ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2015; 11(9):1056-1072. doi:10.7150/ijbs.12408 This issue Cite
Research Paper
Knockout of SRC-1 and SRC-3 in Mice Decreases Cardiomyocyte Proliferation and Causes a Noncompaction Cardiomyopathy Phenotype
Xian Chen1, Li Qin1, Zhaoliang Liu1, Lan Liao1, James F. Martin2, Jianming Xu1, 3, ![]()
1. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX 77030, USA.
2. Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030, USA.
3. Sichuan Medical University, Luzhou, Sichuan 646000, China.
Abstract

Noncompaction cardiomyopathy (NCC) is a congenital heart disease that causes ventricular dysfunction and high mortality rate in children. The mechanisms responsible for NCC are still unknown. The steroid receptor coactivator-1 (SRC-1) and SRC-3 are transcriptional coactivators for nuclear hormone receptors and certain other transcription factors that regulate many genes in development and organ function. However, the roles of SRC-1/3 in heart morphogenesis, function and NCC occurrence are unknown. This study aims to examine the spatial and temporal expression patterns of SRC-1/3 in the heart and investigate the specific roles of SRC-1/3 in heart development, function and NCC occurrence. Immunochemical analysis detected SRC-1/3 expressions in the proliferating cardiomyocytes of mouse heart at prenatal and neonatal stages, while these expressions disappeared within two weeks after birth. Through generating and characterizing mouse lines with global or cardiomyocyte-specific knockouts of SRC-1/3, we found ablation of SRC-1/3 in the myocardial lineage resulted in prominent trabeculae, deep intertrabecular recesses and thin ventricular wall and septum. These developmental defects caused a failure of trabecular compaction, decreased internal ventricular dimension, reduced cardiac ejection fraction and output and led to a high rate of postnatal mortality. Collectively, these structural and functional abnormalities closely simulate the phenotype of NCC patients. Further molecular analysis of cardiomyocytes in vivo and in vitro revealed that SRC-1/3 directly up-regulate cyclin E2, cyclin B1 and myocardin to promote cardiomyocyte proliferation and differentiation. In conclusion, SRC-1/3 are required for cardiomyocyte proliferation and differentiation at earlier developmental stages, and their dysfunction causes NCC-like abnormalities in the hearts of newborn and adult mice.
Keywords: knockout mice, heart diseases, myocyte proliferation, cardiac output, nuclear receptor coactivator
Introduction
Congenital heart disease (CHD) resulted from cardiac developmental defect is a major cause of infant mortality, which affects 0.4-5% babies in United States [1-3]. The noncompaction cardiomyopathy (NCC) is a congenital heart defect characteristic of prominent trabeculae in the ventricular chamber and deep intertrabecular recesses in the ventricular wall [4-6]. Although the noncompaction of myocardium can happen in both ventricular chambers, NCC is also known as left ventricular noncompaction (LVNC). NCC can be grouped into non-isolated NCC with both ventricular noncompaction and other cardiac defects and isolated NCC with only ventricular noncompaction. The current prevalence of NCC is still inaccurate because the American Heart Association only classified this disease recently [6]. Currently, NCC is diagnosed by echocardiography as the thickness ratio of the noncompacted trabeculae to the compacted ventricular wall is larger than 2.0 [4, 5]. Although the children with NCC demonstrate a high rate of lethality because of ventricular systolic dysfunction, thromboemboli, myocardial infarction and heart failure [5-10], the genetic causes and pathogenic mechanisms of the disease are largely unknown [6]. Our lack of knowledge about NCC is largely attributed to the lack of animal models that can faithfully simulate NCC disease from embryonic stage to adulthood.
The p160 steroid receptor coactivator (SRC) family contains SRC-1 (NCOA1), SRC-2 (GRIP1/TIF2/NCOA2) and SRC-3 (AIB1/ACTR/NCOA3) [11, 12]. These SRC transcriptional coactivators robustly enhance gene expression through interacting with nuclear hormone receptors such as estrogen, progesterone and glucocorticoid receptors and other transcription factors (TFs) such as E2F1, Mef2, AP-1, PEA3 and Ets-2 and recruiting chromatin remodeling factors such as CBP (cAMP response element-binding protein (CREB)-binding protein), p300 and CARM1 (coactivator-associated arginine methyltransferase 1) [11, 12]. Because under normal physiological conditions these coactivators exist at rate-limiting concentrations or activities, any changes in their concentrations or activities may significantly affect their target gene expression and thereby result in functional alterations [11, 12]. SRC-1 knockout mice exhibit normal viability, somatic growth and reproductive function, but display a delay in mammary gland growth and Purkinje cell differentiation [13-15]. Most SRC-2 knockout mice survive to adulthood with impaired male and female reproductive capabilities because of abnormal spermatogenesis and compromised uterine decidual response [16, 17]. SRC-3 knockout mice display partial perinatal lethality, somatic growth retardation, female hypofertility and delayed mammary gland growth [12, 18]. These distinct phenotypes may be attributed to both specific functions and distinct spatiotemporal expression patterns of individual SRCs.
