Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(9):1016-1025. doi:10.7150/ijbs.11751 This issue Cite
Research Paper
Evolutionary History of Cathepsin L (L-like) Family Genes in Vertebrates
Jin Zhou1,2,3#, Yao-Yang Zhang4#, Qing-Yun Li4, Zhong-Hua Cai1,2,3 ![]()
1. The Division of Ocean Science & Technology, Graduate School at Shenzhen, Tsinghua University, Shenzhen, 518055, P. R. China
2. Shenzhen Public Platform of Screening & Application of Marine Microbial Resources, Graduate School at Shenzhen, Tsinghua University, Shenzhen, 518055, P. R. China
3. Shenzhen Key Laboratory for Coastal Ocean Dynamic and Environment, Graduate School at Shenzhen, Tsinghua University, Shenzhen, 518055, P. R. China
4. School of Life Science, Tsinghua University, Beijing, 100084, P. R. China
# These two authors contributed equally to this work.
Received 2015-2-2; Accepted 2015-5-22; Published 2015-7-14
Abstract

Cathepsin L family, an important cysteine protease found in lysosomes, is categorized into cathepsins B, F, H, K, L, S, and W in vertebrates. This categorization is based on their sequence alignment and traditional functional classification, but the evolutionary relationship of family members is unclear. This study determined the evolutionary relationship of cathepsin L family genes in vertebrates through phylogenetic construction. Results showed that cathepsins F, H, S and K, and L and V were chronologically diverged. Tandem-repeat duplication was found to occur in the evolutionary history of cathepsin L family. Cathepsin L in zebrafish, cathepsins S and K in xenopus, and cathepsin L in mice and rats underwent evident tandem-repeat events. Positive selection was detected in cathepsin L-like members in mice and rats, and amino acid sites under positive selection pressure were calculated. Most of these sites appeared at the connection of secondary structures, suggesting that the sites may slightly change spatial structure. Severe positive selection was also observed in cathepsin V (L2) of primates, indicating that this enzyme had some special functions. Our work provided a brief evolutionary history of cathepsin L family and differentiated cathepsins S and K from cathepsin L based on vertebrate appearance. Positive selection was the specific cause of differentiation of cathepsin L family genes, confirming that gene function variation after expansion events was related to interactions with the environment and adaptability.
Keywords: cathespin L family gene, evolution, positive selection, functional divergence, environmental adaptability
Introduction
“Cathepsin” originated from the Greek word “katahepsein”, which means “to digest”. Cathepsin L superfamily is a multifunctional cysteine protease enzyme and widely distributed in most animals. Approximately 11 cysteine proteases (cathepsins B, C, F, H, K, L, O, S, V, X, and W), 2 aspartic proteases (D and E), and 1 serine protease (G) have been recognized [1]. Cathepsins are approximately 30 kDa in size and comprise disulfide-linked heavy and light chains [2]. These proteins slightly differ in their amino acid composition and length, but all of them evolved from the same ancestral gene and use a similar mechanism for protein degradation.
As multifunctional enzymes, cysteine cathepsins widely exist particularly in lysosomes. Cathepsins B and B-like proteases are identified in various species [3]. Cathepsins B-like and L-like cysteine proteases are found in Caenorhabditis elegans [4, 5]. Similar proteases are also detected in some invertebrates [6]. Most proteomic research and related studies on cysteine cathepsins have focused on vertebrates, particularly mammals, including primates and rodents [7, 8]. All 11 cysteine cathepsins are detected in Homo sapiens (human) through a bioinformatic study on the human genome [9]. Rodents contain 10 cysteine cathepsins and carry additional genes that express other cathepsins and cathepsin-like proteins [10]. Moreover, several cathepsins and cathepsin-like proteases are revealed through functional and structural analyses in fishes, amphibians, reptiles, and birds in addition to mammals [11].
Currently, cysteine cathepsins should not be solely considered as lysosomal proteases because they are also found in other cellular compartments. These cathepsins participate in many biological processes in addition to protein turnover. The isoforms of cathepsin L are detected in the nucleus and function as a regulator of cell cycle by cutting the histone H3's N-terminus tail [12]. In zebrafish, a cathepsin L variant is involved in developing fish embryos [13]. A series of cathepsin L-like proteases is also discovered in rodents; these proteases perform specific roles in gestation [14]. Cysteine cathepsins are significant signaling molecules and vital regulators in physiological events, as indicated by the experimental evidence accumulated. Their nonendosomal functions also become highly fascinating.
In the field of classification, although most mature enzymes share highly homologous amino acid sequences, their motifs significantly differ on the basis of the sequence analysis in the proregion. Two distinct groups, namely, cathepsin L-like group (cathepsins F, H, K, L, S, V, and W) and cathepsin B-like group, have been classified. The two groups differ in proregion and mature peptide sequence. In cathepsin L subfamily, the propeptide comprises 100 residues and 2 conserved motifs, namely, ERFNIN and GNFD. In cathepsin B subfamily, the propeptide is approximately 60 residues in length and contains the GNFD motif only [15-17].
Evidence shows that cathepsin L family diverges from cathepsin B family even earlier than the differentiation between cysteine cathepsin-like proteases in plants and lower species [18]. Cathepsin L family contains several groups, including cathepsins L and V, S and K, and F and W. Gene localization in chromosomes and sequence analysis reveal that cathepsin H diverges early from cathepsin L family ancestors [19].
