Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):892-900. doi:10.7150/ijbs.11729 This issue Cite
Research Paper
Mild ischemic Injury Leads to Long-Term Alterations in the Kidney: Amelioration by Spironolactone Administration
Jonatan Barrera-Chimal1,2,4, Rosalba Pérez-Villalva1,2, Juan Antonio Ortega1,2, Andrea Sánchez1,2, Roxana Rodríguez-Romo1,2, Marta Durand3, Frederic Jaisser4, Norma A. Bobadilla1,2 ![]()
1. Molecular Physiology Unit, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México
2. Nephrology Department of Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
3. Reproductive Biology Department of Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
4. INSERM U1138, Centre de Recherche de Cordeliers, Team 1, Paris, France
Received 2015-1-29; Accepted 2015-5-5; Published 2015-6-6
Abstract

Administration of the mineralocorticoid receptor antagonist spironolactone prevents the development of chronic kidney disease (CKD) after a severe ischemic injury. However, whether brief periods of ischemia lead to CKD and whether spironolactone administration after ischemia may be a useful therapeutic strategy to prevent the gradual deterioration of structure and function remains unexplored.
Nineteen male Wistar rats were divided into four groups: rats that underwent renal bilateral ischemia for 10, 20, or 45 min were compared with sham operated rats. Additionally, thirteen male Wistar rats that underwent renal bilateral ischemia for 20 min were divided into an untreated ischemic group (I) and two groups receiving spironolactone, 20 mg/kg by gavage, at either 0 (Sp0) or 1.5-h after ischemia (Sp1.5). The rats were followed up and studied after 9 months.
Mild (20 min) and severe (45 min) ischemia induced a progressive increase in proteinuria at varying magnitudes, whereas minor ischemia (10 min) did not modify proteinuria. CKD induced by moderate ischemia was characterized by renal hypertrophy and tubulointerstitial fibrosis. These effects were associated with activation of the transforming growth factor β (TGFβ) signaling pathway and up-regulation of endothelin receptor A (ETA) and alpha smooth muscle actin (αSMA). Spironolactone treatment immediately or 1.5-h after the ischemic insult prevented the onset of these disorders.
Our results show that moderate ischemic insult leads to long-term structural and molecular changes that may compromise renal function in later stages. Additionally, we demonstrate that spironolactone administration after mild ischemia prevents this detrimental effect.
Keywords: Aldosterone, fibrosis, acute kidney injury, chronic kidney disease
Introduction
Ischemia/reperfusion injury is the main cause of acute kidney injury (AKI) occurring in approximately 15% of hospitalized patients, and its incidence rises to 40-60% in intensive care unit (ICU) patients (1) (2) (3). Despite recent advances in new therapies and the knowledge of the mechanisms involved in AKI, this syndrome has high morbidity and mortality rates (4).
More worrisome is the recent accumulating evidence indicating that patients who survive an AKI episode have a higher risk of developing chronic kidney disease (CKD) in the following years (5). Therefore, AKI has been recognized as a risk factor for the development of CKD (6) (7). A recent study showed that 6.6% of AKI patients who had complete recovery of renal function had a greater risk of death and de novo CKD in the following months (8). A recent meta-analysis that included thirteen large studies found that AKI is an independent risk factor for CKD (9). Worldwide, 20% of patients with an AKI episode will develop CKD after 3 years, which represents 300,000 patients in high-income countries, and this value might be higher than 1.8 million in low- and middle-income countries (10).
In support of several epidemiological studies, animal models have shown that after a renal ischemia/reperfusion event, the recovery process may be incomplete, producing progressive renal dysfunction, tubulointerstitial fibrosis and chronic inflammation (for review (11)). We have previously shown that aldosterone plays a key role in the physiopathology of renal injury induced by ischemia. In this regard, we showed that adrenal gland removal or mineralocorticoid receptor (MR) blockade with spironolactone before or even after ischemia prevents the acute (24 h) functional and structural injury induced by I/R (12-14). Interestingly, CKD was prevented when spironolactone was administered upon severe ischemic injury, and the untreated ischemic group developed progressive renal dysfunction, proteinuria, glomerular hypertrophy, glomerulosclerosis, aberrant tubular dilation and tubule-interstitial fibrosis (15). These results suggest that MR blockade is a powerful strategy to prevent CKD induced by a longer period of ischemia in the rat (15). However, this severe ischemic injury model might only be applicable to patients undergoing cardiovascular surgery or renal transplantation.
