ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2015; 11(9):1006-1015. doi:10.7150/ijbs.12456 This issue Cite
Research Paper
Identification and Functional Characterization of Cis-Regulatory Elements Controlling Expression of the Porcine ADRB2 Gene
Alexandra Jaeger, Stephan Fritschka, Siriluck Ponsuksili, Klaus Wimmers, Eduard Muráni ![]()
Institute for Genome Biology, Leibniz-Institute for Farm Animal Biology, Wilhelm-Stahl-Allee 2, D-18196 Dummerstorf, Germany
Abstract

The beta-2 adrenergic receptor (beta-2 AR) modulates metabolic processes in skeletal muscle, liver, and adipose tissue in response to catecholamine stimulation. We showed previously that expression of the porcine beta-2 AR gene (ADRB2) is affected by cis-regulatory polymorphisms. These are most likely responsible for the association of ADRB2 with economically relevant muscle-related traits in pigs. The present study focused on characterization of promoter elements involved in basal transcriptional regulation of the porcine ADRB2 in different cell types to aid identification of its cis-regulatory polymorphisms. Based on in silico analysis, luciferase reporter gene assays and gel shift assays were performed using COS-7, HepG2, C2C12, and 3T3-L1 cells. Deletion mapping of the 5´ flanking region (-1324 to +33) of ADRB2 revealed the region between -307 and -269 to be the minimal promoter, including regulatory elements essential for the basal transcriptional activity in all four tested cell types. Directly upstream (-400 to -323) we identified an important enhancer element required for maximal promoter activity. In silico analysis and gel shift assays revealed that this GC-rich element harbors two evolutionarily conserved binding sites of Sp1, a constitutive transcriptional activator. Significant transcriptional activation of the porcine ADRB2 promoter was demonstrated by overexpression of Sp1. Our results demonstrate, for the first time, an important role of Sp1 and of the responsive enhancer element in the regulation of ADRB2 expression. Polymorphisms located in this domain of the porcine ADRB2 promoter represent candidate causal cis-regulatory variants.
Keywords: beta-2 adrenergic receptor, ADRB2, promoter, Sp1, porcine
Introduction
The adrenergic receptor beta 2 (ADRB2) gene encodes a seven-transmembrane domain receptor, a member of the G protein-coupled receptor superfamily. As a primary signaling receptor of the sympathetic nervous system, the beta-2 adrenergic receptor (beta-2 AR) is responsible for maintaining homeostasis in diverse mammalian tissues. By activation of the cyclic adenosine monophosphate (cAMP) - protein kinase A (PKA) and phosphoinositol 3-kinase (PI3K) - protein kinase B (Akt) signaling pathways, beta-2 ARs mediate catecholamine effects on various health-related physiological processes including myocardial contractility and rate, smooth muscle relaxation in the peripheral vasculature, uterine, and respiratory system, and growth and differentiation of thyroid cells (1,2), as well as glycogenolysis, proteolysis, and lipolysis in skeletal muscle, liver, and adipose tissue (3,4). Accordingly, beta-2 ARs are major pharmacological targets for the treatment of respiratory diseases (5), and their dysfunction is implicated in the pathogenesis of common medical conditions such as obesity (6).
This pleiotropic receptor is under complex regulation. Studies of human and rodent ADRB2 revealed different factors and DNA elements regulating beta-2 AR expression at the transcriptional (1,7-9), and also at the posttranscriptional level (10-12). Most of the studies focused on the regulation of ADRB2 in hepatocytes and smooth muscle cells, or utilized common, easy-to-transfect cell lines such as African green monkey kidney cells (COS-7). Since ADRB2 is ubiquitously expressed, much remains to be discovered regarding its regulation and cis-regulatory elements. Such knowledge can help uncover the mechanisms and variants driving its functional divergence. Indeed, expression of beta-2 AR in humans is diversified by regulatory polymorphisms in the promoter region of ADRB2, contributing to inter-individual variation in responsiveness to beta-2 agonist-based drugs (13) and possibly to obesity risk (14).
Similar to human ADRB2, mRNA expression of the porcine homologue is modulated by multiple polymorphisms in the highly polymorphic 5´ flanking region, as evidenced by the analysis of haplotype-specific expression of the porcine ADRB2 (15). These polymorphisms seem to contribute to variation in economically relevant muscle-related traits in pigs (16). However, pinpointing the causal regulatory polymorphisms is hampered by high sequence variability, unfavourable haplotype structure, and general lack of knowledge of the regulation of porcine ADRB2. Therefore, in this study we set out to explore promoter elements and transcription factors regulating basal transcription of the porcine ADRB2. To this end we performed a luciferase-based reporter gene assay and electrophoretic mobility shift assays (EMSA) in four different cell lines—C2C12 (mouse myoblast cells), HepG2 (human hepatocellular liver carcinoma cells), 3T3-L1 (mouse fibroblast/adipocyte cells), and COS-7 (fibroblast-like)—including established model cells for tissues where ADRB2 expression is affected by cis-regulatory variation. Our results demonstrate that the transcription factor specificity protein 1 (Sp1) serves as a constitutive transcriptional activator of the porcine ADRB2 gene by binding to an evolutionarily conserved GC-rich element in the proximal promoter region. Further, the findings point to polymorphisms associated with the GC-rich element as candidate causal cis-regulatory polymorphisms. To the best of our knowledge, this study provides the first experimental evidence for an involvement of Sp1 in transcriptional regulation of the ADRB2 gene.