Among endopeptidase cysteine proteases, cathepsin L is an important family because of its multifunctional role in many biochemical pathways, including intracellular protein degradation, antigen presentation, and cellular development [20-22]. Although relatively detailed information has been accumulated regarding the structure and function of this enzyme, only fragmentary data are presently available with regard to the evolutionary relationship among vertebrate species. Thus, we combined phylogenetic analysis, selective pressure prediction, relative evolution tests, and functional divergence to interpret the evolutionary process of cathepsin L-like superfamily. This study aimed to provide novel insights into the origin and evolutionary fates of this gene family.
Methods
Sequence source
The protein sequences of cathepsin L family (B, H, K, L, S, V, and W) of 22 species were accessed in the National Center for Biotechnology Information (NCBI) GenBank database or ENSEMBL and University of California, Santa Cruz genome browsers, and the matching cDNA sequences were acquired [23-25]. The retrieved genomes belonged to H. sapiens (human), Pan troglodytes (chimpanzee), Macaca mulatta (macaque), Rattus norvegicus (rat), Mus musculus (mouse), Oryctolagus cuniculus (rabbit), Cavia porcellus (guinea pig), Canis familiaris (dog), Sus scrofa (pig), Bos taurus (cow), Ornithorhynchus anatinus (platypus), Monodelphis domestica (opossum), Gallus gallus (chicken), Anolis carolinensis (lizard), Xenopus tropicalis (frog), Takifugu rubripes (fugu), Oryzias latipes (medaka), Gasterosteus aculeatus (stickleback), Danio rerio (zebrafish), Petromyzon marinus (lamprey), Branchiostoma floridae (lancelet), and Ciona intestinalis (ciona). All assembly genomes were retrieved using the basic local alignment search tool (BLAST) or BLAST-like alignment tool from the NCBI GenBank database or ENSEMBL. The genomes were manually checked and edited. All acquired cDNA sequences were converted to amino acid sequences by using EMBOSS Transeq (http://www.ebi.ac.uk/Tolls/emboss/transeq/index.html).
Gene alignment and phylogenetic analysis
A total of 114 annotated cathepsin L family amino acid sequences from 11 species (we selected 11 representative species from the total 22 species) were aligned using ClustalX v1.83 [26] and then manually adjusted to optimize the alignment. Prottest [27] suggested that the phylogenetic relationship of these sequences can be constructed using Bayesian inference [28] and maximum-likelihood methods under a WAG+I substitution model. In Bayesian inference, Metropolis-coupled Monte Carlo-Markov chain (MC3) searches were performed using three incrementally heated chains and one cold chain in two parallel runs for 1 million generations with distinct random initial trees. Sampling frequency was set every 100 generations. After a burn-in of 2,500 generations, MC3 was removed, and the posterior probabilities were estimated. A maximum-likelihood tree was built using PHYML v3.0 [29], with clade supports assessed at 100 bootstrap replicates. Another two methods, namely, neighbor joining and maximum parsimony, for phylogenetic tree construction were used to build trees with MEGA v4.0 [30] in the Poisson correction model and to assess the clade support with 1,000 bootstrap replicates.
Selective pressure analysis
We performed a site-based analysis with the Codeml program within the PAML v4.3 package to investigate the selective pressure on cathepsin L family in mammals [31, 32]. The program utilized the maximum-likelihood approach to detect selection events. We aligned 47 full-cDNA sequences of the mammal cathepsin L family genes with PAL2NAL [33], whereas the corresponding protein sequences were aligned using ClustalX [26]. The in-tree used was retrieved from the Bayesian inference with the corresponding protein sequences. Evolution of these sequences was evaluated using the ratio of nonsynonymous (dN) and synonymous (dS) substitution rates (dN/dS=ω) as a parameter. We conservatively estimated that ω<1 is the purifying or negative selection, ω=1 is the neutral evolution and ω>1 is in accordance with the positive (Darwinian) selection. In practice, likelihood ratio test (LRT) was conducted to detect codon sites with ω>1 and lineage specificity of ω. LRT required two comparison models, including the null hypothesis pattern. The log-likelihood difference between the null and alternative models was evaluated twice from χ2 distribution. Thus, χ2 test can be applied with degrees of freedom (df) corresponding to the differences in the free parameter numbers between the two paired models. Site-specific models were calculated with discrete model M3, selection model M2a, neutral null model M1a, beta and ω model M8, and beta null model M7; each model was compared with one-ratio null model M0. Branch-specific models were represented with a free ratio model and a one-ratio null model M0.
Structure analysis and putative positively selected sites
The template protein of the H. sapiens cathepsin L1 [Protein Data Bank (PDB accession number 2YJC http://www.rcsb.org/pdb/explore/explore.do?structureId=2YJC] was downloaded from the PDB website (http://www.rcsb.org/pdb/home/home.do). The models were visualized and subjected to positive selection site determination through PyMOL (http://www.pymol.org). ClustalW [34] was utilized to align sequences with strong positive selection sites. The result was presented with GeneDoc (http://www.nrbsc.org/gfx/genedoc/).