The probability of developing CKD or end-stage renal disease (ESRD) over time is proportional to the severity and the duration of the AKI event (16). Until now, studies performed in rats have explored the effects of severe ischemic injury (45 to 60 min of renal ischemia) on long-term renal functional and structural deterioration (15;17-24). Because in most of the patients AKI occurs unexpectedly, and renal injury appears as a result of a lower degree of hypoperfusion, we addressed the following issues in this study: whether mild ischemic injury (20 min) is able to induce chronic renal injury, and whether spironolactone administration post-ischemia is effective in preventing the long-term effects of mild ischemia.
Methods
All experiments involving animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC, 1996) and were approved by the Animal Care and Use Committees at our institutions (Comisión de Investigación en Animales del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubiran and Comisión Institucional para el Cuidado y Uso de Animales del Laboratorio del Instituto de Investigaciones Biomédicas).
To test the impact of various durations of ischemia on the progressive increase of proteinuria, nineteen male Wistar rats (270-300 g) were divided into four groups: sham-operated (n=4), I/R of 10 min (n=5), I/R of 20 min (n=5), and I/R 45 min (n=5). To investigate the efficacy of MR antagonism on the long-term effects of mild ischemia, thirteen male Wistar (270-300 g) rats were divided into three groups: rats that were subjected to bilateral renal ischemia for 20 min (I, n=5) and two groups of rats that underwent bilateral renal ischemia for 20 min receiving only one dose of spironolactone (20 mg/kg by gastric gavage) either immediately or 1.5 h after ischemia (Sp0, n=4 and Sp1.5, n=4, respectively). These groups were compared with the sham-operated group used in the first set of experiments.
Ischemia/reperfusion model
Rats were anesthetized with an intra-peritoneal injection of sodium pentobarbital (30 mg/kg) and placed on a heating pad to maintain rat core body temperature at 37 ºC. Renal pedicles were isolated and bilateral ischemia was induced by the collocation of a non-traumatic clamps during 10, 20 or 45 minutes. Ischemia was verified visually by change in kidney color. Reperfusion was achieved by release of the clips and confirmed by return of blood to the kidney. The incision was closed in two layers with 3-0 sutures. For sham surgery, anesthesia, laparotomy and renal pedicle dissection, without clamp collocation was performed. After the surgery the rats were allowed to recover and followed up for 270 days.
Functional parameters
Urinary protein excretion was determined every 30 days throughout the follow up in all studied groups using the urine collected over a 24-h period. At the end of the experimental periods (9 months), rats were anesthetized with sodium pentobarbital (30 mg/kg) and placed on a homoeothermic table. The left femoral artery was catheterized with polyethylene tubing (PE-50). The mean arterial pressure (MAP) was monitored with a pressure transducer (model p23 db, Gould) and recorded on a polygraph (Grass Instruments, Quincy, MA). An ultrasound transit-time flow probe was placed around the left artery and filled with ultrasonic coupling gel (HR Lubricating Jelly, Carter-Wallace, New York, NY) to record the renal blood flow (RBF). Blood samples were taken at the end of the study. Urine and serum creatinine concentrations were measured with Quantichrom creatinine assay kit (DICT-500), and renal creatinine clearance was calculated by the standard formula C = (U X V)/P, where U is the concentration in urine, V is the urine flow rate, and P is the serum concentration. Urinary protein excretion was measured by the TCA turbidimetric method (25).
Light microscopy analysis
Histopathological analysis was performed in all rats after 9 months. The right kidney was removed and the cortex and medulla were isolated; then, the tissue was frozen in liquid nitrogen and stored at -80° C. The left kidney was perfused through the femoral catheter with a physiological solution. Following blanching of the kidney, the perfusate was replaced by freshly prepared 10% neutral-buffered formalin, and perfusion was continued until fixation was completed. After appropriate dehydration, renal tissue was embedded in paraffin, sectioned at 4 μm and stained with periodic acid-Schiff (PAS) reagent or Sirius red stains. The degree of tubulointerstitial fibrosis was evaluated by morphometry in Sirius red-stained preparations (magnification x400). Accordingly, five to eight subcortical fields per section were randomly selected in kidneys from the groups studied. Tubulointerstitial fibrosis consisted of extra cellular matrix expansion with collagen deposition together with distortion and collapse of the tubules; fibrosis was evidenced by red coloration in Sirius red stained slides. The affected area was delimited, and the percentage of tubulointerstitial fibrosis was calculated by dividing the fibrotic area by the total field area, excluding the glomerular and tubular luminal areas. All of the slides were blindly analyzed.