Materials and Methods
Cell lines and culture conditions
COS-7 cells were cultured in Dulbecco's Modified Eagle's medium (DMEM) containing GlutaMAX-I, 1 g/L D-glucose, and sodium pyruvate (Life Technologies, Darmstadt, Germany) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin (all from PAA, Cölbe, Germany). HepG2, C2C12, and 3T3-L1 cells were cultured in DMEM containing L-glutamine, 4.5 g/L D-glucose, and sodium pyruvate (Life Technologies) supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. C2C12 cells were induced to differentiation upon 80% confluence by serum withdrawal (DMEM with 2% horse serum; PAA) for seven days. 3T3-L1 cells were differentiated as follows: two days after cells reached 80% confluency, adipocyte differentiation was initiated by treatment with growth medium containing 10 μg/mL insulin, 1 μM dexamethasone, and 0.5 mM isobutylmethylxanthine (all from Sigma-Aldrich, Steinheim, Germany) for two days. Cells were then treated with growth medium containing 10 μg/mL insulin for two days. Cells were then maintained in regular growth medium (DMEM/10% FBS) for an additional four days. Differentiation was achieved by day eight. Cell incubation was performed at 37 °C in a humidified 5% CO2 atmosphere.
Preparation of ADRB2 deletion reporter constructs
Progressive deletions of the 5´ flanking sequence of the porcine ADRB2 promoter region (constructs -1324/+33, -1078/+33, -882/+33, -709/+33, -515/+33, -307/+33, -269/+33) were generated by polymerase chain reaction (PCR) amplification using different forward primers, P1-P7, and a common reverse primer, P8, with restriction enzyme sites NheI and HindIII (Supplementary Table S1). Nucleotide numbering of our constructs starts with +1 at the A of the ATG translation initiation codon. Genomic DNA from a homozygous carrier of the major W2 haplotype served as a template. To delete putative AP-2 and NF-kappaB binding sites from the construct -515/+33, primers P11 and P8 (Supplementary Table S1) were combined. Two putative Sp1 binding sites and the intervening GC-rich region were deleted from the -515/+33 construct by fusion PCR using primers P5, P10, P9, and P8 (Supplementary Table S1). Each PCR product was separated on and purified from an agarose gel using the NucleoSpin Extract II Kit (Macherey-Nagel, Düren, Germany). The purified fragments were subcloned into the pGEM®-T Easy vector using the pGEM®-T Easy Vector System (Promega, Mannheim, Germany) and JM109 Escherichia coli competent cells (Agilent Technologies, Böblingen, Germany). The plasmids were extracted using the NucleoSpin Plasmid Kit (Macherey-Nagel) and the promoter fragments were subsequently transferred into the pGL3-enhancer vector (Promega) utilizing the NheI and HindIII restriction sites to produce the final reporter gene plasmids. For transfection endotoxin free reporter plasmids were purified using NucleoBond® Xtra Midi EF Kit (Macherey-Nagel). Fragment orientation and integrity of the inserts were proven by sequencing using an ABI 3130 sequencer (Applied Biosystems Inc, Foster City, CA, USA).
Plasmid transfection and dual luciferase reporter assay
COS-7 cells (8 x 10^4 cells/well) and HepG2 cells (4 x 10^4 cells/well) were seeded into 24-well plates and co-transfected the following day with 800 ng of the respective ADRB2 plasmid reporter constructs or with 800 ng of the empty pGL3-enhancer vector and 40 ng of pRL-TK Renilla (Promega) reporter using Lipofectamine 2000 (Invitrogen, Darmstadt, Germany) according to the manufacturers' recommendations. Cells were harvested 48 h post-transfection and assayed for luciferase activity. C2C12 cells (1 x 10^5 cells/well) were co-transfected directly after seeding as described above, and harvested for luciferase assay after differentiation. Differentiated 3T3-L1 cells were co-transfected using Neon® Transfection System 100 µL Kit (Invitrogen) according to the manufacturer`s protocol. Afterwards, 1.5 x 10^5 cells/well were seeded into 24-well plates coated with collagen R (Menal GmbH, Emmendingen, Germany) and harvested 48 h post-transfection for luciferase assay. Luciferase assays were carried out using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer's instructions, on a DTX 880 multimode detector (Beckman Coulter, Krefeld, Germany). The relative luciferase activity was calculated as the ratio of firefly luciferase activity to Renilla luciferase activity (internal transfection control) and in relation to pGL3-enhancer vector to reflect the ADRB2 promoter activity.
Transcription factor binding site prediction and electrophoretic mobility shift assays (EMSA)
In silico analysis of transcription factor binding sites (TFBS) was performed using MatInspector software (http://www.genomatix.de). Evolutionary conservation of the promoter sequence and of the predicted TFBS was analysed using Mulan and MultiTF online tools, respectively (www.dcode.org). Sequences of human, bovine, and murine ADRB2 promoter region were retrieved from public database (NCBI).
Long double-stranded probes and competitors covering the GC-rich region were produced by PCR amplification. Primer sequences are given in Supplementary Table S1. To generate protruding 5´ ends for labeling, the PCR products were subcloned into pGEM®-T Easy Vector, excised using PdiI and PauI restriction endonucleases (Fermentas, St. Leon-Rot, Germany), and gel-purified as described above. Integrity of the probe sequence was verified by sequencing. Short double-stranded probes were prepared by annealing overhanging or exact complementary oligonucleotides listed in Supplementary Table S1 to generate labeled probes or cold competitors, respectively.