Results
Chromosomal location of cathepsin L family genes
All 22 species contained at least one copy of cathepsin L or L-like (Table 1). Cathepsin L family was retained and expanded from a common ancestor, which indicated that cathepsin L in zebrafish, cathepsins S and K in xenopus, and cathepsin L1 in rats and mice underwent severe tandem-repeat events. Most genes in the tandem-repeat regions in rats and mice were arranged in similar orientation, which suggested that most tandem repeat regions resulted from recent gene duplication. Cathepsin V (L2) was found only in eutherian mammals and always appeared in the near site, with cathepsin L (L1) at the same chromosome (Supplementary Table S1). Cathepsins S and K interlocked on the same strand at the same chromosome in most of the vertebrates, whereas cathepsins S and K sequences were not found in ciona, lancelet, and lamprey (Supplementary Table S1). Cathepsin H contained 12 exons, whereas cathepsins L, V, S, and K comprised only 8 exons. This finding suggested that cathepsin H may diverge earlier from cathepsin L family than the other family members (Supplementary Table S2).
The main gene sequences of cathepsin L and L-like family.
| Cathepsin L-like family sequences by species | ||
|---|---|---|
| Class Mammalia | Species | Sequences |
| Human (Hsa) | 5 | |
| Chimpanzee (Ptr) | 5 | |
| Macaque (Mmul) | 6 | |
| Mouse (Mmu) | 15 | |
| Rat (Rno) | 15 | |
| Guinea pig (Cpo) | 4 | |
| Rabbit (Ocu) | 6 | |
| Pig (Sus) | 7 | |
| Cow (Bta) | 5 | |
| Dog (Cfa) | 6 | |
| Opossum (Mdo) | 8 | |
| Platypus (Oan) | 6 | |
| Aves | Chicken (Gga) | 6 |
| Reptilia | Lizard (Aca) | 6 |
| Amphibia | Frog (Xru) | 16 |
| Actinopterygii | Fugu (Tru) | 6 |
| Medaka (Ola) | 6 | |
| Stickleback (Gac) | 8 | |
| Zebrafish (Dre) | 21 | |
| Agnatha | Lamprey (Pma) | 6 |
| Cephalochordata | Lancelet (Fr1) | 9 |
| Urochordata | Ciona (Cin) | 5 |
| Total | 22 | 177 |
Phylogenetic analysis
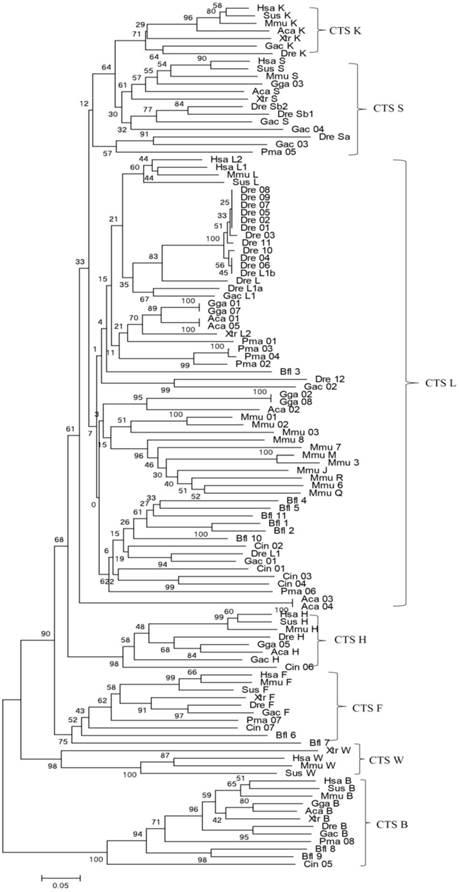
Bayesian inference and maximum-likelihood methods were used to build a phylogenetic tree (Fig. 1). From the phylogenetic tree, the evolutionary order was as follows: cathepsins B, W, F, H, L, and L-like members (S and K). Among them, cathepsin B appeared earliest and as an out-group. The genes of cathepsins S, K, L, V, and H were clustered into independent clades, thereby demonstrating the evolutionary sequence of this family. In each clade, gene evolution was generally consistent with the species evolution order. Cathepsin H diverged earlier from the L family than cathepsins S and K. Considering that a similar protein Cin05 was found in cathepsin H clade, we inferred that cathepsin H diverged from the L family earlier than the appearance of chordate. Cathepsins S and K appeared and diverged from the L family after the emergence of vertebrates. In consideration of the interlocking of cathepsins S and K and the evolutionary tree, these cathepsins stemmed from the same ancestor and diverged because of duplication and mutation events. Cathepsins S and K analogs, such as Pma05, existed in lamprey; hence, these cathepsins possibly originated from the ancestor of vertebrates.
Phylogenetic tree of cathepsin L-like family. The phylogeny of 114 cathepsin L-like family genes from other species was constructed using MrBayes. Numbers at nodes are posterior probabilities from Bayesian inference. Aca (Anolis carolinensis, Lizard), Bfl (Branchiostoma floridae, Lancelet), Ciona (Ciona intestinalis, vase tunicatea), Dre (Danio rerio, Zebrafish), Gac (Gasterosteus aculeatus, Stickleback), Gga (Gallus gallus, Chicken), Hsa (Homo sapiens, Human), Mmu (Mus musculus, Mouse), Pma (Petromyzon marinus, Lamprey), Sus (Sus scrofa, Pig), and Xtr (Xenopus tropicalis, Frog).