Western Blot analysis
Total renal proteins were isolated from renal cortex from each rat and homogenized in lysis buffer (50 mM HEPES ph 7.4, 250 mM NaCl, 5 mM EDTA, 0.1% NP-40, and complete protease inhibitor (Roche). Protein samples containing 50 μg of total protein were resolved by 8.5% SDS-PAGE electrophoresis and electroblotted onto polyvinylidinedifluoride membranes (Millipore). Membranes were then blocked with 5 % blotting-grade non-fat dry milk. After that membranes were incubated in 0.1 % blotting-grade non-fat dry milk with their respective antibodies. Specific antibodies against α-smooth muscle actin (Sigma A2547, 1:5000), Smad3 (sc-101154, 1:500), phosho-Smad3 (Millipore, 1:500), TGF-β (sc-146, 1:500), ETA (Abcam, 1:5000) and ETB (Abcam, 1:5000) were used. After incubation with primary antibody, membranes were washed and incubated with their respective secondary antibody. As a loading control, membranes were incubated overnight at 4 ºC with goat anti-actin antibody (Santa Cruz Biotechnology, 1:5000 dilution). Actin was detected using donkey anti-goat IgG-HRP (1:5000, Santa Cruz Biotechnology). Proteins were detected with an enhanced chemiluminescence kit (Millipore) and autoradiography, following the manufacturer's recommendations. The bands were scanned for densitometric analysis.
Endothelin ELISA
Endothelin-1 levels were analyzed using a commercially available ELISA kit (Endothelin-1 (1-31) Assay kit; Immuno-Biological Laboratories Inc.) according to the manufacturer's instructions. Tissue homogenates and standards were added to the pre-coated wells and incubated overnight at 4°C. Endothelin-1 was captured by the antibody and then detected by adding the labeled antibody and the chromogen. The optical density of the samples was read at 450 nm by a plate reader and was compared to a standard curve generated from known concentrations of endothelin-1 that ranged from 1.56 to 200 pg/mL. The protein concentration in the tissue homogenates was determined by the Lowry method (BioRad). The endothelin-1 concentration was normalized by the amount of protein added to the well.
Statistical analysis
The results are presented as the mean ± S.E. The significance of the differences between the groups was assessed by analysis of variance (ANOVA) using the Bonferroni correction for multiple comparisons. All of the comparisons passed the normality test. Statistical significance was defined as having p values <0.05.
Results
Mild and severe ischemic insult leads to progressive increase in proteinuria
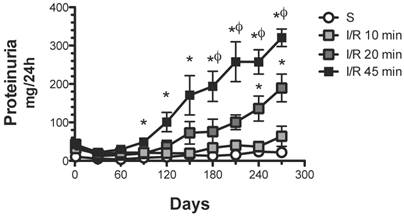
As expected, 45 min of renal ischemia induced a progressive increase in proteinuria from 21.8 ± 3.4 (one month) to 320.4 ± 22.8 mg/24 h (nine months post-ischemia) (Figure 1). A brief period of ischemia (20 min) also induced a progressive increase in urinary protein excretion, although the extent was significantly lesser than in the group with 45 min ischemia, from 12.1 ± 2.2 (one month) to 189.6 ± 36.2 mg/24 h (9 months post-ischemia). In contrast, rats that suffered a minor ischemic insult (10 min) did not develop proteinuria after the nine-month follow-up.
Effect of various durations of ischemia on proteinuria development. Four groups were included: sham (S), and rats that underwent bilateral renal ischemia of 10, 20 or 45 min duration. The urinary protein excretion was determined every 30 days: sham (white circles, n=4), I/R 45 min (black squares, n=5), I/R 20 min (dark gray squares, n=5) and I/R 10 min (gray squares, n=5). *p<0.05 vs. Sham-operated rats and φ p<0.05 vs. I/R 45 group.