For labeling, a Klenow fill-in reaction was performed at 37 °C for 30 min. The reaction mixture contained 5U enzyme (Fermentas), 1× buffer supplied by the manufacturer, 20 μCi [alpha-32P]dCTP (Hartmann Analytic, Braunschweig, Germany) and 100 μM of each cold dNTP. The reaction was completed by incubation with 100 μM cold dCTP for 5 min, and finally stopped at 72 °C for 10 min. Unincorporated nucleotides were removed using ProbeQuant G-50 micro columns (GE Life Sciences, Freiburg, Germany). Nuclear extracts (~3 μg) of COS-7 (Abcam, Cambridge, UK), HepG2, and C2C12 cells (both from Active motif, La Hulpe, Belgium) were incubated with 80 fmol labeled probes at 20 °C for 20 min in a 20 μL binding mixture containing 10 mM HEPES-KOH (pH 7.9), 50 mM KCl, 0.1 mM EDTA, 0.5 mM DTT, 1 μM poly(dI-dC) (Roche, Mannheim, Germany), 10% glycerol, and 1× protease inhibitor cocktail (Roche). For competition experiments 100-fold excess of unlabeled oligonucleotides were incubated with nuclear extract 10 min before addition of labeled probes. For supershift assay 4 μg of primary rabbit polyclonal antibodies (all from Santa Cruz Biotechnology, Heidelberg, Germany) were added to the binding reaction before addition of labeled probe and incubated for 20 min at room temperature: anti-Sp1 (H-225, sc-14027), anti-Egr1 (C-19, sc-189), and anti-MAZ (H-50, sc-28745). After pre-running of the gel for 20 min, samples were subjected to electrophoresis using native 6% polyacrylamide (30:1 AA: Bis; 20 x 20 cm) gels and run at 200 V for 1.5 h in 0.5 × tris-borate-EDTA (TBE) buffer at 20 °C. In the shift experiments using recombinant Sp1 protein (SRP2030; Sigma-Aldrich), the assay conditions were modified according to the manufacturer's recommendations. After electrophoresis, gels were exposed overnight to phosphor storage screens and analysed on a STORM 840 PhosphorImager (Molecular Dynamics, Krefeld, Germany).
Transient overexpression of Sp1
The mammalian expression vectors CMV-Sp1 (CMV-Sp1 was a gift from Robert Tjian, Addgene plasmid # 12097) and pEGFP-Sp1 (pEGFP-Sp1 was a gift from Beatrice Yue, Addgene plasmid # 39325) were used for the study. CMV-Sp1 is based on a modified pBluescript backbone and encodes for a mutant (aa1-87 deleted) Sp1 protein. Sp1 expression is driven by CMV LTR. The plasmid contains restriction sites for BamHI and XhoI and an ampicillin resistance gene. pEGFP-Sp1 is based on the pEGFP-C backbone, contains a CMV promoter, and encodes an Sp1 protein tagged with EGFP fused to its N-terminal end. pEGFP-Sp1 contains restriction sites for XhoI and KpnI and a kanamycin resistance gene.
The GC-rich region (-401/-317) was amplified using primers P12 and P13 with restriction enzyme sites NheI and HindIII, respectively (Supplementary Table S1). The PCR product was cloned into the reporter vector pGL4.23[luc2/minP] (Promega) containing a minimal promoter upstream of the luc2 luciferase gene. For promoter analysis, COS-7 cells were co-transfected with 800 ng of ADRB2 plasmid reporter construct (-401/-317), either 100 ng CMV-Sp1 or 100 ng pEGFP-Sp1 expression vectors, respectively, and 40 ng of pRL-TK Renilla (internal control). Cells were harvested after 48 h of incubation for luciferase assay. The relative luciferase activity was calculated as the ratio of firefly luciferase activity to Renilla luciferase activity and in relation to empty pGL4.23 vector to reflect the ADRB2 promoter activity. We had decided to use the improved pGL4.23 vector (fewer consensus regulatory sequences) instead of the pGL3-enhancer vector for this experiment to avoid potential interactions between the overexpressed Sp1 and vector backbone.
Protein extraction and Western/immunoblotting
For protein extraction, cells were washed twice with cold PBS (PAA) and incubated for 5 min on ice with cell lysis buffer (Cell Signaling, Frankfurt am Main, Germany) according to manufacturer's protocol. Cells were collected by scraping, homogenized by using a 5 mL syringe with a 21-gauge needle to shear DNA, and incubated on ice for 30 min. Homogenates were centrifuged at 14,000 x g and 4 °C for 10 min and supernatants were collected. The total protein concentrations were determined by the Bradford method. Equal amounts (50 µg) of total cell protein were loaded onto 8% polyacrylamide gels and run on SDS-PAGE (Bio-Rad, München, Germany) with a Tris/glycine running buffer at 130 V for 1.5 h. Separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Carl Roth) by semi-dry blotting (Bio-Rad) at 80 mA for 1 h. The blots were washed three times with Tris-buffered saline (TBS; Sigma-Aldrich) for 5 min each. Unspecific protein binding sites were blocked with TBS plus 3% non-fat dried milk powder (ECL advance blocking agent; GE Healthcare, Freiburg, Germany) for 1 h, followed by three washing steps with TBS plus 0.1% Tween 20 (TBS-Tw; Sigma-Aldrich) for 15 min each. For detection we used the primary antibody rabbit anti-Sp1 (H-225, sc-14027, 1:1000, Santa Cruz) and the secondary antibody goat anti-rabbit-HRP (#7074, 1:1000, Cell Signaling). In addition, we used beta-Actin (rabbit anti-beta-Actin, 13E5, #4970, 1:1000, Cell Signaling) as an internal loading control. All antibodies were diluted in TBS-Tw plus 1% non-fat dried milk powder. The blots were developed with Amersham ECL Prime Western Blotting Detection Reagent (GE Life Sciences) as described by the manufacturer and scanned using a Bio-Rad Scanner.
Statistical Analysis
Relative luciferase activity was expressed as the mean of three independent experiments each done in at least triplicate measures, ± SEM. Mean values between groups were compared using the two-tailed Student's t-test. Differences with a p-value ˂ 0.05 were considered statistically significant.
