Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):781-793. doi:10.7150/ijbs.11619 This issue Cite
Research Paper
Identification and Characterization of Novel Maize Mirnas Involved in Different Genetic Background
Lei Sheng*, Wenbo Chai*, Xuefeng Gong, Lingyan Zhou, Ronghao Cai, Xiaoyu Li, Yang Zhao, Haiyang Jiang ![]() , Beijiu Cheng
, Beijiu Cheng ![]()
Key Laboratory of Crop Biology of Anhui Province, Anhui, Agricultural University, Hefei 230036, China
* These authors contributed equally to this work.
Received 2015-1-17; Accepted 2015-4-1; Published 2015-5-22
Abstract

MicroRNAs (miRNAs) are a class of small, non-coding regulatory RNAs that regulate gene expression by guiding target mRNA cleavage or translational inhibition in plants and animals. At present there is relatively little information regarding the role of miRNAs in the response to drought stress in maize. In this study, two small RNA libraries were sequenced, and a total of 11,973,711 and 14,326,010 raw sequences were generated from growing leaves of drought-tolerant and drought-sensitive maize seedlings, respectively. Further analysis identified 192 mature miRNAs, which include 124 known maize (zma) miRNAs and 68 potential novel miRNA candidates. Additionally, 167 target genes (259 transcripts) of known and novel miRNAs were predicted to be differentially expressed between two maize inbred lines. Of these, three novel miRNAs were up-regulated and two were down-regulated under drought stress. The expression of these five miRNAs and nine target genes was confirmed using quantitative reverse transcription PCR. The expression of three of the miRNAs and their putative target genes exhibited an inverse correlation, and expression analysis suggested that all five may play important roles in maize leaves. Finally, GO annotations of the target genes indicated a potential role in photosynthesis, may therefore contribute to the drought stress response. This study describes the identification and characterization of novel miRNAs that are the differentially expressed in drought-tolerant and drought-sensitive inbred maize lines. This provides the foundation for further investigation into the mechanism of miRNA function in response to drought stress in maize.
Keywords: Maize, Drought stress, MicroRNA, Target genes, High-throughput sequencing, qRT-PCR
Introduction
MicroRNAs (miRNAs) are small, endogenous RNAs that regulate gene expression in plants and animals [1-5] at the post-transcriptional level by translational repression or target degradation and gene silencing. Plant miRNAs function in diverse processes associated with growth and development such as developmental transitions, organ polarity, auxin signaling, boundary formation or organ separation, leaf and stem growth, floral organ identity and reproductive development [6]. Several miRNAs are involved in the regulation of root development in Arabidopsis, including miR167 that targets auxin response factors (ARFs) in order to regulate the emergence of shoot-borne roots [7-9], and miR164 that directly cleaves transcription factor NAC1 which inhibits lateral root development. Other miRNAs such as miR156 and miR172 affect flowering time when over-expressed in Arabidopsis [9-12], and miR172 exhibits a similar temporal expression pattern in maize, where it targets Glossy15, a gene required for the expression of juvenile epidermal traits [13]. Moreover, miR395 regulates sulfur assimilation and translocation by adjusting the mRNA levels of ATP sulfurylase and a low affinity sulfur transporter [14, 15], while miR399 regulates phosphate homeostasis in Arabidopsis by suppressing the ubiquitin-conjugating E2 enzyme, PHO2 [16, 17]. In addition, accumulating data demonstrate that miRNAs play major roles in response to biotic and abiotic stress [18, 19, 20], such as low nitrate condition [21, 22], low phosphorus stress [23], salt stress [24] and so on.
Bioinformatics approaches have been successful for the discovery of conserved miRNAs in numerous plant species including cotton [25], maize [26], caster [27], sorghum [28], and soybean [29]. At the time of writing, there are 18,226 mature miRNAs deposited in miRBase [30]. As an important model system for basic biological research, maize has contributed significantly to our understanding of plant development and evolution, and this knowledge has been employed to elucidate the regulatory functions of miRNA genes [26]. The total number of miRNAs in an individual organism has been estimated to represent approximately 1% of the total number of coding genes [31, 32]. To date, only 150 genes within 26 miRNA families have been identified in maize [26], and many more remain to be discovered.
Maize is one of the most important food crops worldwide, and is also used for animal feed, silage and in industrial products. Drought stress is often the limiting factor for plant growth and agricultural productivity, and this is especially true for maize. Plant microRNAs have been shown to play important functions in plant growth and development. However, role of miRNAs in the response to drought stress is poorly understood in maize. In recent years, high-throughput sequencing technologies and bioinformatics approaches have together identified many novel miRNAs [33, 34, 47]. In the present work, drought-tolerant (Hz4) and drought-sensitive (3189) inbred maize lines [35] were investigated using Illumina sequencing to identify novel drought-responsive miRNAs. Expression of the identified miRNAs was confirmed using qRT-PCR and target genes were elucidated. Probing of two small RNA libraries from young leaves led to the identification of 124 known maize (zma) miRNAs, and a further 68 novel maize miRNAs were discovered. Of these, five novel miRNAs that were differentially expressed in the two inbred lines were characterized, and the results suggest they may play an important role in response to drought stress.
Materials and methods
Plant materials and drought stress treatment
Drought-tolerant (Hz4) and drought-sensitive (3189) maize inbred lines were germinated in a greenhouse and grown under standard conditions until seedlings had developed three leaves. Seedlings were then subjected to drought conditions. Specifically, the soil water content was reduced from 90% to 70%, and approximately 5 g of leaves was collected from each seedling and immediately frozen in liquid nitrogen.
Construction of small RNA libraries for high-throughput sequencing
To construct small RNA libraries, total RNA was extracted from leaves using Trizol reagent (Invitrogen, USA) according to the manufacturer's protocol as modified by LC Sciences (Houston, USA). The small RNA fraction (10-40 nt) was isolated by polyacrylamide gel electrophoresis and ligated with proprietary adaptors (Illumina). Short RNAs were then reverse-transcribed into cDNA by RT-PCR. Small RNA libraries were constructed for each maize line and sequenced using a Genome Analyzer GA-IIX (Illumina) following the manufacturer's instructions. Small RNA sequencing data was processed with the in-house program ACGT101-miR (LC Sciences, USA). Program parameters were set as previously described [36] with modifications to adapt the program for plant miRNA prediction.
Sequencing data processing
To begin with, we compared the filtered small RNA sequences with known miRNAs in miRBase 20.0 [http://www.mirbase.org [37] and a 0-2 nucleotide mismatch was considered. Meanwhile, the filtered small RNA sequences were compared with ZmB73_4a.53 (ZmB73_AGPv1) [38] using BLASTn and no mismatches were permitted. Then ESTs that contained small RNA sequences were selected and their secondary structures were predicted using MFOLD (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) for identifying potential novel miRNAs. At last, small RNAs with corresponding star (miRNA*) sequences were counted to be novel miRNAs according to the criteria for miRNA definition [39].
Prediction and validation of miRNA target genes
The target genes of miRNAs were predicted using the method of target prediction proposed by Allen et al. (2005) [40] and Schwab et al. (2005) [41]. The plant small RNA target analysis server psRNATarget [42] was employed for prediction through searching against the maize full-length cDNA sequences. In addition, the rules of target prediction were referred to universal criteria previously suggested [40, 41] and the parameters of psRNATarget server were the default.