The progressive increase in proteinuria induced by a brief ischemic period is reduced by spironolactone administration after the ischemic insult
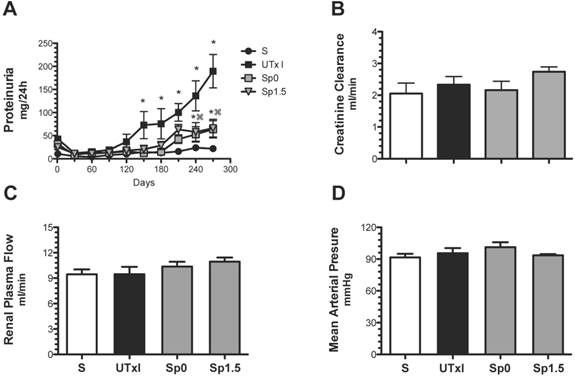
We next evaluated the effectiveness of post-ischemic treatment with spironolactone to prevent long-term functional, structural and molecular damage. The progressive increase of proteinuria induced by 20-min of ischemia was significantly lessened after nine months in the groups treated with spironolactone immediately (Sp0) or 1.5-h (Sp1.5) after ischemia (63.7 ± 18.5 and 66.1 ± 19.2 mg/day, respectively) (Figure 2A). Despite the presence of proteinuria in the untreated ischemic group, the rats did not exhibit renal dysfunction at the end of the experiment; similar values of renal blood flow and creatinine clearance were observed among the groups (Figures 2B and 2C). None of the rats presented with an increase in mean arterial pressure, indicating that phenotypic changes are directly related to the duration of ischemia and not secondary to systemic hypertension (Figure 2D).
Mild acute kidney injury leads to the development of proteinuria, and the effect was ameliorated by spironolactone administration. Four groups were included: sham (S, n=4), rats that underwent bilateral renal ischemia for 20 min (UTxI, n=5) and rats that received spironolactone (20 mg/kg) at 0 or 1.5 hours after ischemia (Sp0, and Sp1.5, respectively, n=4). A) Urinary protein excretion was determined every 30 days during the follow-up: sham (black circles), A-C (black squares), Sp0 (gray squares) and Sp1.5 (gray triangles). At the end of the 9-month period, B) creatinine clearance, C) renal blood flow and D) mean arterial pressure were determined in the sham (white bars), untreated ischemic group (black bars), and spironolactone-treated groups (gray bars). *p<0.05 vs. sham operated rats and ⌘P<0.05 vs. the UTxI group.
Long-term renal structural changes induced by a brief ischemic period
Representative light microscopy sections from rat kidneys stained with periodic acid-Schiff are shown in Figure 3A-D. IR induced structural changes characterized by glomerular hypertrophy, glomerulosclerosis, and cast formation (Figure 3B). In contrast, the Sp0 and Sp1.5 groups exhibited glomerular and tubular architecture similar to those observed in sham-operated rats (Figure 3C-D). Accordingly, the untreated ischemic group exhibited an increase in the percentage of glomerulosclerosis (14.4%), which was not observed in rats treated with spironolactone (Figure 3E). Renal hypertrophy was evaluated by kidney weight. Despite similar body weights among the groups, the kidney weight and body weight ratio (KW/BW) was 40% higher in the IR group than in the sham operated group (0.0042 ± 0.0004 vs. 0.00291 ± 0.0001, p=0.03), as shown in Figure 3F. This renal hypertrophy was not observed in any of spironolactone-treated groups (0.0030 ± 0.0001 and 0.0032 ± 0.0001 for Sp0 and Sp1.5, respectively).
Twenty minutes of bilateral renal I/R led to renal structural injury, which can be prevented by spironolactone administration. Representative images of periodic acid-Schiff (PAS)-stained sections from A) Sham (n=4), B) untreated ischemic group (n=5), C) Sp0 (n=4) and D) Sp1.5 (n=4) groups. The main effects observed were: Tubular dilation, tubular cast formation and glomerular sclerosis. Original magnification: X100. E) Glomerulosclerosis percentage and F) Ratio between kidney weight and body weight (KW/BW). *p<0,05 vs. all the groups.