Results
Identification of the minimal promoter and cis-acting regulatory region of ADRB2
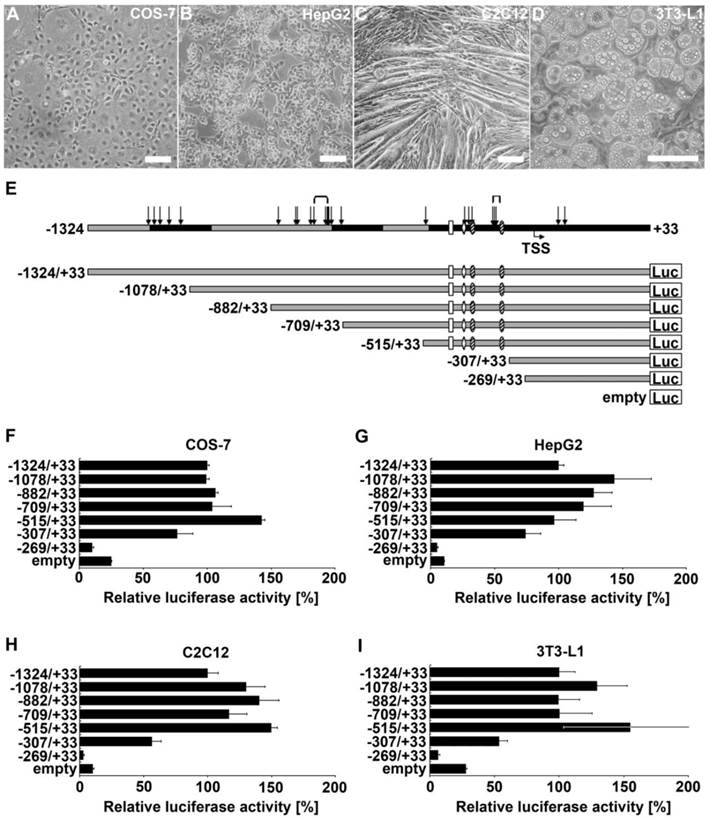
To determine which regions of the ADRB2 promoter are involved in its transcriptional regulation in myofibers, hepatocytes, and adipocytes, we performed luciferase reporter gene assays in corresponding model cell lines C2C12, HepG2, and 3T3-L1, respectively (Fig. 1B-D). In addition, we used COS-7 cells due to their high transfection efficiency and widespread use in ADRB2 promoter studies (Fig. 1A). The reporter deletion constructs were designed based on prior in silico analysis, distribution of polymorphisms, and consideration of the position of promoter elements of ADRB2 identified in other species (Fig. 1E). The relative luciferase activity of the longest ADRB2 construct (-1324/+33) in the respective cell line was set to 100%. While promoter activity of the shortest construct (-269/+33 ) was very low across all tested cell lines, promoter activity of the -307/+33 construct was 3-fold increased in COS-7, 7-fold increased in HepG2, 5-fold increased in C2C12, and 2-fold increased in 3T3-L1 compared with the empty pGL3-enhancer reporter vector. Because the -307/+33 construct appears to include regulatory elements that are able to drive the basal promoter activity in all four tested cell types, we considered the region between -307 and -269 to contain the minimal promoter of ADRB2 (Fig. 1F-I). Furthermore, the region between -515 and -307 is important for the basal promoter activity of ADRB2 and showed the strongest relative luciferase activity of approximately 150% in all tested cell types except for HepG2 cells, which showed 100% relative luciferase activity (Fig. 1F-I). After deletion of this fragment the promoter activity was decreased in COS-7 (-66%), in HepG2 (-22%), in C2C12 (-93%), and in 3T3-L1 (-101%) cells, suggesting the presence of an enhancer element in this promoter region. In contrast, deletion of the sequence between -709 and -515 increased the promoter activity in COS-7 (+39%), in C2C12 (+33%), and in 3T3-L1 (+55%) cells, indicating the presence of a repressor element in this promoter region. Additional repressor elements are proposed to be localized in the region between -1324 and -1078 because a deletion of this fragment increased the relative luciferase activity in HepG2 (+44%), in C2C12 (+30%), and in 3T3-L1 (+29%) cells.
Localization and characterization of regulatory elements in ADRB2 promoter region. (A-D) 5´ deletion constructs were analyzed with respect to their transcriptional activity in COS-7, HepG2, C2C12, and 3T3-L1 cells (phase contrast microscopy, bars = 100 µm). (E) Schematic illustration of ADRB2 deletion constructs between -1324 and +33 showing the positions of evolutionarily conserved regions (black), single nucleotide polymorphisms (SNP; arrows), microsatellites (brackets), predicted transcription start site (TSS), and putative binding sites of Sp1 (shaded oval), AP-2 (rhomb), and NF-kappaB (rectangle). (F-I) Luciferase activity of the seven 5´ deletion constructs was determined relative to the activity of the backbone vector pGL3-enhancer. Values are means ± SEM of data from at least three independent experiments, each performed in triplicate.
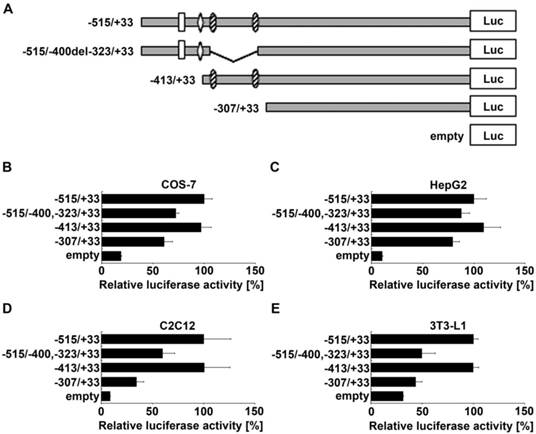
Deletion of potential Sp1 binding sites reduces ADRB2 promoter activity
To narrow down the position of enhancer elements in the region between -515 and -307 of the ADRB2 promoter, two additional deletion constructs were generated (Fig. 2A). Deletion of the GC-rich region ranging from -400 to -323, containing two predicted evolutionarily conserved Sp1 binding sites (Supplementary Table S2), reduced the ADRB2 promoter activity by about 32.2% in COS-7, 12.3% in HepG2, 40.6% in C2C12, and 53.7% in 3T3-L1 cells, which was approximately the activation level of the minimal ADRB2 promoter (corresponding to -307/+33 construct) in each cell line (Fig. 2B-E). In contrast, deletion of the sequence from -515 to -413 with potential binding sites for activating protein 2 (AP-2) and nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-kappaB), described in the rat ADRB2 promoter (1), had no effect on ADRB2 promoter activity in any of the four cell lines (Fig. 2B-E). These results show that the GC-rich region spanning from -400 to -323 of the ADRB2 promoter plays a critical role in the regulation of the basal expression of the porcine ADRB2.