Validations of 18 novel miRNAs which low abundance and more than 24 nt in length were carried out by stem-loop reverse transcription-PCR (RT-PCR). The stem-loop RT-PCR was using the Applied Bio systems 7300 Real-Time PCR System (Applied Bio systems, Foster City, CA) and all reactions were run in triplicate. 18S rRNA was used as the internal control for stem-loop RT-PCR. In addition, quantitative real-time RT-PCR (qRT-PCR) was performed to validate the target genes of five novel miRNAs that were differentially expressed between the two inbred lines using three biological replicates. Total RNA from the leaves of the two maize inbred lines was extracted using Trizol reagent (Invitrogen, USA) according to the manufacturer's protocol as modified by LC Sciences (Houston, USA). Samples were collected at the same time as those for miRNAs sequencing. The RNase-free DNase (Promega, USA) was used to remove contaminating DNA from total RNA and first strand cDNA was synthesized according to the manufacturer's instructions (Takara, Japan). The PCR system contained 2 μL cDNA, 12.5 μL Platinum SYBR Green qPCR SuperMix-UDG (Takara, Japan), 2 μL 10 μM primer mixture and 8.5 μL RNase-free water. Sample cycle threshold (Ct) values were standardized for each template based on a GAPDH control reaction and the comparative Ct method (2-ΔΔCt) was used to determine the relative transcript abundance of each gene [43]. Gene accession and primer sequences are listed in Additional File 3: Table S4.
Gene ontology (GO) analysis
Target genes were subjected to Gene Ontology (GO) analysis to uncover the miRNA-gene regulatory network on the basis of biological process and molecular function. GO annotations were extracted using AgriGO (http://bioinfo.cau.edu.cn/agriGO/) and included alignments of individual transcripts representing diverse functions, processes or components. A graphical representation of the GO ontology distribution of the biological processes, cellular component and molecular function categories can be found in Online Resource 8.
Results
Overview of sequencing results
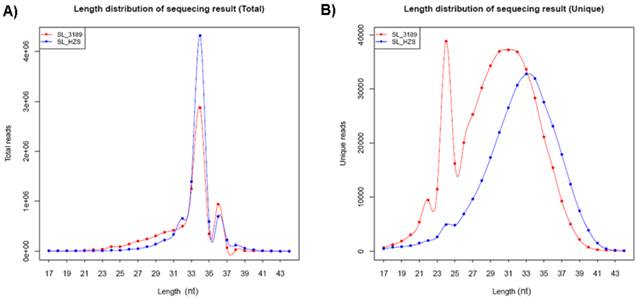
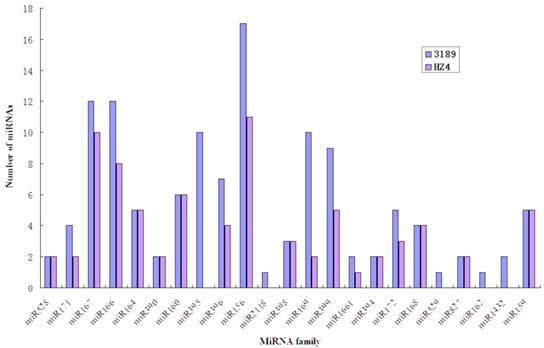
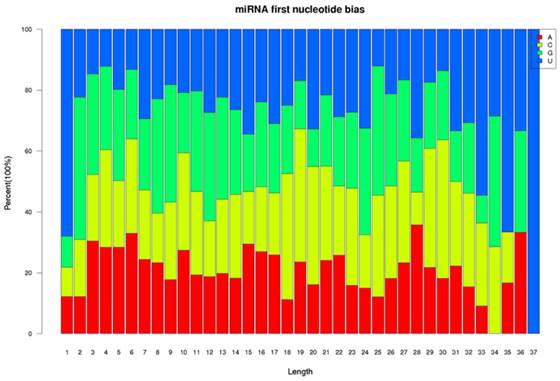
Two small RNA libraries constructed from the leaves of drought-tolerant and drought-sensitive maize seedlings were sequenced using high-throughput methods, and a total of 11,973,711 and 14,326,010 raw sequences were generated from the drought-tolerant and drought-sensitive libraries, respectively. After removing low quality reads and clipping adaptor sequences, 8,983,222 and 7,948,375 clean reads were remained with lengths ranging from 17 to 44 nt (Table 1). The reads of 34 nt in length were the most abundant among total reads of each small RNA library (Figure 1A). In addition, a length of 33 nt was the most abundant among unique reads of SL_HZ4 and a length of 24 nt was the most abundant among unique reads of SL_3189 (Figure 1B). To identify known miRNAs, small RNA sequences were used with BLASTn to search the miRBase database (release 20.0, June 2013). A total of 124 known maize (zma) miRNAs were identified, of which 76 were detected in Hz4 and 124 were detected in 3189. These miRNAs belong to different miRNA families such as the miR156, miR166, and miR167 families (Figure 2). The miR156 family was the most represented, indicating a higher level of expression in maize seedlings, as was previously reported [26]. Uridine (U) was the most common nucleotide at the 5′ end (>65%), whereas this nucleotide was comparatively rare at positions 2-4 (Figure 3).
Summary of small RNA sequencing.
| Category | type | Total | % of Total | uniq | % of uniq | Total | % of Total | uniq | % of uniq |
|---|---|---|---|---|---|---|---|---|---|
| Raw reads | NA | 14326010 | 100 | 817844 | 100 | 11973711 | 100 | 576650 | 100 |
| 3ADT&length filter | Sequence type | 67178 | 0.47 | 184566 | 22.57 | 89273 | 0.75 | 142047 | 24.63 |
| Junk reads | Sequence type | 9707 | 0.07 | 4933 | 0.6 | 7494 | 0.06 | 3474 | 0.6 |
| Rfam | RNA class | 5976056 | 41.71 | 59780 | 7.31 | 2742169 | 22.9 | 45778 | 7.94 |
| mRNA | RNA class | 585726 | 4.09 | 146304 | 17.89 | 297523 | 2.48 | 83734 | 14.52 |
| Repeats | RNA class | 1280 | 0.01 | 951 | 0.12 | 320 | 0 | 237 | 0.04 |
| rRNA | RNA class | 457557 | 3.19 | 34204 | 0.24 | 596924 | 4.99 | 30120 | 0.25 |
| tRNA | RNA class | 5491075 | 38.33 | 19683 | 0.14 | 2131079 | 17.8 | 11951 | 0.1 |
| snoRNA | RNA class | 2407 | 0.02 | 937 | 0.01 | 2061 | 0.02 | 775 | 0.01 |
| snRNA | RNA class | 11645 | 0.08 | 1663 | 0.01 | 5958 | 0.05 | 1202 | 0.01 |
| other Rfam RNA | RNA class | 13372 | 0.09 | 3293 | 0.02 | 6147 | 0.05 | 1730 | 0.01 |
| Clean reads | Sequence type | 7948375 | 55.48 | 424202 | 51.87 | 8983222 | 75.02 | 303613 | 52.65 |
Overview of reads from raw data to cleaned sequences.
3ADT&length filter: reads removed due to 3ADT not found and length with <17 nt and >25 nt were removed (for plants); length with<16 and >29 were remove(for animals)
Junk reads:Junk: >=2N, >=7A, >=8C, >=6G, >=7T, >=10Dimer, >=6Trimer, or >=5Tetramer
Rfam:Collection of many common non-coding RNA families except micro RNA; http://rfam.janelia.org
Repeats:Prototypic sequences representing repetitive DNA from different eukaryotic species; http://www.girinst.org/repbase.