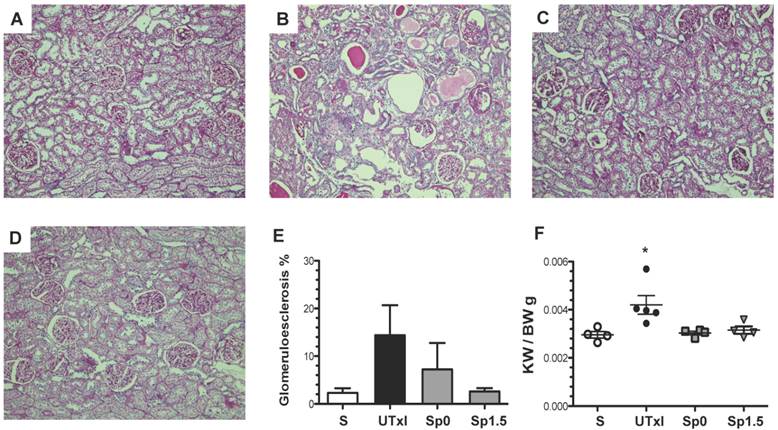
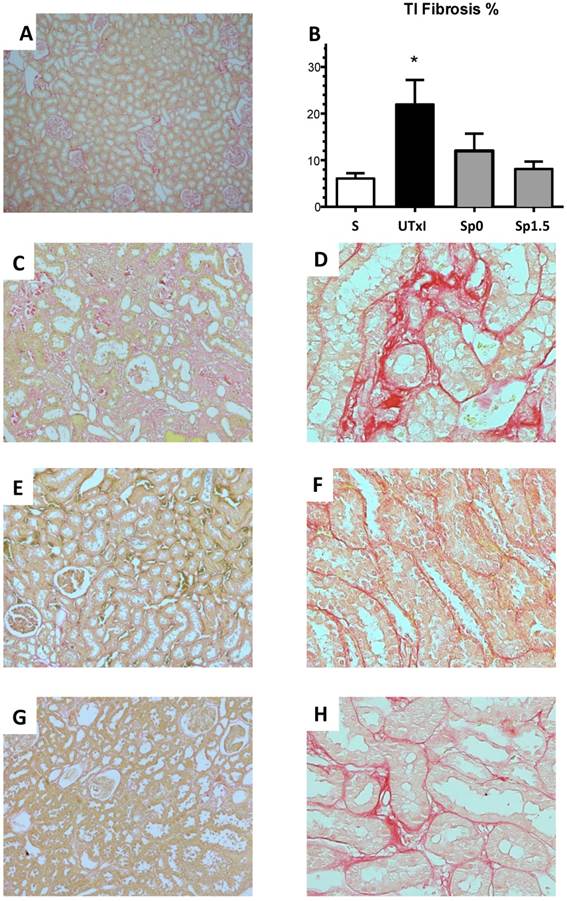
Figure 4 shows the representative microphotographs from kidney slides stained with Sirius red and the morphometric analysis of the various groups. The untreated ischemic group exhibited a significant area affected by tubulointerstitial fibrosis (Figure 4C-4D). In contrast, the spironolactone-treated groups showed practically no staining for Sirius red (Figure 4E-4H). These observations were confirmed by the morphometric analysis presented in Figure 4B.
Tubulointerstitial fibrosis development after 9 months of renal ischemia. Representative light micrographs after Sirius red staining showing the presence of fibrosis (in red) from the A) Sham (n=4), C and D) ischemic untreated ischemic group (n=5), E and F) Sp0 (n=4) and G and H) Sp1.5 (n=4). B) The percentage of tubulointerstitial fibrosis in each of the five groups at the end of the 270-day experiment was quantified by morphometric analysis for sham (white bars), untreated ischemic group (black bars), and spironolactone-treated groups (gray bars). Original magnification: X100 (A, C, E, G) and X400 (D, F, H). *p<0,05 vs. all the groups.
Long-term molecular changes induced by a brief ischemic period and prevention by spironolactone
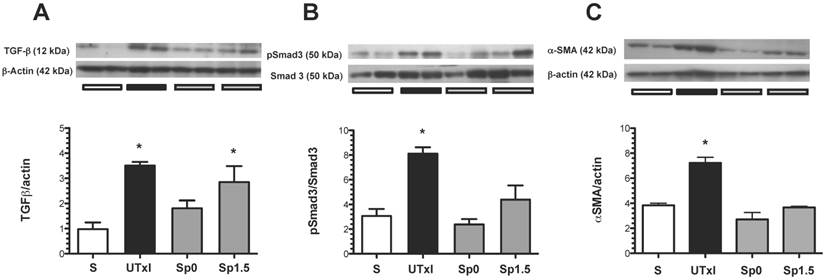
The role of the TGF-β pathway in promoting the observed fibrosis was assessed. The ischemic untreated group exhibited a significant increase in TGF-β protein levels (Figure 5A). To assess the activation of this pathway, the renal levels of a downstream effector of the TGF-β pathway, phospo-Smad-3, were evaluated by Western blot analysis (Figure 5B). Accordingly, phospho-Smad3 was significantly elevated in the untreated ischemic group. Similar to the effect observed at the TGF-β level, the increased phosphorylation of Smad-3 was not observed in the spironolactone-treated groups. Additionally, the structural injury was associated with a significant up-regulation of renal α-SMA protein levels. This progressive increase was prevented in the spironolactone-treated groups (Figure 5C).