Selection analysis
We performed site-specific and branch-specific model analyses with PAML to identify the selective pressure on cathepsins L1 and L2 in eutherian mammals. According to the site-specific models of LRT, the discrete model M3 was notably higher than the one-ratio model M0 (2ΔlnL=1569.28, p<0.001, df=4), whereas the beta and ω model M8 was significantly higher than the beta-null M7 (2ΔlnL=74.72, p<0.001, df=2) (Table 2). These findings indicated a distinct heterogeneous selection among amino acid sites. The log-likelihood values of M1a and M2a models were equal (2ΔlnL=0). The model M3 exhibited three types of sites with values of 0.05, 0.43, and 1.23, which suggested that specific amino acid sites underwent positive selection. Thus, positive selection can be assumed from the single sites of 47 cathepsin L family genes.
Results of LRT for selection of cathepsin L-like family in vertebrates.
| Model | np | estimates of parameters | lnL | LRT pairs | df | 2flnL | positively selected sites (BEB) |
|---|---|---|---|---|---|---|---|
| M0:one ratio | 1 | ω0:one | -21586.13 | ||||
| M3:discrete | 5 | p0=0.31,p1=0.46,p2=0.23,p0=0.05,p1=0.43,ω2=1.23 | -20801.49 | M0/M3 | 4 | 1569.28*** | |
| M1a:neutral | 2 | p0=0.56,p1=0.44,p0=0.17,p1=1.00 | -20956.12 | ||||
| M2a:selection | 4 | p0=0.56,p1=0.34,p2=0.10,p0=0.17,p1=1.00,p2=1.00 | -20956.12 | M1a/M2a | 2 | 0 | 3 site p<0.01: 159Q,284E,337E;4 site p<0.05: 238S, 260K, 291E, 305D |
| M7:beta | 2 | p=0.48,q=0.63 | -20817.70 | ||||
| M8:beta&0 | 4 | p0=0.92,p=0.56,q=0.88,(p1=0.08),ω0.08) | -20780.34 | M7/M8 | 2 | 74.72*** | 8 site p<0.01:159Q, 202E, 238S, 260K,284E, 291E,305D,337E; 3 site p<0.05: 211Y,359A,364T |
| Fr:free ratios | 92 | see Figure | -21416.49 | M0/Fr | 91 | 339.28*** |
Selection analysis by three types of models was performed using Codeml implemented in PAML. np: number of free parameters, InL: log likelihood. LRT: likelihood ratio test. df: degrees of freedom. 2∆LnL: twice the log-likelihood difference of the models compared. The significant tests at 5% cutoff are labeled with *, and those at 1% cutoff are labeled with ***.
Given that positive selection does not affect all amino acid sites through prolonged time, it may only work in specific stages of evolution or in specific sites. Thus, a branch-specific model was utilized to determine the positive selection that works on specific branches. The free-ratio model was distinctly higher than the one-ratio model M0 (2ΔlnL=339.28, p<0.001, df=91) (Table 2), which suggested a heterogeneous selection among these branches. From the 91 branches of the analyzed phylogeny, 13 branches exhibited ω>1 (Fig. 2), which was a strong evidence for positive selection; the highest ω values were observed in branches A (Ocu01 in rodent; ω=infinite) and B (human HsaL2; ω=infinite). The estimated numbers of nonsynonymous (N*dN) values in A and B were 16.7 and 2.2, respectively; the estimated synonymous (S*dS) changes were zero for each branch (Table 2).
Selection of cathepsin L-like family estimated by the free ratio model. Branches with ω > 1 are shown as thick lines. The estimated ω ratios are given above the branches and numbers of nonsynonymous, and synonymous changes are given under the branches. Bta (Bos Taurus, Cow), Cfa (Canis familiaris, Dog), Cpo (Cavia porcellus, Guinea pig), Hsa (Homo sapiens, Human), Mmu (Mus musculus, Mouse), Ocu (Oryctolagus cuniculus, Rabbit), Ptr (Pan troglodytes, Chimpanzee), Rno (Rattus norvegicus, Rat), and Sus (Sus scrofa, Pig).
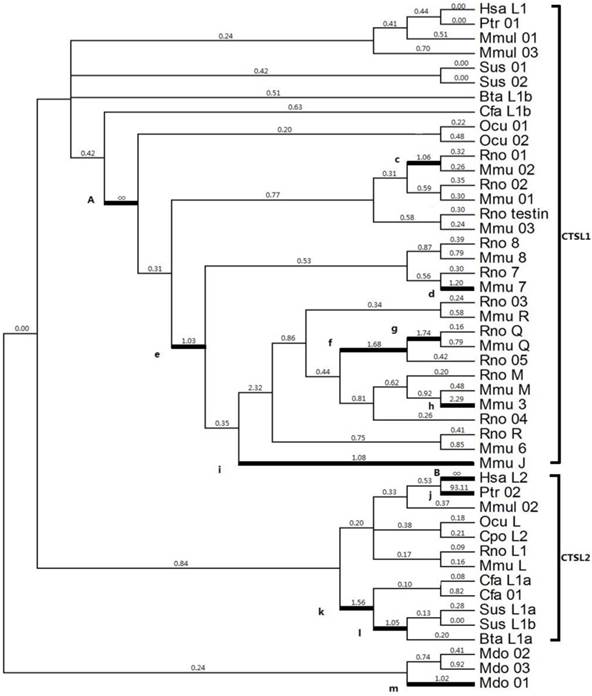
According to M2a and M8 models, only 8% to 10% of the sites underwent positive selection. Naive empirical Bayes and empirical Bayes methods were used to calculate the posterior probability of the sites that underwent positive selection (Table 2). Seven sites (159Q, 238S, 260K, 284E, 291E, 305D, and 337E) were identified as positively selected sites with p<0.05 through both models (M2a and M8). Three sites (159Q, 284E, and 337E) exhibited p<0.01, which was a strong indication of the positive selection for the seven amino acids. Another three sites (211Y, 359A, and 364T) were identified through M8. However, conservation played a major role in the evolution of the eutheria cathepsin L because most branches presented values with ω<1, indicative of negative selection.