Notes:There is overlap in mapping of reads with mRNA, rRNA, tRNA, snRNA, snoRNA and repeats.
Length distribution of small RNAs from sequencing of the two inbred maize libraries. (A) Size distribution of total sequences. (B) Size distribution of unique sequences.
Abundance of conserved miRNA families in the two inbred maize libraries.
Relative nucleotide bias at each position of the known maize miRNAs.
Identification of novel miRNAs in the maize genome
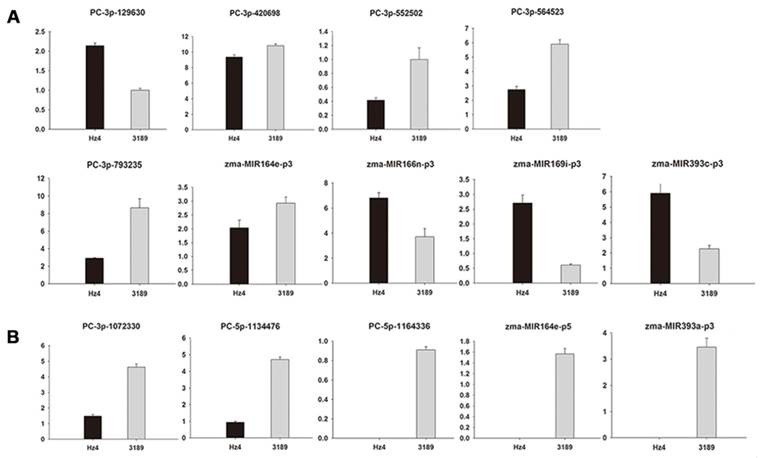
A computational prediction method was employed to identify novel miRNAs in the maize genome. After excluding sRNAs that matched known miRNAs, rRNAs, tRNAs, snRNAs, and snoRNAs, as well as those overlapping protein-coding genes, remaining sRNAs exhibiting a perfect match to maize genomic sequences were used to predict potential novel miRNAs. The selection of novel miRNAs was based on the ability of flanking sequences to fold back in a hairpin structure [44]. Predicted secondary structures of the flanking sequences were generated using Mfold. These putative miRNA precursors were then used to extract miRNA*s, which are considered strong evidence for DICER-LIKE-1 (DCL1)-derived products [30]. A total of 68 regions satisfied these criteria and were designated as novel miRNA gene candidates (Table 2). The lengths of novel miRNAs varied from 20 to 37 nt, with 24 nt the most abundant length (18 out of 68), which is typical of DCL digestion products. Of these, 24 were found to be expressed in both maize inbred lines, with 10 weakly expressed, as indicated by a normalized sequencing read of less than 1 in both samples. In addition, the sequence frequency of most novel miRNA candidates was extremely low. It has been reported that miRNAs may be expressed at very low levels and may be present only in particular cell types and/or under particular circumstances [45]. None of the novel miRNAs were previously reported in other species and are therefore likely to be maize-specific. Then 18 novel miRNAs which low abundance and more than 24 nt in length were selected for validation by stem-loop real-time PCR (Additional File 1: Table S1). According to our RT-PCR analysis, 4 novel miRNAs (PC-3p-201205, PC-3p-359801, PC-5p-864621, zma-MIR166b-p5) had no expression quantity in both the leaves of two maize inbred lines. In addition, 5 novel miRNAs (PC-3p-201205, PC-5p-1164336, zma-MIR164e-p5, zma-MIR166b-p5, zma-MIR393a-p3) had no expression quantity in the leaves of Hz4 and 1 novel miRNA (PC-5p-864621) had no expression quantity in the leaves of 3189, which was consistent with the results of the high-throughput sequencing (Additional File 1: Table S1). Expression levels of a few members—PC-3p-129630, PC-3p-420698, PC-3p-552502, PC-3p-564523, PC-3p-793235, zma-MIR164e-p3, zma-MIR166n-p3, zma-MIR169i-p3 and zma-MIR393c-p3—were slightly different (Figure 4A). This difference may be due to variations in Hz4 and 3189 sampling times, or differences in sensitivity and specificity of the two technologies. Expression patterns of the remaining novel miRNAs (PC-3p-1072330, PC-5p-1134476, PC-5p-1164336, zma-MIR164e-p5, zma-MIR393a-p3) obtained by stem-loop real-time RT-PCR were in accordance with the sequencing data (Figure 4B).
Novel maize miRNAs identified in this study.
| PC-3p-687149_1 | AGATGAGAAATGAAGGCACCAGAT | 24 | tag_chr3 | + | 3' | -169.7 | 49.6 | 1.3 | 0 | 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| PC-5p-691043_1 | TTAGGCTCGGGGACTACGGT | 20 | tag_chr3 | + | 5' | -97.3 | 59.3 | 0.9 | 0.5 | 0 |
| PC-3p-711926_1 | CCGTGGCTCCTGCTCCTGAT | 20 | tag_chr3 | + | 3' | -97.3 | 59.3 | 0.9 | 0 | 0.5 |
| PC-3p-687149_1 | AGATGAGAAATGAAGGCACCAGAT | 24 | tag_chr3 | - | 3' | -154.7 | 49.1 | 1.2 | 0 | 2 |
| PC-5p-1134476_1 | TCTTACTTTTGGCATTTGTGACATTGACTT | 30 | tag_chr3 | - | 5' | -58.2 | 31.3 | 0.8 | 0 | 2 |
| PC-3p-1072330_1 | ATCGCCCTGATCGATGCCTAATCGCG | 26 | tag_chr4 | - | 3' | -33.5 | 57.6 | 1 | 0 | 2 |
| PC-5p-431452_1 | TCGTGTTTTTTCCTCAGCTGTGCC | 24 | tag_chr4 | - | 5' | -91.1 | 40.7 | 1.5 | 0 | 1 |
| PC-3p-1160150_1 | AGGACACAGCTGAGTAAAAAACAC | 24 | tag_chr4 | - | 3' | -91.1 | 40.7 | 1.5 | 0 | 1 |
| PC-5p-366433_1 | TGCCTTTAGGGCTGATTTGGTGC | 23 | tag_chr5 | + | 5' | -118.2 | 49.7 | 1.6 | 0 | 1 |
| PC-3p-564523_1 | TTCTCCCCCATGGATCCCTTTGGGA | 25 | tag_chr5 | + | 3' | -118.2 | 49.7 | 1.6 | 0 | 1 |
| PC-5p-12469_78 | ATAGTTTTTTCTACCACACTTTAGATTCTT | 30 | tag_chr5 | + | 5' | -69.6 | 29 | 0.9 | 68.5 | 184.5 |
| PC-3p-359801_1 | AGAATAGACTAGAATAGATTATAGTAAAAG | 30 | tag_chr5 | + | 3' | -69.6 | 29 | 0.9 | 1 | 3 |
| PC-3p-1033669_1 | ATAGATGAGCACACTACCAAAACT | 24 | tag_chr6 | - | 3' | -123.9 | 45.5 | 1.8 | 0 | 2.5 |
| PC-3p-201205_3 | AGAAAAGATTGAGCCGAATTGAATTA | 26 | tag_chr6 | - | 3' | -33.2 | 30 | 1.8 | 0 | 6.5 |
| PC-5p-1123590_1 | TCGCAGTCGGCCGTGTCCTCGGAG | 24 | tag_chr6 | - | 5' | -62.7 | 58.8 | 0.5 | 0 | 2 |
| PC-3p-237604_2 | AATATGGAAACGGGACGGAAACGG | 24 | tag_chr7 | - | 3' | -32.7 | 37.9 | 1 | 0 | 2 |
| PC-5p-691043_1 | TTAGGCTCGGGGACTACGGT | 20 | tag_chr8 | + | 5' | -99 | 53.3 | 1 | 0.5 | 0 |
| PC-3p-711926_1 | CCGTGGCTCCTGCTCCTGAT | 20 | tag_chr8 | + | 3' | -99 | 53.3 | 1 | 0 | 0.5 |
Validation of 18 novel miRNAs which low abundance and more than 24 nt in length using stem-loop real-time PCR. (A) different expression levels of miRNAs compared with the sequencing data. (B) similar expression levels of miRNAs compared with the sequencing data. Ordinates indicate relative expression levels.