The homologous molecular structures of the SRC family members and their interactions with a group of common TFs suggest that SRCs may share redundant functions. Indeed, in cultured cells each SRC could coactivate multiple nuclear hormone receptor-mediated transcriptions [11, 19-23]. Double SRC-1 and SRC-2 null mice exhibit impaired lung maturation and neonatal lethality and SRC-1+/-;SRC-2-/- mice are completely infertile [24]. Double knockout of SRC-1 and SRC-3 also results in placental defects and embryonic lethality [25]. However, the specific and redundant roles of SRCs in heart development and function have not been investigated.
Herein, we investigated the spatiotemporal expression patterns of SRC-1 and SRC-3 during mouse heart development and generated many global and cardiomyocyte-specific knockout mouse lines for SRC-1, SRC-3 and their combinations. We report that SRC-1 and SRC-3 are highly expressed in the developing heart at embryonic and early postnatal stages and they cooperatively regulate cardiomyocyte proliferation and heart morphogenesis. Cardiomyocyte-specific knockout of both SRC-1 and SRC-3 results in a severe phenotype that closely simulates NCC. Similar to young patients, these mice with NCC also displayed a high rate of postnatal mortality. Our findings not only revealed the functions of SRC-1 and SRC-3 during heart development, but also provided new insight into NCC development and a new animal model for studying NCC from embryonic stage to adulthood.
Methods
Mice
Wild type (WT), SRC-1 null (SRC-1-/-), SRC-3 null (SRC-3-/-) and SRC-1 and SRC-3 double null (SRC-1-/-;SRC-3-/-) mice and their embryos with a backcrossed FVB strain background were described previously [25]. The floxed SRC-3 (SRC-3f/f) mouse line was previously established in our laboratory [26, 27]. These mice were backcrossed with SRC-1-/- mice for four generations before their offspring were interbred to generate SRC-3f/f and SRC-1-/-;SRC-3f/f mice with about 93.7% FVB background. The Nkx2.5cre mouse line harboring Cre sequence at one of the two Nkx2.5 alleles [28] was purchased from MouseBook, and crossbred with SRC-1-/- and SRC-3-/- mice for generating Nkx2.5Cre;SRC-3+/-, Nkx2.5cre;SRC-1+/-;SRC-3+/-, and Nkx2.5cre;SRC-1-/-;SRC-3+/- mice with more than 90% FVB background. These mice were further bred with SRC-3f/f and SRC-1-/-;SRC-3f/f mice to generate “SRC-3f/+” mice (Control), “SRC-1-/-;SRC-3f/+” mice with SRC-1 knockout and a floxed SRC-3 allele (S1KO;S3F), “Nkx2.5cre;SRC-3f/-” mice with conditional myocardial SRC-3 knockout (S3MKO), and “Nkx2.5cre;SRC-1-/-;SRC-3f/-” mice with SRC-1 knockout and conditional myocardial SRC-3 knockout (S1KO;S3MKO). The day when the mating plug was observed was considered as embryonic day 0.5 (E0.5). Pregnant mice were sacrificed by cervical dislocation after deeply anesthetized by intraperitoneal injection of Avertin (2.5% in saline, 45 ml/kg body weight). The embryos were dissected and examined on different pregnant days. Genotypes were analyzed by PCR using allele-specific primer pairs and genomic DNA isolated from ear tips of 3-week-old mice or the tails of embryos after proteinase K digestion [29, 30]. The survival percentage for each genotype group was calculated by using the formulas, (the number of pups at weaning time/the expected number of pups) × 100. The data were statistically analyzed by Chi-Square test between the observed and expected numbers of progeny. Animal protocols were approved by the Animal Care and Use Committee of Baylor College of Medicine.
Western blotting
Frozen tissue powders and harvested cells were extracted with RIPA buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.25% Na-deoxycholate. The tissue extracts were analyzed by Western blotting as described previously [31]. The primary antibodies were against SRC-1 (#2191, Cell Signaling, Danvers, MA), SRC-3 (#2126, Cell Signaling, Danvers, MA) and α-tubulin (#T8203, Sigma-Aldrich, St. Louis, MO). Appropriate secondary antibodies conjugated with horseradish peroxidase were used to visualize the signals with ECL reagent kit.