Structure analysis and putative positively selected sites
Given that the spatial structure of cathepsin L family members is highly conserved, we considered the protein human (H. sapiens) cathepsin L1 as the template to show the positive selection sites. The mature protein is composed of two domains, namely, the left (L-) and right (R-) domains [3]. Each domain contains two loops, and these four loops form the active-site surface of the enzyme (Figs. 3A and 3B).
Protein structure of cathepsin L-like family. (a) Model of CL protein based on homology modeling. (b) Positions of type-1 sites in the model. Type-1 sites are shown as spheres; SRS, red; helix F-G, green. (c) Positions of type-II sites in the model. Type-II sites are shown as spheres colored as in (B). (D) Example of multialignment of CL family amino acid sequences. Conserved sites are shaded, and the meaning of each symbol is given in the box.
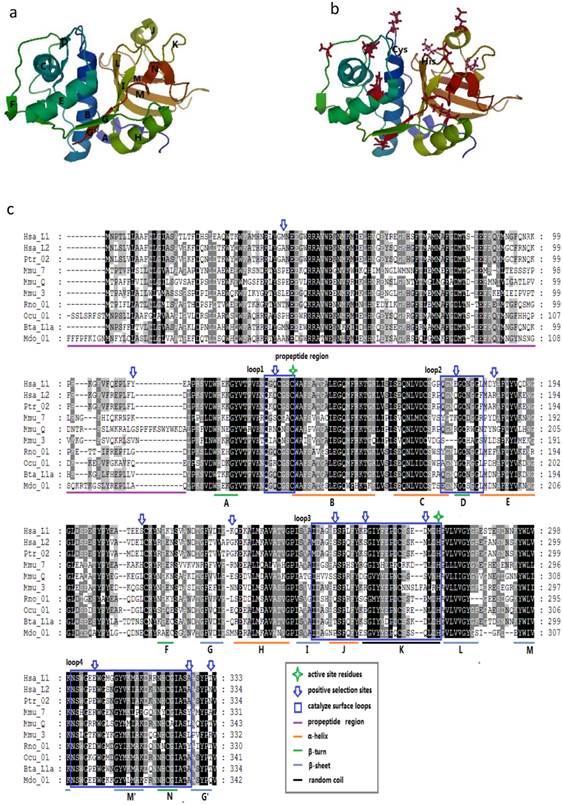
We mapped the sites in the enzyme structure and in the sequence alignment to confirm some insights into the positive selection sites (Fig. 3C). In M3, M2a, and M8 models, most of the positively selected sites appeared in the mature enzyme, whereas M3 showed a few sites in the propeptide region. These results suggested the presence of positive selection pressure in the propeptide of the family members. Moreover, seven positively selected sites were detected on the four loops of the active-site surface. These sites were unevenly distributed in the four loops, and most of them were concentrated along loops 3 and 4. In the entire enzyme, these sites approximately emerged between two secondary structures, including 159Q between A β-turn and B α-helix, 284E between I β-sheet and J α-helix, and 337E between M β-sheet and M' β-sheet (Figs. 3A and 3B). This finding indicated that most of these sites may slightly change the spatial structure but do not enormously modify the secondary structure. The critical amino acids of the active-site residues, such as Cys in loop 1 and His in loop 3, were not located under positive selection. Therefore, the overall spatial structure and the major function of the family members were highly conserved.
Discussion
Cathepsins are lysosomal proteases that are remarkably widespread and present in most animals. They are primarily defined as “digestive enzymes”, which can catalyze hydrolysis of many proteins with different specificities and play an important role in intracellular protein degradation [2]. These molecules slightly differ in their amino acid composition and length, but all of them use a similar mechanism for protein degradation [11]. The increasing research on cathepsins has revealed many new characteristics, such as expression patterns and functional differentiation, regarding the gene family. Some cathepsins (e.g., cathepsins B, H, and L) are ubiquitously expressed, whereas other enzymes (e.g., cathepsins S and K) show tissue or cell specificity. In terms of biological functions, except digestive role, cathepsins exhibit an array of new functions, including antigen presentation [1], apoptosis [35], bone resorption [36], proenzyme and hormone processing [37], sperm maturation [38], and general protein degradation and turnover [39]. From the evolutionary biology perspective, these changes or functional fates may be related to the origin and evolutionary process.