Clustering of miRNAs
Clusters of miRNAs are often present in the genome where multiple miRNAs are aligned in the same orientation and transcribed as a polycistronic structure to facilitate cooperative action [46]. A total of 69 of the miRNAs identified in this research were located within 22 miRNA clusters, with chromosomes 1, 2, 3, 4, 5, 6, 7, 8 and 10 containing 5, 1, 5, 1, 4, 2, 1, 2 and 1 clusters, respectively (Table 3). The largest miRNA cluster contained 7 miRNAs, all of which belonged to the same miRNA family (miRNA395). Some miRNAs in the same cluster may be transcribed by the same primary miRNA, although their expression levels can differ, as was observed with PC-3p-1105743/PC-3p-897448, PC-3p-552502/PC-5p-62968 and PC-5p-1164336/PC-3p-104764. This is presumably due to these miRNAs undergoing different transcriptional regulation during maturation.
Genome location clusters of pre-miRNAs.
| Pre-miRNA Cluster ID | Chromosome | Start | End | Strand | miRNA Name |
|---|---|---|---|---|---|
| 1 | tag_chr1 | 6415393 | 6415814 | + | bdi-MIR528-p3_1ss21GT |
| 1 | tag_chr1 | 6415393 | 6415814 | + | zma-miR528a-5p_R+1 |
| 1 | tag_chr1 | 6415592 | 6415714 | + | zma-miR528a-3p |
| 1 | tag_chr1 | 6415592 | 6415714 | + | zma-miR528a-5p |
| 2 | tag_chr1 | 19971673 | 19972096 | - | PC-3p-1105743_1 |
| 2 | tag_chr1 | 19971742 | 19972165 | + | PC-3p-897448_1 |
| 3 | tag_chr1 | 114311615 | 114312042 | + | PC-3p-46539_19 |
| 3 | tag_chr1 | 114311615 | 114312042 | + | PC-5p-62968_15 |
| 3 | tag_chr1 | 114311584 | 114312011 | - | PC-3p-552502_1 |
| 3 | tag_chr1 | 114311584 | 114312011 | - | PC-5p-97360_9 |
| 4 | tag_chr1 | 203925757 | 203926180 | - | PC-3p-270024_3 |
| 4 | tag_chr1 | 203925757 | 203926180 | - | PC-5p-1164336_1 |
| 4 | tag_chr1 | 203925786 | 203926210 | + | PC-3p-104764_7 |
| 5 | tag_chr1 | 274716509 | 274716929 | - | osa-miR166m |
| 5 | tag_chr1 | 274716709 | 274716784 | - | zma-miR166a-3p |
| 5 | tag_chr1 | 274716709 | 274716784 | - | zma-miR166h-5p_L-1R+4 |
| 6 | tag_chr10 | 144744354 | 144744457 | + | zma-miR395a-3p_L-1R-1 |
| 6 | tag_chr10 | 144744531 | 144744772 | + | zma-miR395a-3p_L-1R-1 |
| 6 | tag_chr10 | 144744844 | 144744970 | + | zma-miR395a-3p_L-1R-1 |
| 7 | tag_chr2 | 6321644 | 6321709 | - | zma-miR395a-3p_L-1R-1 |
| 7 | tag_chr2 | 6322300 | 6322369 | - | zma-miR395a-3p_L+11R-1 |
| 7 | tag_chr2 | 6331941 | 6332012 | - | zma-miR395a-3p_L+11R-1 |
| 7 | tag_chr2 | 6331941 | 6332012 | - | zma-miR395i-5p_R+11 |
| 7 | tag_chr2 | 6332583 | 6332667 | - | zma-miR395a-3p_L-1R-1 |
| 7 | tag_chr2 | 6332742 | 6332855 | - | zma-miR395a-3p_L+11R-1 |
| 7 | tag_chr2 | 6333397 | 6333545 | - | zma-miR395a-3p_L-1R-1 |
| 8 | tag_chr3 | 7774272 | 7774395 | - | zma-miR156a-5p_L+1 |
| 8 | tag_chr3 | 7774558 | 7774696 | - | zma-miR156a-5p |
| 9 | tag_chr3 | 25490777 | 25491197 | - | ath-miR159a |
| 9 | tag_chr3 | 25490976 | 25491190 | - | zma-MIR159f-p5 |
| 9 | tag_chr3 | 25490976 | 25491190 | - | zma-miR159a-3p_R-1 |
| 10 | tag_chr3 | 27314699 | 27315122 | - | PC-3p-687149_1 |
| 10 | tag_chr3 | 27314819 | 27315242 | + | PC-3p-687149_1 |
| 11 | tag_chr3 | 37610192 | 37610611 | + | PC-3p-711926_1 |
| 11 | tag_chr3 | 37610192 | 37610611 | + | PC-5p-691043_1 |
| 11 | tag_chr3 | 37610280 | 37610409 | + | zma-MIR169a-p5_1ss22TC |
| 12 | tag_chr3 | 119175685 | 119175874 | + | zma-miR167a-5p |
| 12 | tag_chr3 | 119177648 | 119177890 | + | zma-miR167e-5p_R+1 |
| 13 | tag_chr4 | 173295127 | 173295263 | + | zma-miR396a-3p_R-1 |
| 13 | tag_chr4 | 173295127 | 173295263 | + | zma-miR396a-5p |
| 13 | tag_chr4 | 173300108 | 173300273 | - | zma-miR396e-5p |
| 14 | tag_chr5 | 21933496 | 21933916 | - | cme-miR166i_L+2R-1 |
| 14 | tag_chr5 | 21933694 | 21933797 | - | zma-miR166a-3p |
| 15 | tag_chr5 | 146894716 | 146894809 | + | zma-miR399a-3p |
| 15 | tag_chr5 | 146903662 | 146903752 | + | zma-miR399e-3p |
| 16 | tag_chr5 | 210001500 | 210001929 | + | PC-3p-359801_1 |
| 16 | tag_chr5 | 210001500 | 210001929 | + | PC-5p-12469_78 |
| 16 | tag_chr5 | 210008725 | 210008868 | - | bdi-MIR5056-p3 |
| 17 | tag_chr5 | 210632198 | 210632365 | - | zma-miR166j-3p |
| 17 | tag_chr5 | 210632469 | 210632624 | - | zma-miR166l-3p |
| 17 | tag_chr5 | 210632469 | 210632624 | - | zma-miR166m-5p |
| 18 | tag_chr6 | 84226293 | 84226713 | - | osa-miR166m |
| 18 | tag_chr6 | 84226489 | 84226701 | - | zma-miR166a-3p |
| 19 | tag_chr6 | 159686859 | 159686963 | - | zma-miR399f-3p_L+10R-1 |
| 19 | tag_chr6 | 159686859 | 159686963 | - | zma-miR399f-5p_R+11 |
| 19 | tag_chr6 | 159694634 | 159694840 | + | zma-miR399a-3p |
| 19 | tag_chr6 | 159694634 | 159694840 | + | zma-miR399c-5p |
| 20 | tag_chr7 | 9830090 | 9830510 | - | cpa-miR167c |
| 20 | tag_chr7 | 9830212 | 9830330 | - | zma-miR167e-5p_R+1 |
| 21 | tag_chr8 | 4791774 | 4792193 | + | PC-3p-711926_1 |
| 21 | tag_chr8 | 4791774 | 4792193 | + | PC-5p-691043_1 |
| 21 | tag_chr8 | 4791975 | 4792130 | + | zma-miR169a-3p_L+8R-1 |