Identification of specific transcription factor binding to the GC-rich enhancer element of ADRB2 promoter in vitro
We performed EMSA experiments to identify transcription factor binding to the GC-rich enhancer element located in the region from -400 to -323 of the ADRB2 promoter. In silico analysis revealed, in addition to the two predicted evolutionarily conserved Sp1 binding sites (GC-boxes), potential recognition sites for transcription factors binding GC-rich sequences such as early growth response gene 1 (Egr1), Myc-associated zinc finger protein (MAZ), and CCCTC-binding factor (CTCF) (Supplementary Table S3). Using the radiolabeled probe W2 covering the GC-rich region (-415/-304; Fig. 3A), cold W2 probe as a competitor, and nuclear extracts from COS-7, HepG2, and C2C12 cells, several specific complexes were detected; in contrast, no complex was formed in the absence of nuclear extracts (Fig. 3B). Competition and supershift assays using cold Sp1 consensus oligonucleotide as a competitor and Sp1 antibody, respectively, resulted in a competed band shift (Fig. 3B) or produced a prominent supershifted band accompanied by a considerable reduction in the shifted band (Fig. 3C). These results demonstrate that Sp1 participates in the formation of specific complexes between the W2 probe and nuclear proteins of COS-7, HepG2, and C2C12 cells, thus indicating specific binding of Sp1 to the GC-rich region of the ADRB2 promoter in vitro (Fig. 3B-C). To corroborate these results and to examine whether Sp1 specifically binds to the two evolutionarily conserved GC-boxes, cross-competition experiments were performed whereby recombinant Sp1 was incubated with radiolabeled Sp1 consensus probe, and radiolabeled probes containing the two conserved Sp1 binding sites (-403/-382; -336/-315), respectively. In all three assays a distinct shifted band was observed, which was nearly abolished after incubation with the respective cold probes as competitors (Fig. 3D). Taken together, the in silico analysis and the EMSA experiments provide strong evidence for specific binding of Sp1 to the two evolutionarily conserved GC-boxes in the GC-rich enhancer element of ADRB2 promoter.
Localization of enhancer elements in the region between -515 and -307 of the ADRB2 promoter. (A) Schematic illustration of ADRB2 deletion constructs between -515 and -307 showing the positions of putative binding sites of Sp1 (shaded oval), AP-2 (rhomb), and NF-kappaB (rectangle). (B-E) Relative luciferase activity of the four presented 5´ deletion constructs was determined in COS-7, HepG2, C2C12, and 3T3-L1 cells. Values are means ± SEM of data from at least three independent experiments, each performed in triplicate.
Electrophoretic mobility shift assays. (A) Schematic illustration of the GC-rich enhancer region (-400/-323) showing the two evolutionarily conserved Sp1 binding sites (403/-382; -336/-315), evolutionarily conserved sequence (*), single nucleotide polymorphisms (SNP; in bold letter, IUPAC code), microsatellite (in bold gray letter), and positions of EMSA probes. (B) EMSAs using nuclear extracts from COS-7, HepG2, and C2C12 cells, radiolabeled probe W2 (-415/-304) and unlabeled Sp1, Egr1, CTCF, and MAZ consensus oligonucleotides as competitors. (C) Supershift assay using nuclear extracts from COS-7, HepG2, and C2C12 cells, radiolabeled probe W2 (-415/-304) and antibodies against Sp1, Egr1, and MAZ. (D) Cross-competition experiments using recombinant Sp1 and either radiolabeled Sp1 consensus oligonucleotide, or two conserved Sp1 oligonucleotides (-403/-382; -336/-315), and the corresponding cold oligonucleotides as competitors.
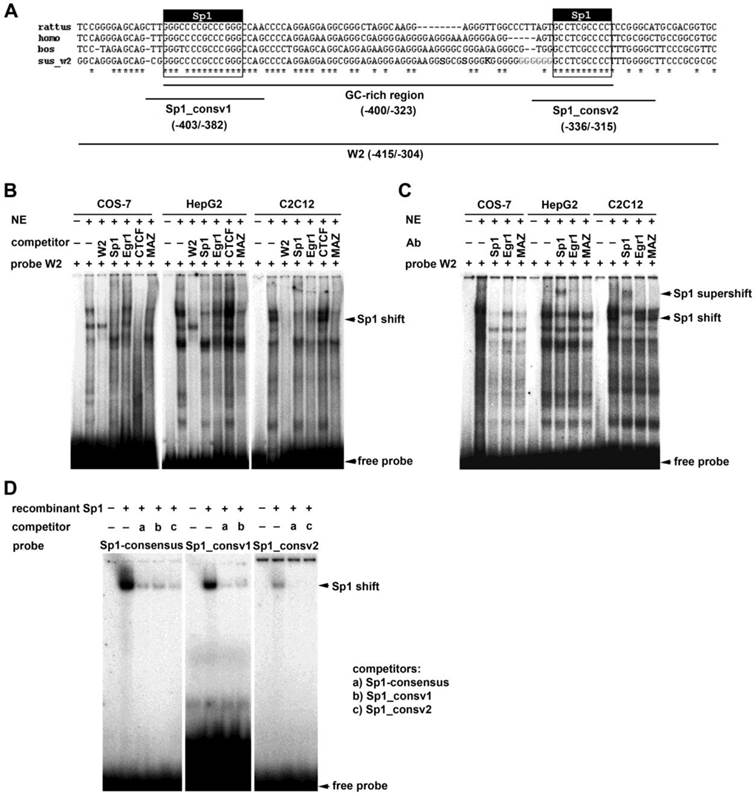
To examine binding of MAZ, CTCF, and Egr1 to the GC-rich region, we performed additional competition experiments using cold probes containing corresponding binding motifs. The MAZ competitor showed a competition profile very similar to that of the consensus Sp1 oligonucleotide (Fig. 3B). However, a supershift assay using an MAZ antibody provided no clear supporting evidence for binding of MAZ to the W2 probe (Fig. 3C). Addition of the cold CTCF consensus oligonucleotide eliminated all binding complexes of the W2 probe with nuclear proteins from COS-7 cells, but did not affect binding of nuclear proteins from HepG2 or C2C12 cells (Fig. 3B). Addition of the cold Egr1 consensus oligonucleotide or Egr1 antibody had no apparent effect on binding of nuclear proteins to the labeled W2 probe (Fig. 3B-C).