Molecular changes associated with the pro-fibrotic TGF-β pathway activation. A) TGF-β protein levels were quantified by Western blot, B) p-Smad3 and C) α-smooth muscle actin (α-SMA); the densitometric analysis is depicted in the graphs for the sham (white bars, n=4), untreated ischemic group (black bars, n=5), and spironolactone groups (gray bars, n=4). In each panel, the upper insets depict representative blots of the corresponding proteins. *p<0.05 vs. sham operated rats.
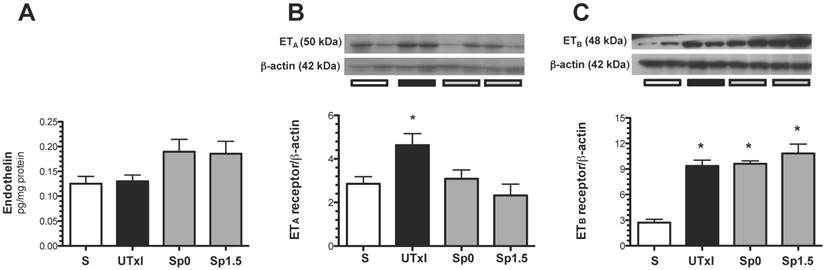
Recently, it was suggested that endothelin-1 activation and enhanced ETA expression may contribute to the progression of AKI to CKD (18). For this reason, the protein levels of ET-1 and its receptors ETA and ETB were measured. Although no difference in the intra-renal content of ET-1 was observed (Figure 6A), we found an up-regulation of ETA receptors in the untreated ischemic group, and the effect was prevented by spironolactone treatment (Figure 6B). Regarding the ETB receptor, an up-regulation in all ischemic groups was observed (Figure 6C).
Molecular changes associated with the endothelin pathway. A) Protein levels of endothelin were quantified in renal tissue by an ELISA assay, B) endothelin receptor A (ETA), and C) endothelin receptor B (ETB) protein levels. Sham (white bars, n=4), untreated ischemic group (black bars, n=5), and spironolactone-treated groups (gray bars, n=4). * p<0.05 vs. sham operated rats.
Discussion
The development of CKD in the years following an AKI episode is a major public health issue, and it is associated with poor quality of life in the patients and increased expenses to the health system. The association between AKI and CKD has been consistently recognized in several epidemiological studies. For example, complete recovery of renal function after an episode of AKI in patients with normal baseline kidney function is associated with increased risk of the development of incident stage 3 CKD (5). Moreover, Chawla LS et al. showed that the severity of the AKI episode is strongly associated with the risk of CKD progression (16). However, most of the experimental studies, including a study from our laboratory (15;19;26-29), investigating the possible association between AKI and CKD have focused on the long-term effects of a severe ischemic lesion; therefore, more information on the effects of a brief period of ischemia on chronic renal injury is required. In this study, we first characterized whether the severity of the ischemic insult was associated with the presence and intensity of chronic renal injury induced by renal bilateral ischemia. Rats that underwent a longer period of ischemia (45 min) developed heavy proteinuria beginning in the third month after ischemia, as we previously reported (15). A mild period of ischemia (20-min) also induced a progressive increase in proteinuria, but this was evident only after 5 months of the initial insult and was less severe compared with the longer-period ischemia group.
In contrast, a brief period of ischemia (10 min) did not lead to proteinuria by the conclusion of the experiment (9 months after ischemia); we cannot exclude the possibility, however, that rats would develop CKD after a longer period of time. This finding is important because many patients in whom AKI may not be accurately diagnosed may be at risk of developing chronic renal injury that may compromise renal function. Indeed, a recent study from Linder A. et al. (30) showed that patients who underwent a mild AKI episode have significantly decreased long-term survival than critically ill patients with no signs of AKI. Although proteinuria developed in rats that underwent 20 min of ischemia, the renal function of the rats was normal nine months after ischemia. These data suggest that the structural injury might be masked by the glomerular compensatory hypertrophy and the nephron functional reserve at this time after mild ischemia. Therefore, although renal dysfunction was not present at this time, these animals exhibited structural and molecular damage, evidenced by glomerular and tubular injury, as well as by effects on pro-fibrotic signaling pathways.