| 21 | tag_chr8 | 4791975 | 4792130 | + | zma-miR169a-5p_R+14 |
| 22 | tag_chr8 | 10528867 | 10529066 | + | zma-MIR159h-p3 |
| 22 | tag_chr8 | 10532710 | 10532961 | + | zma-MIR159i-p3 |
| 22 | tag_chr8 | 10544675 | 10544926 | + | zma-miR159a-3p_R-1 |
| 22 | tag_chr8 | 10585048 | 10585247 | + | zma-miR159a-3p_R-1 |
| 22 | tag_chr8 | 10588904 | 10589324 | + | ath-miR159a |
| 22 | tag_chr8 | 10588923 | 10589143 | + | zma-miR159a-3p_R-1 |
Differentially expressed miRNAs
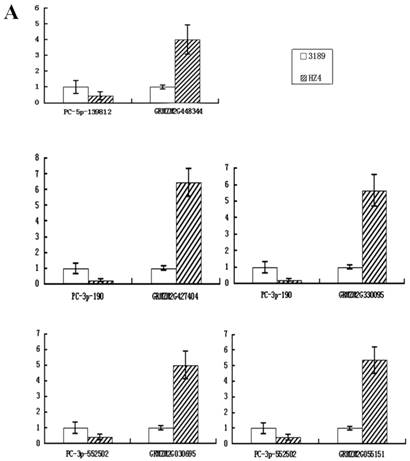
To identify miRNAs involved in the response to drought stress, differential expression in the two libraries was estimated from the read counts of the high-throughput sequencing. In the two libraries, miRNAs exhibiting a log2 (Hz4/3189) fold change higher than 1 were designated up-regulated, while a log2 (Hz4/3189) fold change less than -1 was designated as down-regulated. In total, 29 miRNAs were differentially expressed between the two inbred lines, with 18 down-regulated and 11 up-regulated (Table 4). Among these differentially expressed miRNAs, five novel miRNAs were selected for expression analysis using qRT-PCR. These were PC-3p-190, PC-3p-104764, PC-3p-129630, PC-3p-552502 and PC-5p-139812. Of these, two were up-regulated and three were down-regulated. Secondary structures were predicted to be of a typical hairpin shape, which is different from siRNAs and piRNAs (Figure 5). The expression patterns of these five miRNAs, obtained by qRT-PCR experiments, were consistent with the results of the high-throughput sequencing reads (Figure 6). However the fold changes obtained from the qRT-PCR data were much lower than those estimated from the high-throughput sequencing data, presumably due to differences in sensitivity and specificity between the experimental approaches [47].
miRNAs differentially expressed in the two maize inbred lines.
| miR name | miR seq | 3189(norm) | HZS(norm) | Fisher exact test | Chis quare | Log2(Hz4/3189) |
|---|---|---|---|---|---|---|
| zma-miR167e-5p_R+1 | TGAAGCTGCCAGCATGATCTGA | 0.850384 | 3.271683 | 0.629352 | 0.94767 | down |
| zma-miR390a-5p | AAGCTCAGGAGGGATAGCGCC | 0.831897 | 5.708753 | 0.823974 | 0.56767 | down |
| zma-miR160a-5p | TGCCTGGCTCCCTGTATGCCA | 0.277299 | 1.101689 | 1 | 0.95794 | down |
| zma-miR396c_L-1 | TCCACAGGCTTTCTTGAACTG | 11.64656 | 43.5668 | 0.591401 | 0.87501 | down |
| zma-miR160f-5p_1ss21GA | TGCCTGGCTCCCTGTATGCCA | 0.277299 | 1.101689 | 1 | 0.95794 | down |
| zma-miR399a-3p | TGCCAAAGGAGAATTGCCCTG | 0.554598 | 2.203378 | 0.524953 | 0.94054 | down |
| zma-miR168a-5p | TCGCTTGGTGCAGATCGGGAC | 7.487077 | 17.72718 | 0.30102 | 0.34827 | down |
| zma-miR168b-3p_R+1_1ss12TC | CCCGCCTTGCACCAAGTGAAT | 7.487077 | 15.02304 | 0.191204 | 0.19522 | down |
| zma-miR168a-5p | TCGCTTGGTGCAGATCGGGAC | 7.487077 | 17.72718 | 0.30102 | 0.34827 | down |
| zma-miR827-5p_L+1 | TTTTGTTGGTGGTCATTTAACC | 11.64656 | 25.83962 | 0.110772 | 0.17769 | down |
| zma-miR827-3p | TTAGATGACCATCAGCAAACA | 38.26728 | 141.8175 | 0.638664 | 0.81446 | down |
| zma-miR164a-5p | TGGAGAAGCAGGGCACGTGCA | 0.332759 | 1.201843 | 1 | 0.99321 | down |
| zma-miR167h-3p_L+1R+1 | AGATCATGTTGCAGCTTCACT | 1.663795 | 10.81659 | 0.814904 | 0.46092 | down |
| zma-miR408a | CTGCACTGCCTCTTCCCTGGC | 4.991384 | 35.45436 | 0.957678 | 0.13853 | down |
| zma-miR408b-5p | CAGGGACGAGGCAGAGCATGG | 4.991384 | 0.600921 | 0.002503 | 0.000121 | up |
| zma-miR398a-3p_L+9R-1 | GATCTTGCATGTGTTCTCAGGTCGCCCCC | 0.831897 | 3.004607 | 0.629352 | 0.98926 | down |
| zma-miR398a-3p_L+7R-1 | TGCTGCATGTGTTCTCAGGTCGCCCCC | 64.0561 | 3.004607 | 9.53E-39 | 3.33E-48 | up |
| zma-miR172a_R+1 | AGAATCTTGATGATGCTGCAT | 1.663795 | 0.400614 | 0.048259 | 0.041885 | up |
| zma-miR396f-3p_L+3 | GAAGGTCAAGAAAGCTGTGGGAAG | 1.663795 | 0.600921 | 0.123582 | 0.061209 | up |
| zma-MIR397b-p3 | TCACCAGCGCTGCACTCAATT | 1.663795 | 0.600921 | 0.123582 | 0.061209 | up |
| zma-miR399f-3p_L+10R-1 | GTGCCACTGCTGCCAAAGGAAATTTGCCCC | 6.655179 | 0.600921 | 0.000159 | 5.64E-06 | up |
| zma-miR169a-5p_R+14 | CAGCCAAGGATGACTTGCCGATCTATCGTCGATCA | 3.32759 | 1.201843 | 0.035379 | 0.008106 | up |
| zma-miR159a-3p_R-1 | TTTGGATTGAAGGGAGCTCT | 3.535564 | 1.547373 | 0.023761 | 0.009544 | up |
| PC-5p-139812_4 | GAAGGGTAGAAAAAGTTATTAGATAGCGA | 48.25005 | 3.605528 | 1.89E-27 | 2.99E-35 | up |
| PC-3p-793235_1 | TCCAATGCTATCTAGTAATTTTTCTACCTACA | 1.663795 | 0.600921 | 0.123582 | 0.061209 | up |
| PC-3p-552502_1 | ACTAGAATGAACAATGCTGTAGCAATAAATGCGAGAA | 8.318974 | 3.004607 | 0.000464 | 2.82E-05 | up |
| PC-3p-129630_5 | TTAGAAAAGATTGAGCCGAATTGAATTA | 1.663795 | 5.107832 | 0.476801 | 0.87006 | down |
| PC-3p-104764_7 | AGAAAAGATTGAGCCGAATTGAATT | 1.663795 | 17.42672 | 0.943169 | 0.16161 | down |
| PC-3p-190_11180 | CCAACAGGATATTGGGTATTTCTT | 1435.855 | 8141.283 | 1 | 0 | down |
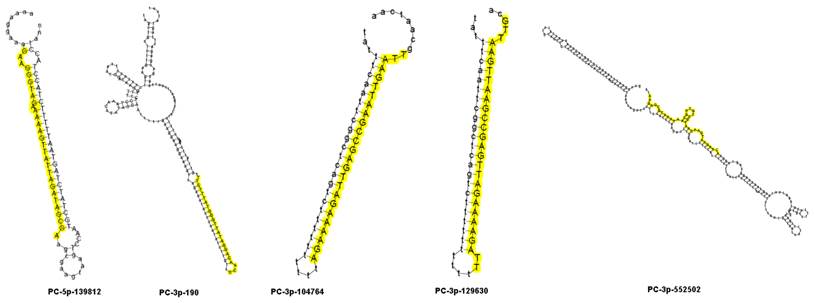
Secondary structure of five novel miRNA precursors. Mature miRNA sequences are shown in yellow.