Overexpression of Sp1 increases ADRB2 promoter activity
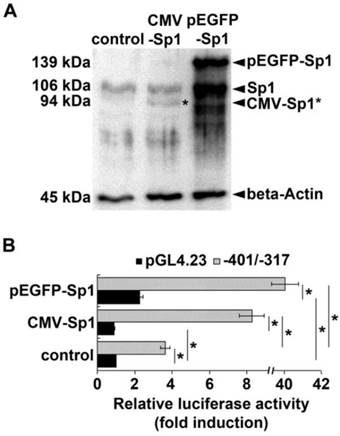
To explore if Sp1 is involved in enhancer function of the GC-rich region of the ADRB2 promoter we examined the effect of Sp1 overexpression on porcine ADRB2 promoter activity in COS-7 cells. The ADRB2 plasmid reporter construct (-401/-317) containing the GC-rich region including the two Sp1 binding sites and the corresponding empty vector pGL4.23 were co-transfected in parallel with Sp1 expression vectors cytomegalovirus (CMV)-Sp1 or enhanced green fluorescent protein (pEGFP)-Sp1. Western blotting confirmed that exogenous Sp1 was expressed in COS-7 cells after transient transfection with CMV-Sp1 as well as pEGFP-Sp1 (Fig. 4A). Luciferase reporter gene assay revealed that basal promoter activity of the -401/-317 reporter construct was 3.6-fold higher compared to the empty vector pGL4.23 (Fig. 4B), thus providing additional evidence that the GC-rich region has enhancer function. Co-transfection of either 100 ng of CMV-Sp1 or pEGFP-Sp1 expression vector significantly (p ˂ 0.05) increased promoter activity of the -401/-317 construct by 2.3- and 11-fold, respectively (Fig. 4B). In contrast, luciferase activity of the empty vector pGL4.23 was either not stimulated (0.9-fold), or was stimulated to a significantly lower extent (2.3-fold) by co-transfection with 100 ng of CMV-Sp1 or pEGFP-Sp1 expression vector, respectively (Fig. 4B). These results demonstrate that the GC-rich enhancer is indeed Sp1-responsive.
Effect of Sp1 overexpression on ADRB2 promoter activity. (A) Protein expression of endogenous Sp1 and exogenous Sp1 (pEGFP-Sp1, CMV-Sp1) was determined by Western blotting. Beta-Actin was used as internal loading control. Both pEGFP-Sp1 (139 kDa) and CMV-Sp1 (94 kDa) were detected in COS-7 cells after transient transfection. (B) Luciferase reporter gene assay shows that the promoter activity of the ADRB2 plasmid reporter construct (-401/-317) containing the GC-rich region including the two Sp1 binding sites (gray bars) was significantly higher increased by overexpression of exogenous Sp1 than the promoter activity of the empty pGL4.23 vector (black bars) when compared to control without exogenous Sp1. Values are means ± SEM of data from at least three independent experiments, each performed in triplicate, *p ˂ 0.05.
Discussion
The present study focused on functional characterization of the porcine ADRB2 promoter and provides a first catalogue of general and cell type-specific regulatory domains controlling basal expression of ADRB2 in the pig.
Reporter gene analysis of progressively truncated promoter fragments, performed with HepG2, C2C12, 3T3-L1, and COS-7 cells, revealed a lack of promoter activity for the shortest construct, -269/+33, terminating ~20 bp upstream of the predicted transcription start site (located at -248). In rat, a second, more proximal (relative to start codon) promoter and transcription start site were described in a corresponding segment (1). Our results, however, do not support the existence of a second promoter in the porcine 5´ untranslated region. Notably, promoter activity of the -269/+33 construct was even lower than the relative luciferase activity of the promoterless pGL3-enhancer vector. One explanation for this observation could be the presence of a short open reading frame (sORF) in the 5´ untranslated region (located between -112 and -53). This highly evolutionarily conserved sequence feature of ADRB2, also termed the 5´ leader cistron (5´LC), was first described in the human gene (17). Short upstream ORFs repress expression of corresponding genes by interfering with translational initiation or by triggering mRNA decay (18). In fact, mutational inhibition of the sORF increased beta-2 AR expression in vitro (10). It is thus conceivable that this element repressed expression of the downstream luciferase reporter gene and that the sORF modulates expression of the porcine ADRB2 in vivo by posttranscriptional mechanisms.