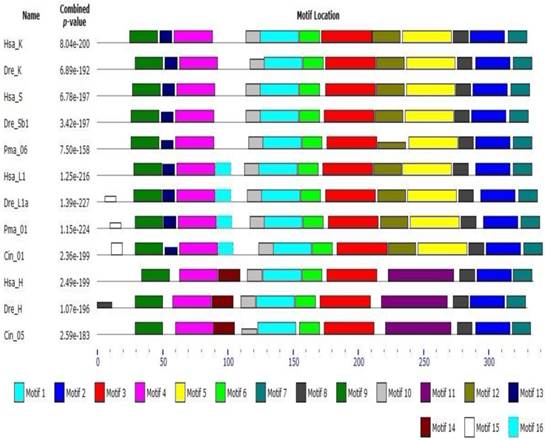
Early studies have shown that cathepsin L superfamily originated during eukaryotic evolution and may predate the eukaryote/prokaryote divergence [15]. Sequence alignment and phylogeny construction demonstrate that cathepsin L family diverges from cathepsin B family even earlier than the differentiation between plants and fungus [40]. Berti [41] confirmed that the L, S, and K members of cathepsin L family evolved from a common ancestral gene prior to mammalian divergence; the sequence conservation among the orthologs of different mammals was higher than that among the paralogs. In the present work, phylogenetic analysis of cathepsin L family members showed that cathepsins H, S, K, and V chronologically diverged from cathepsin L with an order of evolution as B, W, F, H, L, S, and K (Fig. 1). The results were consistent with those of earlier studies [11, 41]. Classification analysis showed that cathepsin L-like family can be classified into cathepsins L, V, S, K, H, F, and W [15]. The different genes among the members exhibited different evolutionary speeds and individual features. Cathepsin F presented a longer propeptide than the other genes in cathepsin L family, and the mature enzymes shared similar structure to the other members. BLAST whole genome of cathepsins L and F in some species (nematode, fruitfly, zebrafish, and human) revealed that they shared different counterparts in these organisms. This finding indicated that cathepsin F diverged from cathepsin L earlier than H, V, S, and K. Cathepsin H contained 12 exons in ciona (Ciona intestinalis), whereas cathepsins S, K, L, and V contained only 8 exons (Supplementary Table S2). Thus, cathepsin H diverged from cathepsin L within or prior to chordate appearance. Cathepsins S and K originated from cathepsin L-like family on the basis of chromosomal localization analysis. The evolutionary tree (Fig. 1) demonstrated that cathepsins S and K diverged from cathepsin L after the appearance of vertebrate. The lamprey (P. marinus) gene Pma06 clustered in cathepsins S and K gene clades, which suggested that these cathepsins possibly originated from the ancestors of jawless vertebrates. Motif analysis provided further evidence for the functional differentiation among cathepsins (Fig. 4). The motif test program MEME showed that Pma06 shared a distinct motif with cathepsins S and K, whereas the motifs of cathepsins L and H were apparently different.
Motif analysis of cathepsin L family genes. Motif type and length are represented by different colors and box sizes.
Gene duplications in particular gene families are regarded as an important source of evolutionary novelties that contribute to innovative phenotypic traits and biological functions [42]. With possible relevance to biological requirements, genes encoding digestive proteases are remarkably amplified through gene duplication in some vertebrates [43]. For example, cathepsin K gene is highly expressed in osteoclasts and plays an essential role in bone resorption [44], whereas cathepsin S gene is prevalently expressed in antigen-presenting cells and participates in adaptive immunity processes, such as major histocompatibility complex-II-mediated antigen presentation [21]. These results indicate that functional divergence occurs during the evolutionary process of cathepsin genes, which could be attributed to chromosome replication or gene duplication (Fig. 2). Kutsukake et al. (2008) showed that gene duplication and accelerated molecular evolution comprise a general and important evolutionary process that enables the acquisition of novel functions [45]. In the present study, cathepsin L in zebrafish, cathepsins S and K in xenopus, and cathepsin L1 in mice and rats underwent severe tandem duplications (Figs. 2 and 3), which was in accordance with Kutsukake's notion [45]. During the tandem-repeat events of cathepsins, the products are differentially regulated spatially and temporally and can perform various unique functions [40]. Rispe et al. (2008) also demonstrated that the dynamic evolutionary patterns of cathepsin L genes are probably relevant to the relaxed functional constraints in the multigene family; these constraints were generated through massive gene amplification in vertebrates [43]. In addition, the functional divergence of cathepsin L gene coincides with the structure formation of the ancestors of vertebrates (such as endoskeleton appearance and immune system occurrence) [46]. From the perspective of Darwin's evolution theory, gene duplication and functional fates may be an acquired mechanism of environmental adaptability under long-term evolution. Similarly, Thomas (2007) revealed that phylogenetic genes exhibit accessory functions associated with unstable environmental interactions [47].
Selection pressure is a powerful force and a universal phenomenon during evolution and contributes to the functional stability of genes [48]. Selection is categorized into three methods (positive, negative, and neutral), and four main types (stabilizing selection, directional selection, disruptive selection, and balancing selection) [49]. Among these methods, positive interactions have received more attention. This work aimed to determine whether duplication and differentiation of cathepsin L genes underwent enormous environmental change, particularly Darwinian selection. Positive selection was detected using the branch-specific model in cathepsin L family in rodents. The results showed that cathepsin L family was subjected to positive selection during the course of their evolution (Fig. 1), which indicated that recent environment changes may specifically affect the gene evolution of rodents. Hence, positive selection induced functional diversity and stability within cathepsin L family members. Similar evolutionary patterns were detected in the primate cathepsin V; this finding may coincide with the special function of cathepsin V. In addition, cathepsin family members exhibited accelerated molecular evolution caused by positive selection among molecules. Some researchers have reported that positive selection plays important roles in functional divergence [50], gene fitness and stability [51], and purification [48]. These results can provide a basis that cathepsins S and K specifically originated with the appearance of vertebrates, and positive selection contributed to the sequence diversity and functional stability in cathepsin L superfamily.