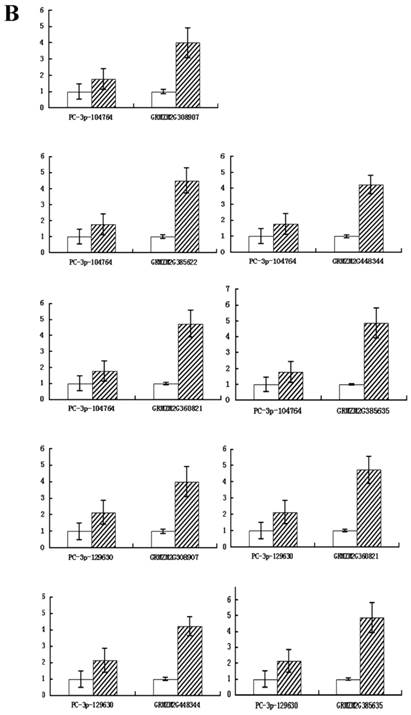
Quantitative real-time RT-PCR analysis of five novel miRNAs and their target genes. Expression levels of miRNAs were normalized against 18S rRNA. Fold changes in expression level were estimated using the 2-ΔΔCT method. Data are reported as mean ± SE for three independent experiments. (A) The inverse relationship between three novel miRNAs (PC-3p-190, PC-3p-552502 and PC-5p-139812) and their putative target genes. (B) The uniform relationship between two novel miRNAs (PC-3p-104764 and PC-3p-129630) and their putative target genes.
Identification and confirmation of miRNA targets using qRT-PCR
In most cases, the identified miRNAs were predicted to cleave two or more different targets. With a specific value of 0.85 and a signal-to-noise value of 6.28, a total of 167 genes were predicted to be potential targets for 23 of the 29 differentially expressed miRNAs. Among these, nine genes were predicted to be targets of the five novel miRNAs (Table 5). Interestingly, PC-3p-104764, PC-3p-129630, PC-3p-552502 and PC-5p-139812 shared a common target (GRMZM2G448344), and PC-3p-104764 and PC-3p-129630 shared three common targets (GRMZM2G360821, GRMZM2G385635, GRMZM2G308907), suggesting differnet miRNAs may function in combination in gene regulatory networks.
Gene Ontology (GO) analysis of potential targets of the five novel miRNAs.
| miR name | Target gene | GO annotation |
|---|---|---|
| PC-3p-190 | GRMZM2G427404 | GO:0015934 large ribosomal subunit |
| GO:0015935 small ribosomal subunit | ||
| GO:0003735 structural constituent of ribosome | ||
| GO:0016740 transferase activity | ||
| GO:0003723 RNA binding | ||
| GO:0006412 translation | ||
| GO:0015934 large ribosomal subunit | ||
| GO:0015935 small ribosomal subunit | ||
| GO:0003735 structural constituent of ribosome | ||
| GO:0016740 transferase activity | ||
| GRMZM2G330095 | GO:0003735 structural constituent of ribosome | |
| GO:0006412 translation | ||
| GO:0005840 ribosome | ||
| PC-3p-104764 | GRMZM2G360821 | GO:0015977 carbon fixation |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G448344 | GO:0015977 carbon fixation | |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G308907 | GO:0015977 carbon fixation | |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G385622 | GO:0004176 ATP-dependent peptidase activity | |
| GO:0045261 proton-transporting ATP synthase complex, catalytic core F(1) | ||
| GO:0015986 ATP synthesis coupled proton transport | ||
| GO:0004252 serine-type endopeptidase activity | ||
| GO:0046933 hydrogen ion transporting ATP synthase activity, rotational mechanism | ||
| GO:0000166 nucleotide binding | ||
| GO:0046961 proton-transporting ATPase activity, rotational mechanism | ||
| GO:0006508 proteolysis | ||
| GO:0008553 hydrogen-exporting ATPase activity, phosphorylative mechanism | ||
| GRMZM2G385635 | Unknown | |
| PC-3p-129630 | GRMZM2G308907 | GO:0015977 carbon fixation |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G360821 | GO:0015977 carbon fixation | |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G385635 | Unknown | |
| GRMZM2G448344 | GO:0015977 carbon fixation | |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| PC-3p-552502 | GRMZM2G448344 | GO:0015977 carbon fixation |
| GO:0016984 ribulose-bisphosphate carboxylase activity | ||
| GRMZM2G030695 | GO:0019684 photosynthesis, light reaction | |
| GO:0016021 integral to membrane | ||
| GO:0009523 photosystem II | ||
| GRMZM2G055151 | Unknown | |
| PC-5p-139812 | GRMZM2G448344 | GO:0015977 carbon fixation |
| GO:0016984 ribulose-bisphosphate carboxylase activity |
To confirm whether the miRNAs regulated their potential targets under drought stress conditions, the expression patterns of putative target genes were analyzed using qRT-PCR. It is known that miRNAs regulate gene expression at the post-transcriptional level by translational repression or target degradation and gene silencing. As expected, an inverse relation was observed between the expression of three miRNAs (PC-3p-190, PC-3p-552502 and PC-5p-139812) and their putative target genes, suggesting miRNA-mediated regulation of their potential targets (Figure 6A). The other two novel miRNAs (PC-3p-104764 and PC-3p-129630) did not exhibit this inverse relationship with their putative target genes (Figure 6B). On account of the regulatory mechanism between miRNAs and their target genes is so complicated that one target gene may be correspond to multiple miRNAs. Therefore, we speculated that these two novel miRNAs had little effect on their target genes and the expression of the target genes might be affected by other regulators.
