al. Effects of epoxyeicosatrienoic acids on levels of eNOS phosphorylation and relevant signaling transduction pathways involved. Sci China C Life Sci. 2005;48:495-505
16. Jiang JG, Chen RJ, Xiao B. et al. Regulation of endothelial nitric-oxide synthase activity through phosphorylation in response to epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 2007;82:162-74
17. Rajendran P, Rengarajan T, Thangavel J. et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9:1057-69
18. Ching LC, Zhao JF, Su KH. et al. Activation of transient receptor potential vanilloid 1 decreases endothelial nitric oxide synthase phosphorylation at Thr497 by protein phosphatase 2B-dependent dephosphorylation of protein kinase C. Acta Physiol (Oxf). 2013;209:124-35
19. Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853-63
20. Moran MM, McAlexander MA, Bíró T. et al. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601-20
21. Xu X, Wang P, Zhao Z. et al. Activation of transient receptor potential vanilloid 1 by dietary capsaicin delays the onset of stroke in stroke-prone spontaneously hypertensive rats. Stroke. 2011;42:3245-51
22. Freichel M, Suh SH, Pfeifer A. et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat Cell Biol. 2001;3:121-7
23. Tominaga M, Tominaga T. Structure and function of TRPV1. Pflugers Arch. 2005;451:143-50
24. Loot AE, Popp R, Fisslthaler B. et al. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc Res. 2008;80:445-52
25. Vriens J, Owsianik G, Fisslthaler B. et al. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908-15
Author Biography
 Dr. Tzong-Shyuan Lee serves as a Professor at the Department of Physiology, National Yang-Ming University, Taipei, Taiwan. His areas of expertise include pathophysiology of cardiovascular diseases and physiology of vascular cells. Dr. Lee is interested in the molecular mechanism of early-stage atherosclerosis including endothelial dysfunction and the formation of macrophage foam cells in recent years. By use of clinical therapeutic reagents, food nutrition, and traditional herbal medicine in vitro and in vivo, Dr. Lee's team have discovered several crucial molecules and important molecular mechanisms in the promotion of endothelial function and cholesterol clearance of macrophages.
Dr. Tzong-Shyuan Lee serves as a Professor at the Department of Physiology, National Yang-Ming University, Taipei, Taiwan. His areas of expertise include pathophysiology of cardiovascular diseases and physiology of vascular cells. Dr. Lee is interested in the molecular mechanism of early-stage atherosclerosis including endothelial dysfunction and the formation of macrophage foam cells in recent years. By use of clinical therapeutic reagents, food nutrition, and traditional herbal medicine in vitro and in vivo, Dr. Lee's team have discovered several crucial molecules and important molecular mechanisms in the promotion of endothelial function and cholesterol clearance of macrophages.
![]() Corresponding author: Tzong-Shyuan Lee, DVM, PhD, Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, 11221 Taiwan. E-mail: tsleeedu.tw. Jeng Wei, MD, PhD, Heart Center, Cheng-Hsin General Hospital, Taipei, 11221 Taiwan. E-mail: chghjwcom.tw.
Corresponding author: Tzong-Shyuan Lee, DVM, PhD, Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, 11221 Taiwan. E-mail: tsleeedu.tw. Jeng Wei, MD, PhD, Heart Center, Cheng-Hsin General Hospital, Taipei, 11221 Taiwan. E-mail: chghjwcom.tw.