Conclusions
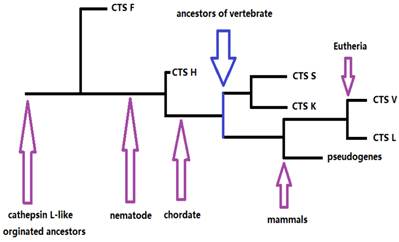
Our results provided information on the phylogeny and functional divergence of cathepsin L-like family. Cathepsin L family genes originated from successive evolutionary events (such as those shown in the simplified chart in Fig. 5). The possible evolutionary order was F, H, S, K, and L. Gene duplication and accelerated molecular evolution may play potential roles in gene evolutionary history and functional diversity formation. Positive selection was the main force driving gene stability and environmental adaptability. Overall, this work provided an evolutionary view of cathepsin L families, thereby facilitating further functional analyses and elucidating cathepsin L family genes within the vertebrate lineage.
The skeleton of evolutionary process of cathepsin L (L-like) family.
Supplementary Material
Tables S1-S2.
Acknowledgements
This study was supported by Marine Public Welfare Project of China (201305021), National Natural Science Foundation of China (41476092), S&T Project of Shenzhen Science and Technology Innovation Committee (JSGG20140519113458237), S&T Plan of Economy, Trade and Information Commission of Shenzhen Municipality (NYSW20130329010054), and National Natural Science Foundation of Guangdong Province (2014A030313774).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zavasnik-Bergant T, Tur B. Cystiene cathepsins in the immune response. Tissue Antigens. 2007;67(5):349-55
2. Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20(17):4629-33
3. Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta. 2000;1477(1):98-111
4. Jasmer DP, Roth J, Myler PJ. Cathepsin B-like cysteine proteases and Caenorhabditis elegans homologues dominate gene products expressed in adult Haemonchus contortus intestine. Mol Biochem Parasitol. 2001;116(2):159-69
5. Ultaigh SN, Carolan JC, Britton C, Murray L, Ryan MF. A cathepsin L-like protease from Strongylus vulgaris: an orthologue of Caenorhabditis elegans CPL-1. Exp Parasitol. 2009;121(4):293-99
6. Hu X, Hu X, Hu B, Wen C, Xie Y, Wu D. et al. Molecular cloning and characterization of cathepsin L from freshwater mussel, Cristaria plicata. Fish Shellfish Immun. 2014;40(2):446-54
7. Fonović M, Turk B. Cysteine cathepsins and their potential in clinical therapy and biomarker discovery. Proteom Clin Appl. 2014;8(5-6):416-26
8. Shahinian H, Tholen S, Schilling O. Proteomic identification of protease cleavage sites: cell-biological and biomedical applications. Expert Rev Proteomic. 2013;10(5):421-33
9. Rossi A, Deveraux Q, Turk B, Sali A. Comprehensive search for cysteine cathepsins in the human genome. Biol Chem. 2004;385(5):363-72
10. Conus S, Simon HU. Cathepsins and their involvement in immune responses. Swiss Med Wkly. 2010;140:w13042
11. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B. et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824(1):68-88
12. Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF. et al. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell. 2008;135(2):284-94
13. Tingaud-Sequeira AL, Cerdà J. Phylogenetic relationships and gene expression pattern of three different cathepsin L (Ctsl) isoforms in zebrafish: Ctsla is the putative yolk processing enzyme. Gene. 2007;386(1-2):98-106
14. Song G, Bailey DW, Dunlap KA, Burghardt RC, Spencer TE, Bazer FW. et al. Cathepsin B, cathepsin L, and cystatin C in the porcine uterus and placenta: potential roles in endometrial/placental remodeling and in fluid-phase transport of proteins secreted by uterine epithelia across placental areolae. Biol Report. 2010;82(5):854-64
15. McDonald JK. An overview of protease specificity and catalytic mechanisms: aspects related to nomenclature and classification. Histochem J. 1985;17(7):773-85
16. Guo YL, Kurz U, Schultz JE, Lim CC, Wiederanders B, Schilling K. The alpha1/2 helical backbone of the prodomains defines the intrinsic inhibitory specificity in the cathepsin L-like cysteine protease subfamily. FEBS Lett. 2000;469(2-3):203-07
17. Dacks JB, Kuru T, Liapounova NA, Gedamu L. Phylogenetic and primary sequence characterization of cathepsin B cysteine proteases from the oxymonad flagellate Monocercomonoides. J Eukaryot Microbiol. 2008;55(1):9-17
18. Wex T, Levy B, Wex H, Brömme D. Human cathepsins W and F form a new subgroup of cathepsins that is evolutionary separated from the cathepsin B- and L-like cysteine proteases. Adv Exp Med Biol. 2000;477:271-80
19. Fontanesi L, Davoli R, Yerle M, Zijlstra C, Bosma AA, Russo V. Regional localization of the porcine cathepsin H (CTSH) and cathepsin L (CTSL) genes. Anim Genet. 2001;32(5):321-23
20. Zwad O, Kübler B, Roth W, Scharf JG, Saftig P, Peters C. et al. Decreased intracellular degradation of insulin-like growth factor binding protein-3 in cathepsin L-deficient fibroblasts. FEBS Lett. 2002;510(3):211-15