We previously reported that spironolactone administration before or after ischemia may be a useful strategy to prevent the development of CKD induced by a longer period of ischemia. However, this ischemic insult can only be extrapolated to renal transplant or cardiac surgery patients. To investigate a model closer to the common clinical situation, such as patients receiving contrast media or hospitalized patients with lower degree of hypoperfusion, in this study, we investigated spironolactone efficacy in preventing chronic renal injury induced by mild ischemic injury when administered after ischemia. A brief period of ischemia induced structural changes, such as glomerular hypertrophy, glomerulosclerosis, and extensive tubulointerstitial fibrosis. All of these changes were completely prevented by spironolactone administration immediately or up to 1.5-h after ischemia. Various mechanisms underlying the association between AKI and CKD have been proposed and include chronic hypoxia, abnormalities in the cell cycle of epithelial cells, inflammation, endothelial injury and capillary rarefaction (19;21;29;31). Recently, Kramann et al. (32) showed a reduction in the total number of capillaries and the single capillary area and its perimeter in mice that underwent a severe ischemia, whereas in moderate ischemia, although the number of cortical capillaries did not change, the size of the capillaries was significantly smaller, suggesting that mice with moderate ischemia may also be a target of chronic hypoxia cycles that eventually induce capillary rarefaction after a longer period of time. Indeed, we observed that a mild ischemic insult led to chronic renal injury.
After an ischemic injury, many pathways are activated to promote tubule regeneration; however, some of these pathways remain activated even if the repair is complete, typical of the TGF-β pathway. This mechanism is of particular importance because TGF-β may promote fibroblast trans-differentiation into myofibroblasts and the consequent pro-fibrotic phenotype (33). Indeed, we found that the protein levels of TGF-β, as well as the downstream effector p-Smad3, are increased in rats that underwent a brief period of ischemia. Moreover, an increase in α-SMA, a myofibroblast marker, was also observed. These effects were prevented in the spironolactone-treated rats except for the TGF-β elevation in the Sp1.5 group, however the SMAD-3 phosphorylation was lesser than the untreated ischemic group, suggesting inhibitory SMADs could be inhibiting SMAD-3 phosphorylation.
A recent report suggested that endothelin-1 activation and enhanced ETA expression may be implicated in the progression from AKI to CKD (18). Therefore, we studied the levels of the endothelin-1, ETA and ETB receptors. We could not detect an increase in endothelin-1 levels; however, this discrepancy may be explained by the stage of CKD in which endothelin expression is assessed. In severe CKD, when an extensive area is injured, these cells do not contribute to endothelin production. The differences between rat vs. mice and the effect of bilateral ischemia vs. unilateral ischemia may also account for these results. However, we cannot exclude the idea that endothelin is indeed participating in the progression of CKD, because enhanced expression of the ETA and ETB receptors in the rats that underwent mild ischemia was found. Interestingly, the spironolactone treated rats did not present an up-regulation of the ETA receptor. These data suggest that after ischemia, ETA remains active and the balance of the receptors may favor the vasoconstrictor effect, which may enhance the chronic hypoxia. In contrast, in spironolactone treated rats, this balance may favor the ETB receptor, thereby promoting normal perfusion of the kidney and preventing chronic renal injury.
In summary, we provide evidence that in an experimental model of AKI, the duration of ischemia is correlated with the strength and timing of the onset of proteinuria. A mild ischemic lesion was enough to produce progressive proteinuria, renal hypertrophy, glomerular injury, and tubulointerstitial fibrosis. The structural changes were associated with increased TGF-β pathway activation and ETA receptor up-regulation. These changes were prevented or reduced by spironolactone even if administered after the ischemic insult had occurred. Our data demonstrate the effectiveness of MR antagonism in preventing chronic renal injury induced by a mild ischemic insult in a model of a clinical situation.