Our data suggest that the region between -307 and -269 serves as a minimal promoter that is responsible for basal promoter activity of the porcine ADRB2 gene in all four tested cell types. Accordingly, DNaseI footprinting and EMSA analyses of a corresponding regulatory domain of the rat ADRB2 revealed a CCAAT box, a typical core promoter element, and a cAMP response element (CRE) (1), which are evolutionarily conserved in the porcine sequence. The CRE is highly conserved among mammals and confers autoregulatory control of ADRB2 transcription (7). The CRE located at -275 to -268 in the human ADRB2 gene mediates the transcriptional enhancing activity of cyclic AMP (7) in response to agonists that act through the PKA signal transduction pathway (19). Thus, both elements most likely drive basal and regulated promoter activity of ADRB2 in the pig, as well.
Basal ADRB2 promoter activity was further enhanced in all used cell lines by a GC-rich segment located directly upstream of the minimal promoter. This segment, located between -400 and -323, is also able to enhance activity of a heterologous minimal promoter (minP in pGL4.23 reporter vector), which suggests that it contains a context-independent enhancer element. In silico analysis revealed that this promoter segment harbors two evolutionarily conserved Sp1 binding sites (GC-boxes) separated by a less well-conserved, repetitive sequence. The repetitive character of the sequence complicates functional characterization and precise localization of regulatory elements in this promoter segment. We confirmed the ability of the two evolutionarily conserved GC-boxes to bind Sp1 in vitro experimentally by supershift and cross-competition EMSA using nuclear extracts as well as recombinant Sp1. Moreover, we demonstrated responsiveness of the GC-rich enhancer element to Sp1 by overexpression experiments in COS-7 cells using two different Sp1 expression constructs, CMV-Sp1 and pEGFP-Sp1. Sp1 is generally considered a constitutive transcriptional activator for genes with TATA-less, GC-rich promoters, but it is also involved in the activation of distally located enhancer elements of many genes (20,21). An Sp1 binding site located in a similar position in a GC-rich segment of the rat ADRB1 promoter is required for its full activity (22). Our results demonstrate, for the first time, a similar critical role of Sp1 and the responsive GC-rich enhancer element in the regulation of ADRB2 expression in mammals. In the rat ADRB1 promoter, the Sp1 binding site partially overlaps with an Egr1 binding site, and the two transcription factors reciprocally regulate ADRB1 expression by competing for their binding sites (22). Although in silico analysis predicted Egr1 binding sites in the GC-rich segment, we found no experimental evidence confirming Egr1 binding in this region. EMSA using a probe covering the whole GC-rich element revealed, besides specific binding of Sp1, other specific complexes with nuclear proteins. Binding of additional transcription factors could be responsible for the observed differential activity of the enhancer element in different cell types. We examined binding of two additional transcription factors specific for GC-rich regions, MAZ (23) and CTCF (24). Competition experiments provided some evidence that they bind in the GC-rich segment of the porcine ADRB2 promoter in vitro. However, the results were either not supported by supershift assay (MAZ) or differed between nuclear extracts from different cell types (CTCF). Thus, further experiments are required to prove cell type-specific binding of these two transcription factors and for clarification of factors responsible for the cell type-specific activity of the GC-rich enhancer.
Using deletion mapping we also explored functional significance of a region (-515 to -413) in which AP-2 and NF-kappaB binding sites were described in the rat ADRB2 promoter (1). Our data suggest that this promoter segment is not involved in the regulation of basal activity of the porcine ADRB2 promoter. In contrast, deletion of a segment between -709 and -515 increased promoter activity in COS-7, C2C12, and 3T3-L1, but not in HepG2 cells, suggesting a cell type-specific repressor element. Phylogenetic footprinting showed that this promoter region is evolutionarily conserved in mammals; however, so far no repressor elements or cognate transcription factor were characterized at this position. An additional cell type-specific negative regulatory domain was revealed by deletion of the segment between -1324 and -1078, which increased promoter activity in HepG2, C2C12, 3T3-L1, but not in COS-7 cells. Deletion of an overlapping segment of the rat promoter (between -1122 and -911, corresponding to -1218 and -1007 in the porcine sequence) likewise increased reporter gene activity (1). However, a CCAAT-enhancer-binding protein alpha (C/EBP-alpha) binding site that has been suggested as the most likely responsible repressor element in the rat lies just outside of the overlapping region (between -1033 and -1015 in the porcine sequence), excluding this binding site as the responsible repressor element in the pig.
Taken together, regulatory elements located directly upstream of the transcriptional start site (between -400 and -269) are essential to drive maximal basal promoter activity of the porcine ADRB2 promoter. This is modulated in a cell type-specific manner by two negative regulatory domains located more distally. Human (25,26) and rat (1) ADRB2 promoters show similar functional organization.
The results obtained in the present study help to narrow the list of potential regulatory polymorphisms of the porcine ADRB2 to those clustered in the distal negative regulatory domain between -1324 and -1078, and those clustered in the GC-rich enhancer element between -400 and -323. We can essentially exclude polymorphisms located in the 5´ untranslated region and in the segment between -882 and -709 because these show no promoter activity. Since polymorphisms in the GC-rich enhancer element are associated with lower haplotype-specific expression of the porcine ADRB2, these represent the most likely causal variants. This hypothesis warrants investigation of the functional impact of polymorphisms clustered in the GC-rich enhancer element on activity of the porcine ADRB2 promoter.
Conclusions
The high degree of conservation of a large stretch (1 kb) of the 5' flanking region of the ADRB2 promoter from across species suggests that important regulatory elements are necessary for proper control of receptor expression, and have therefore been preserved.
Our results indicate that Sp1 directs basal transcription through binding to GC-rich sites of the ADRB2 promoter. The putative AP-2 and NF-kappaB binding sites are not active in the four tested cell lines. Polymorphisms of the evolutionarily conserved GC-rich element in the proximal region of the porcine ADRB2 promoter are suggested as candidate causal cis-regulatory polymorphisms responsible for tissue-specific ADRB2 expression.
Supplementary Material
Supplementary Tables S1-S3.