21. Maehr R, Hang HC, Mintern JD, Kim YM, Cuvillier A, Nishimura M. et al. Asparagine endopeptidase is not essential for class II MHC antigen presentation but is required for processing of cathepsin L in mice. J Immunol. 2005;174(11):7066-74
22. Morin V, Sanchez-Rubio A, Aze A, Iribarren C, Fayet C, Desdevises Y. et al. The protease degrading sperm histones post-fertilization in sea urchin eggs is a nuclear cathepsin L that is further required for embryo development. PLoS One. 2012;7(11):e46850
23. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215(3):403-10
24. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM. et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996-1006
25. Birney E, Andrews TD, Bevan P, Caccamo M, Chen Y, Clarke L. et al. An overview of ensembl. Genome Res. 2004;14(5):925-28
26. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876-82
27. Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104-05
28. Ronquist F, Huelsenbeck J. MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572-74
29. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696-704
30. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596-99
31. Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13(5):555-56
32. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586-91
33. Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609-W612
34. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673-80
35. Stoka V, Turk B, Turk V. Lysosomal cysteine proteases: structural features and their role in apoptosis. IUBMB Life. 2005;57(4-5):347-53
36. Chapman HA, Riese RJ, Shi GP. Emerging roles for cysteine proteases in human biology. Annu Rev Physiol. 1997;59:63-88
37. Tepel C, Bromme D, Herzog V, Brix K. Cathepsin K in thyroid epithelial cells: sequence, localization and possible function in extracellular proteolysis of thyroglobulin. J Cell Sci. 2000;113(24):4487-98
38. Okamura N, Tamba M, Uchiyama Y, Sugita Y, Dacheux F, Syntin P. et al. Direct evidence for the elevated synthesis and secretion of procathepsin L in the distal caput epididymis of boar. Biochim Biophys Acta. 1995;1245(2):221-26
39. Nagler DK, Menard R. Family C1 cysteine proteases: biological diversity or redundancy? Biol Chem. 2003;384(6):837-43
40. Rispe C, Kutsukake M, Doublet V, Hudaverdian S, Legeai F, Simon JC. et al. Large gene family expansion and variable selective pressures for cathepsin B in aphids. Mol Biol Evol. 2008;25(1):5-17
41. Berti PJ, Storer AC. Alignment/phylogeny of the papain superfamily of cysteine proteases. J Mol Biol. 1995;246(2):273-83
42. Ohno S. Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin Cell Dev Biol. 1999;10(5):517-22
43. Rispe C, Kutsukake M, Doublet V, Hudaverdian S, Legeai F, Simon JC. et al. Large gene family expansion and variable selective pressures for cathepsin B in aphids. Mol Biol Evol. 2008;25(1):5-17
44. Watanabe R, Okazaki R. Reducing bone resorption by cathepsin K inhibitor and treatment of osteoporosis. Clin Calcium. 2014;24(1):59-67
45. Kutsukake M, Nikoh N, Shibao H, Rispe C, Simon JC, Fukatsu T. Evolution of soldier-specific venomous protease in social aphids. Mol Biol Evol. 2008;25(12):2627-41
46. Vieira FA, Thorne MA, Stueber K, Darias M, Reinhardt R, Clark MS. et al. Comparative analysis of a teleost skeleton transcriptome provides insight into its regulation. Gen Comp Endocrinol. 2013;191:45-58
47. Thomas JH. Rapid birth-death evolution specific to xenobiotic cytochrome P450 genes in vertebrates. PLoS Genet. 2007;3(5):e67
48. Añez G, Grinev A, Chancey C, Ball C, Akolkar N, Land KJ. et al. Evolutionary dynamics of West Nile virus in the United States, 1999-2011: phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl Trop Dis. 2013;7(5):e2245
49. Loewe L. Negative selection. Nature Education. 2008;1(1):59
50. Song W, Qin Y, Zhu Y, Yin G, Wu N, Li Y. et al. Delineation of plant caleosin residues critical for functional divergence, positive selection and coevolution. BMC Evol Biol. 2014;14:124
51. Firnberg E, Labonte JW, Gray JJ, Ostermeier M. A comprehensive, high-resolution map of a gene's fitness landscape. Mol Biol Evol. 2014;31(6):1581-92
Author contact
![]() Corresponding author: Zhong-Hua Cai, Room 304, L-Building, Graduate School at Shenzhen, Tsinghua University, Shenzhen University Town, Xili Town, Shenzhen City, Guangdong Province, P R. China. E-mail address: caizhtsinghua.edu.cn; Tel: +86 755 26036108; Fax: +86 755 26036108
Corresponding author: Zhong-Hua Cai, Room 304, L-Building, Graduate School at Shenzhen, Tsinghua University, Shenzhen University Town, Xili Town, Shenzhen City, Guangdong Province, P R. China. E-mail address: caizhtsinghua.edu.cn; Tel: +86 755 26036108; Fax: +86 755 26036108