Acknowledgements
The results presented in this paper have not been published previously in whole or in part. This project was supported by grants from the Mexican Council of Science and Technology (CONACyT) (101030, 181267, 235855 to NAB and 229524 to NAB and FJ) and from the National University of Mexico (IN223915 to NAB). RRR is a graduate student in the Biomedical Science Ph.D. Program at Universidad Nacional Autónoma de México and is supported by a scholarship from CONACyT-Mexico.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, Paganini EP, Chertow GM. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66(4):1613-1621
2. Ali T, Khan I, Simpson W, Prescott G, Townend J, Smith W, MacLeod A. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J Am Soc Nephrol. 2007;18(4):1292-1298
3. Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, Himmelfarb J, Collins AJ. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17(4):1135-1142
4. Wu I, Parikh CR. Screening for kidney diseases: older measures versus novel biomarkers. Clin J Am Soc Nephrol. 2008;3(6):1895-1901
5. Jones J, Holmen J, De GJ, Jovanovich A, Thornton S, Chonchol M. Association of complete recovery from acute kidney injury with incident CKD stage 3 and all-cause mortality. Am J Kidney Dis. 2012;60(3):402-408
6. Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010
7. Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol. 2012;23(6):967-969
8. Bucaloiu ID, Kirchner HL, Norfolk ER, Hartle JE, Perkins RM. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012;81(5):477-485
9. Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442-448
10. Lewington AJ, Cerda J, Mehta RL. Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int. 2013;84(3):457-467
11. Leung KC, Tonelli M, James MT. Chronic kidney disease following acute kidney injury-risk and outcomes. Nat Rev Nephrol. 2013;9(2):77-85
12. Mejia-Vilet JM, Ramirez V, Cruz C, Uribe N, Gamba G, Bobadilla NA. Renal ischemia-reperfusion injury is prevented by the mineralocorticoid receptor blocker spironolactone. Am J Physiol Renal Physiol. 2007;293(1):F78-F86
13. Sanchez-Pozos K, Barrera-Chimal J, Garzon-Muvdi J, Perez-Villalva R, Rodriguez-Romo R, Cruz C, Gamba G, Bobadilla NA. Recovery from ischemic acute kidney injury by spironolactone administration. Nephrol Dial Transplant. 2012;27(8):3160-3169
14. Ramirez V, Trujillo J, Valdes R, Uribe N, Cruz C, Gamba G, Bobadilla NA. Adrenalectomy prevents renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2009;297(4):F932-F942
15. Barrera-Chimal J, Perez-Villalva R, Rodriguez-Romo R, Reyna J, Uribe N, Gamba G, Bobadilla NA. Spironolactone prevents chronic kidney disease caused by ischemic acute kidney injury. Kidney Int. 2013;83(1):93-103
16. Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011;79(12):1361-1369
17. Conger JD, Robinette JB, Hammond WS. Differences in vascular reactivity in models of ischemic acute renal failure. Kidney Int. 1991;39(6):1087-1097
18. Zager RA, Johnson AC, Andress D, Becker K. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int. 2013;84(4):703-712
19. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535-43 1p
20. Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13(1):1-7
21. Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281(5):F887-F899
22. Horbelt M, Lee SY, Mang HE, Knipe NL, Sado Y, Kribben A, Sutton TA. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293(3):F688-F695
23. Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and "end-stage" kidney disease. Am J Physiol Renal Physiol. 2011;301(6):F1334-F1345
24. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2):172-183
25. Henry RJ, Sobel C, Segalove M. Turbidimetric Determination of Proteins with Sulfosalicylic and Trichloracetic Acids. Proceedings of the Society for Experimental Biology and Medicine. 1956;92(4):748-751
26. Stroo I, Stokman G, Teske GJ, Raven A, Butter LM, Florquin S, Leemans JC. Chemokine expression in renal ischemia/reperfusion injury is most profound during the reparative phase. Int Immunol. 2010;22(6):433-442
27. Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544-550
28. Basile DP, Leonard EC, Beal AG, Schleuter D, Friedrich JL. Persistent oxidative stress following renal ischemia reperfusion injury increases Ang II hemodynamic and fibrotic activity. Am J Physiol Renal Physiol. 2012
29. Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300(3):F721-F733
30. Linder A, Fjell C, Levin A, Walley KR, Russell JA, Boyd JH. Small acute increases in serum creatinine are associated with decreased long-term survival in the critically ill. Am J Respir Crit Care Med. 2014;189(9):1075-1081
31. Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82(5):516-524
32. Kramann R, Tanaka M, Humphreys BD. Fluorescence Microangiography for Quantitative Assessment of Peritubular Capillary Changes after AKI in Mice. J Am Soc Nephrol. 2014
33. Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, Smith RD, Hayashida T. TGF-beta signal transduction in chronic kidney disease. Front Biosci. 2009;14:2448-2465
Author contact
![]() Corresponding author: Norma A. Bobadilla, PhD, Unidad de Fisiología Molecular, Vasco de Quiroga No. 15, Tlalpan, 14000, México City. Tel.: 5255-5485-2676. Fax: 5255-5655-0382. nabunam.mx; norma.bobadillasmx
Corresponding author: Norma A. Bobadilla, PhD, Unidad de Fisiología Molecular, Vasco de Quiroga No. 15, Tlalpan, 14000, México City. Tel.: 5255-5485-2676. Fax: 5255-5655-0382. nabunam.mx; norma.bobadillasmx