Acknowledgements
The authors are grateful for the excellent technical support from Angela Garve, Janine Wetzel, and Marlies Fuchs. This research was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, Grant MU2965/2-1) and by matched funding from the FBN.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jiang L, Gao B, Kunos G. DNA elements and protein factors involved in the transcription of the beta 2-adrenergic receptor gene in rat liver. The negative regulatory role of C/EBP alpha. Biochemistry. 1996;35:13136-13146
2. Hen R, Axel R, Obici S. Activation of the beta 2-adrenergic receptor promotes growth and differentiation in thyroid cells. Proc Natl Acad Sci U S A. 1989;86:4785-4788
3. Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000;43:533-549
4. Navegantes LC, Resano NM, Migliorini RH, Kettelhut IC. Catecholamines inhibit Ca(2+)-dependent proteolysis in rat skeletal muscle through beta(2)-adrenoceptors and cAMP. Am J Physiol Endocrinol Metab. 2001;281:E449-454
5. Waldeck B. Beta-adrenoceptor agonists and asthma--100 years of development. Eur J Pharmacol. 2002;445:1-12
6. Schiffelers SL, Saris WH, Boomsma F, van Baak MA. beta(1)- and beta(2)-Adrenoceptor-mediated thermogenesis and lipid utilization in obese and lean men. J. Clin. Endocrinol. Metab. 2001;86:2191-2199
7. Collins S, Altschmied J, Herbsman O, Caron MG, Mellon PL, Lefkowitz RJ. A cAMP response element in the beta 2-adrenergic receptor gene confers transcriptional autoregulation by cAMP. J Biol Chem. 1990;265:19330-19335
8. Baeyens DA, McGraw DW, Jacobi SE, Cornett LE. Transcription of the beta2-adrenergic receptor gene in rat liver is regulated during early postnatal development by an upstream repressor element. J Cell Physiol. 1998;175:333-340
9. Cornett LE, Hiller FC, Jacobi SE, Cao W, McGraw DW. Identification of a glucocorticoid response element in the rat beta2-adrenergic receptor gene. Mol Pharmacol. 1998;54:1016-1023
10. Parola AL, Kobilka BK. The peptide product of a 5' leader cistron in the beta 2 adrenergic receptor mRNA inhibits receptor synthesis. J Biol Chem. 1994;269:4497-4505
11. Tholanikunnel BG, Malbon CC. A 20-nucleotide (A + U)-rich element of beta2-adrenergic receptor (beta2AR) mRNA mediates binding to beta2AR-binding protein and is obligate for agonist-induced destabilization of receptor mRNA. J Biol Chem. 1997;272:11471-11478
12. Wang WC, Juan AH, Panebra A, Liggett SB. MicroRNA let-7 establishes expression of beta2-adrenergic receptors and dynamically down-regulates agonist-promoted down-regulation. Proc Natl Acad Sci U S A. 2011;108:6246-6251
13. Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A. 2000;97:10483-10488
14. Leineweber K, Büscher R, Bruck H, Brodde OE. Beta-adrenoceptor polymorphisms. Naunyn. Schmiedebergs. Arch Pharmacol. 2004;369:1-22
15. Muráni E, Ponsuksili S, Reyer H, Wittenburg D, Wimmers K. Expression variation of the porcine ADRB2 has a complex genetic background. Mol Genet Genomics. 2013;288:615-625
16. Muráni E, Ponsuksili S, Srikanchai T, Maak S, Wimmers K. Expression of the porcine adrenergic receptor beta 2 gene in longissimus dorsi muscle is affected by cis-regulatory DNA variation. Anim Genet. 2009;40:80-89
17. Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ. cDNA for the human beta 2-adrenergic receptor: a protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc Natl Acad Sci U S A. 1987;84:46-50
18. Barbosa C, Peixeiro I, Romão L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013:9 doi: 10.1371/journal.pgen.1003529
19. De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol. 2000;64:343-369
20. Li L, He S, Sun JM, Davie JR. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82:460-471
21. Vizcaíno C, Mansilla S1, Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol Ther. 2015 pii: S0163-7258(15)00099-6
22. Bahouth SW, Beauchamp MJ, Vu KN. Reciprocal regulation of beta(1)-adrenergic receptor gene transcription by Sp1 and early growth response gene 1: induction of EGR-1 inhibits the expression of the beta(1)-adrenergic receptor gene. Mol Pharmacol. 2002;61:379-390
23. Komatsu M, Li HO, Tsutsui H, Itakura K, Matsumura M, Yokoyama KK. MAZ, a Myc-associated zinc finger protein, is essential for the ME1a1-mediated expression of the c-myc gene during neuroectodermal differentiation of P19 cells. Oncogene. 1997;15:1123-1131
24. Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13:7612-7624
25. Scott MG, Swan C, Wheatley AP, Hall IP. Identification of novel polymorphisms within the promoter region of the human beta2 adrenergic receptor gene. Br J Pharmacol. 1999;126:841-844
26. Higgins TJ. Molecular Characterization of Beta(2) Adrenergic Receptor Haplotypes. Master Thesis, University of North Carolina, Chapel Hill, NC, USA. 2008
Author contact
![]() Corresponding author: Institute for Genome Biology, Leibniz-Institute for Farm Animal Biology, Wilhelm-Stahl-Allee 2, D-18196 Dummerstorf, Germany. Tel.: ++49 (0) 3820868726; fax: ++49 (0) 3820868702; E-mail address: muranide
Corresponding author: Institute for Genome Biology, Leibniz-Institute for Farm Animal Biology, Wilhelm-Stahl-Allee 2, D-18196 Dummerstorf, Germany. Tel.: ++49 (0) 3820868726; fax: ++49 (0) 3820868702; E-mail address: muranide
Received 2015-4-20
Accepted 2015-6-7
Published 2015-7-3