GO functional and transcriptomics analysis of target genes
The results of GO analysis demonstrated that most of the target genes were involved in cellular or metabolic processes such as response to stimuli or biological regulation among other categories (Table 5). Of the nine target genes characterized, seven could be classified into 21 biological processes including carbon fixation, photosynthesis and proteolysis, while the remaining two were of unknown function. For example, PC-3p-190 targets a gene involved in RNA-binding and PC-3p-129630 targets a gene involved in carbon fixation. PC-3p-552502 targets a gene involved in the light reaction of photosynthesis and the target genes of PC-3p-104764 are ATP-dependent peptidase and serine-type endopeptidase. Interestingly, the common target (GRMZM2G448344) of four of the five novel miRNAs, and the common targets (GRMZM2G360821 and GRMZM2G308907) of PC-3p-104764 and PC-3p-129630 are all involved in photosynthesis.
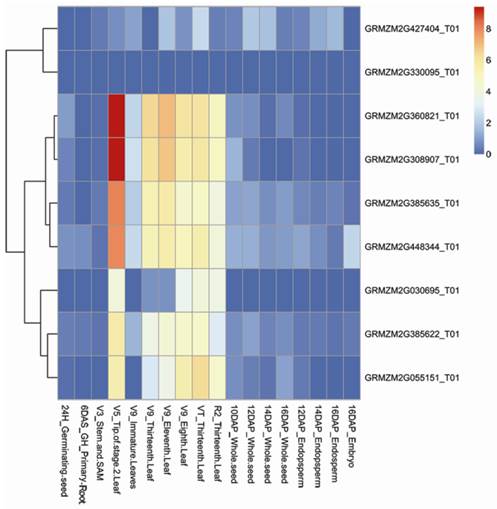
To gain more insight into the expression patterns of the target genes of the five novel miRNAs, a comprehensive expression analysis was performed using the publicly available maize transcriptome data. A total of 18 tissues from five organs have been analyzed using RNA sequencing (RNA-Seq) [48]. Distinct expression profiles were identified for nine target genes from the transcriptome data (Additional File 2: Table S2). Transcriptome data were imported into R and Bioconductor (http://www.bioconductor.org/) for expression analysis to generate a heat map (Figure 7). Based on the heat map, all nine target genes were appeared to be expressed only in the leaves. GRMZM2G360821 and GRMZM2G308907 exhibited the highest expression levels, suggesting important roles for these target genes. These results indicated that these genes may participate in photosynthesis, which was consistent with the GO analysis.
Expression profiles of nine target genes. A heat map was generated by hierarchical clustering using a dedicated heat map package [57]. Expression data were normalized and hierarchically clustered with average linkage. The color scale in the top right corner represents the relative gene expression level, where red, yellow and blue indicate high, medium and low levels of gene expression, respectively.
Discussion
Identification of maize miRNAs by high-throughput sequencing
Identification of miRNAs in model plants using high-throughput sequencing or miRNA arrays has been previously reported. High-throughput sequencing has proven particularly successful for identifying plant miRNAs involved abiotic stress responses [49, 50, 51]. In this study libraries constructed from the leaves of two maize inbred lines were used to identify novel drought-associated miRNAs and their mechanisms of action were subsequently investigated.
Of the miRNAs identified using high-throughput sequencing, 85.48% of those already known were expressed at low levels (less than 10 raw reads; Table 1). This suggests that high-throughput sequencing is a powerful strategy for identifying poorly expressed miRNAs in plants. The miR159, miR167 and miR396 families, which are believed to target MYB transcription factors, auxin responsive factors and GRF transcription factors, respectively, were abundantly represented in both libraries. By comparing the expression levels of all members of an miRNA family, dominant members could be found, such as zma-MIR396d in the miR396 family, zma-MIR167h in the miR167 family and zma-MIR169a in the miR169 family. These dominant members may perform key regulatory roles in response to abiotic stress. Some family members exhibited comparable expression levels, such as zma-MIR166b/c/e/f/g/h/I in the miR166 family and zma-MIR168a and zma-MIR168b in the miR168 family. This indicates that several members of a family may have a synergistic effect in the regulation network.
Potential targets of differentially expressed miRNAs
Earlier research in Arabidopsis demonstrated that many of the potential targets of candidate miRNAs were transcription factors [52] and the majority of miRNAs were involved in multiple diverse biological processes [49]. In this study, 29 differentially expressed miRNAs were identified using high-throughput sequencing, and potential targets of 23 of these were successfully predicted (Additional File 3: Table S3). No targets were identified for the remaining 6 miRNAs, and this may be due to differences in the spatial or temporal expression of a miRNA and its target that could result in insufficient degradation of the target [50]. As anticipated, many of the potential targets were involved in transcription regulation, including MYB, CRP, PPA, HRT, GRF, SR, C2H2, SLS, SMH, RAV, GLH and Phytocyanin-type transcription factors. These transcription factor families are conserved in other plant species [53-56] and are known to regulate plant development.
Potential targets of drought-associated miRNAs
This study identified five novel miRNAs that were differentially expressed in drought-resistant and drought-sensitive maize inbred lines. GO analysis was performed to infer whether further investigation into these miRNAs would be appropriate, and many of the potential target genes play important roles in diverse biological processes, but especially in photosynthesis. Transcriptomics analysis suggested that the nine target genes were expressed only in leaves, which are the primary location for photosynthesis. The chemical constituents of leaves are known to change in response to drought stress, therefore we propose that these target genes are involved in drought stress. The results presented here will guide further functional analysis of the role of these novel drought-associated miRNAs and their target genes in the response to drought stress in maize.
Conclusion
Differentially expressed miRNAs were detected in the leaves of two maize inbred lines, and 124 known and 68 novel miRNAs were identified using a high-throughput sequencing approach. The sRNA-seq data has been deposited into NCBI GEO database and the GEO accession number is GSE61700. The novel drought-associated miRNAs identified and their predicted target genes may play important roles in the regulation of stress responses in this important food plant. These findings have enriched the maize miRNA databases and provide the foundation for further analysis of their roles in maize growth and development.
Supplementary Material
Additional File 1Supplementary Table S1.
Supplementary Table S2.
Supplementary Tables S3-S4.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (201303001), National Key Technology Support Program (2012BAD20B00), National Natural Science Foundation of China (31371285, 31201217) and Natural Science Foundation of Anhui Province (1508085QC64).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297
2. Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Ann Rev Plant Biol. 2006;57:19-53
3. Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669-687
4. Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y. Rice microRNA effector complexes and targets. Plant Cell. 2009;21:3421-3435
5. Chuck G, Candela H, Hake S. Big impacts by small RNAs in plant development. Plant Biology. 2009;12:81-86
6. Sun G. MicroRNAs and their diverse functions in plants. Plant Mol Biol. 2012;80:17-36
7. Yang JH, Han SJ, Yoon EK, Lee WS. Evidence of an auxin signal pathway, microRNA167-ARF8-GH3, and its response to exogenous auxin in cultured rice cells. Nucleic Acids Research. 2006;34:1892-1899
8. Gutierrez L, Bussell JD, Pacurar DI, Schwambach J, Pacurar M, Bellini C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell. 2009;21:3119-3132
9. Wang JW, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138:738-749
10. Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303:2022-2025
11. Wu G, Poethig RS. Temporal regulation of shoot development in Arabidopsis thaliana by MIR156 and its target SPL3. Development. 2006;133:3539-3547
12. Wang JW, Schwab R, Czech B, Mica E, Weigel D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell. 2008;20:1231-1243
13. Lauter N, Kampani A, Carlson S, Goebe M, Moose SP. microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9412-9417
14. Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress induced miRNA. Mol Cell. 2004;14:787-799
15. Kawashima CG, Yoshimoto N, Maruyama-Nakashita A, Tsuchiya YN, Saito K, Takahashi H, Dalmay T. Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J. 2009;57:313-321
16. Fujii H, Chiou TJ, Lin SI, Aung K, Zhu JK. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr Biol. 2005;15:2038-2043
17. Chiou TJ, Aung K, Lin SI, Wu CC, Chiang SF, Su CL. Regulation of phosphate homeostasis by microRNA in Arabidopsis. Plant Cell. 2006;18:412-421
18. Chen X. MicroRNA biogenesis and function in plants. FEBS Lett. 2005;579:5923-5931
19. Qin Y, Duan Z, Xia X, Yin W. Expression profiles of precursor and mature microRNAs under dehydration and high salinity shock in Populus euphratica. Plant Cell Rep. 2011;30:1893-1907
20. Zhang B, Wang Q, Pan X. MicroRNAs and their regulatory roles in animals and plants. J Cell Physiol. 2007;210:279-289
21. Xu Z, Zhong S, Li X, Li W, Rothstein SJ, Zhang S, Bi Y. and Xie C: Genome-Wide Identification of MicroRNAs in Response to Low Nitrate Availability in Maize Leaves and Roots. PLoS ONE. 2011;6(11):e28009
22. Zhao Y, Xu Z, Mo Q, Zou C, Li W, Xu Y, Xie C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Annals of Botany. 2013;112:633-642
23. Zhang Z, Lin H, Shen Y, Gao J, Xiang K, Liu L, Yuan G, Lan H, Zhou S, Gao S, Rong T, Pan G. Cloning and characterization of miRNAs from maize seedling roots under low phosphorus stress. Mol Biol Rep. 2012;39:8137-8146
24. Zhu J, Li W, Yang W, Qi L. Identification of microRNAs in Caragana intermedia by highthroughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013
25. Ruan MB, Zhao YT, Meng ZH, Wang XJ, Yang WC. Conserved miRNA analysis in Gossypium hirsutum through small RNA sequencing. Genomics. 2009;94:263-268
26. Zhang LF, Chia JM, Kumari S, Stein JC, Liu ZJ, Narechania A, Maher CA, Guill K, McMullen MD, Ware D. A genomewide characterization of microRNA genes in Maize. PLoS Genet. 2009;5:11
27. Zeng CY, Wang WQ, Zheng Y, Chen X, Bo WP, Song S, Zhang WX, Peng M. Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants. Nucleic Acids Res. 2010;38:981-995
28. Calvino M, Bruggmann R, Messing J. Characterization of the small RNA component of the transcriptome from grain and sweet sorghum stems. BMC Genomics. 2011;12:356
29. Turner M, Yu O, Subramanian S. Genome organization and characteristics of soybean microRNAs. BMC Genomics. 2012;13:169
30. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154-D158
31. Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001-2019
32. Grad Y, Aach J, Hayes GD, Reinhart BJ, Church GM, Ruvkun G, Kim J. Computational and experimental identification of C. elegans microRNAs. Mol Cell. 2003;11:1253-1263
33. Zhang Z, Wei L, Zou X, Tao Y, Liu Z, Zheng Y. Submergence-responsive MicroRNAs are Potentially Involved in the Regulation of Morphological and Metabolic Adaptations in Maize Root Cells. Annals of Botany. 2008;102(4):509-519
34. Liu H, Qin C, Chen Z, Zuo T, Yang X, Zhou H, Xu M, Cao S, Shen Y, Lin H, He X, Zhang Y, Li L, Ding H, Lübberstedt T, Zhang Z, Pan G. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics. 2014;15:25
35. Zhang HY, Cui LN, Dong ST, Gao RQ, Sun AQ. Selection for Drought Resistance of 37 Maize Inbred Lines. SHANDONG AGRICULTURAL SCIENCES. 2010 (1)
36. Li M, Xia Y, Gu Y, Zhang K, Lang Q, Chen L, Guan J, Luo Z, Chen H, Li Y, Li Q, Li X, Jiang AA, Shuai S, Wang J, Zhu Q, Zhou X, Gao X. MicroRNAome of porcine pre- and postnatal development. PLoS ONE. 2010;5:e11541
37. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152-D157
38. Schnable PS, Ware D, Fulton RS, Stein JC, Wei F. et al. The B73 maize genome: complexity, diversity and dynamics. Science. 2009;326:1112-1115
39. Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20:3186-3190
40. Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207-221
41. Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNA on the plant transcriptome. Dev Cell. 2005;8:517-527
42. Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155-W159
43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCT Method. Methods. 2001;25:402-408
44. Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, Matzke M, Ruvkun G, Tuschl T. A uniform system for microRNA annotation. RNA. 2003;9(3):277-279
45. Chen X, Li QB, Wang J, Guo X, Jiang XR, Ren ZJ, Weng CY, Sun GX, Wang XQ, Liu YP, Ma LJ, Chen JY, Wang J, Zen K, Zhang JF, Zhang CY. Identification and characterization of novel am-phioxus microRNAs by Solexa sequencing. Genome Biology. 2009;10(7):R78
46. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297
47. Zeng QY, Yang CY, Ma QB, Li XP, Dong WW, Nian H. Identification of wild soybean miRNAs and their target genes responsive to aluminum stress. BMC Plant Biology. 2012;12:182
48. Sekhon RS, Lin H, Childs KL, Hansey CN, Robin Buell C, De Leon N, Kaeppler SM. Genome-wide atlas of transcription during maize development. Plant J. 2011;66:553-563
49. Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JDG. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006;312:436-439
50. Kurihara Y, Kaminuma E, Matsui A, Kawashima M, Tanaka M, Morosawa T, Ishida J, Mochizuki Y, Shinozaki K, Toyoda T, Seki M. Transcriptome Analyses Revealed Diverse Expression Changes in ago1 and hyl1 Arabidopsis Mutants. Plant and Cell Phydiology. 2009;50:1715-1720
51. Chen L, Wang T, Zhao M, Tian Q, Zhang WH. Identification of aluminumresponsive microRNAs in Medicago truncatula by genome-wide highthroughput sequencing. Planta. 2012;235(2):375-386
52. Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001-2019
53. Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110(4):513-520
54. Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787-799
55. Sunkar R, Girke T, Jain PK, Zhu JK. Cloning and characterization of microRNAs from rice. Plant Cell. 2005;17(5):1397-1411
56. Buhtz A, Springer F, Chappell L, Baulcombe DC, Kehr J. Identification and characterization of small RNAs from the phloem of Brassica napus. Plant J. 2008;53(5):739-749
57. Sekhon RS, Briskine R, Hirsch CN, Myers CL, Springer NM, Buell CR, de Leon N, Kaeppler SM. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS One. 2013:8 (4)
Author contact
![]() Corresponding authors: Email: hyjiangedu.cn or beijiuchengedu.cn
Corresponding authors: Email: hyjiangedu.cn or beijiuchengedu.cn