Immunohistochemistry (IHC)
IHC was performed as described previously [14, 32]. Primary antibodies were against SRC-1, SRC-3, phosphorylated-Ser10 Histone H3 (P-H3) (#06-570, Millipore, Billerica, MA), Ki67 (#550609, BD biosciences, San Jose, CA) and MF 20 (DSHB, Iowa City, IA). Appropriate biotinylated secondary antibodies were used.
Quantitative RT-PCR (qPCR)
Mouse tissues were quickly isolated, frozen in dry ice and granulated into fine powder under frozen conditions with liquid nitrogen. Total RNA was extracted from the tissue powder by using TRIzol reagent (Invitrogen, Carlsbad, CA). After RNA was reversely transcribed into cDNA by using the reverse transcriptase core kit (Eurogenec, San Diego, CA), TaqMan qPCR was performed with 50 ng cDNA and qPCR MasterMix Plus kit (Eurogenec, San Diego, CA). Parallel measurements of 18S RNA or GAPDH RNA served as internal controls. The qPCR primers and probes for SRC-1 and SRC-3 mRNA analyses were described previously [14, 32]. The primers and probes for measuring mRNA concentrations of mouse cyclin E2, cyclin B1, ACTC1, TNNT2, myocardin, TAZ, LDB3, DTNA, MYH7, RXRα and FKBP12 were designed by using the online software of Universal ProbeLibrary Assay Design Center (Roche Applied Science, Switzerland).
Histological examination
The dissected mouse hearts were fixed overnight in 4% paraformaldehyde (PFA) at 4oC. The fixed specimens were embedded in paraffin. Five μm-thick paraffin sections were stained with hematoxylin and eosin (H&E) for light microscopy.
Echocardiograph
Visualsonic Vevo 770 Imaging System and GE Vivid 7 Dimension BT05 Ultrasound machine were used. Mice were placed in an induction box and anesthetized using isoflurane. Mice were positioned on the electrocardiography (ECG) platform with appropriate delivery of isoflurane and oxygen via a nose cone. During imaging, animals were monitored by the physiological monitoring equipment coupled with the imaging system. B-mode real-time imaging and M-mode imaging were performed. Following the imaging, the animals were moved away from the isoflurane anesthesia and allowed to recover on a heating pad. The recorded images were analyzed by Visualsonic Software.
Preparation and culture of primary heart cells
The neonates (P0) of SRC-3f/f and SRC-1-/-;SRC-3f/f mice were euthanized using CO2 and briefly soaked in 70% ethanol. Mouse hearts were isolated, rinsed in saline and temporarily kept in AD buffer containing 116 mM NaCl, 20 mM HEPES, 1 mM NaH2PO4, 5.5 mM Glucose, 5.4 mM KCl and 0.8 mM MgSO4. For each experiment, at least 10 hearts were collected, and digested for 10 minutes at 37oC in AD buffer containing 75 U/ml of collagenase and 0.6 mg/ml of pancreatin. The digested tissues were vigorously pipetted up and down for 5 minutes, and incubated for another 5 minutes at 37oC. The cell suspension was transferred and mixed with 1 ml of horse serum in a new tube. Cells were centrifuged down and resuspended in horse serum. New digestion buffer was added to the remaining tissue and the above procedure was repeated 4 times until all tissue was digested and all cells were collected. The harvested cells were centrifuged down, resuspended in 10 ml of plating medium (66% DMEM, 17% M199, 5% FBS, 10% horse serum, 100 U/ml penicillin and 100 μg/ml streptomycin), and plated in a 10-cm culture dish. After a 2-hour incubation at 37°C, the unattached cells were collected from the plate, centrifuged down, resuspended in fresh plating medium, and plated in 35-mm culture dishes or 24-well culture plates coated with 1% gelatin. After 24 hours of incubation at 37°C, the plating medium was replaced with fresh medium containing 70.8% DMEM, 17.7% M199, 10% horse serum, 100 U/ml penicillin and 100 μg/ml streptomycin. For adenoviral infection, the culture medium was replaced with serum-free medium (78.7% DMEM, 19.1% M199, 100 U/ml of penicillin and 100 μg/ml of streptomycin). Ad5-CMV-Cre-GFP or Ad5-CMV-GFP adenoviral particles (9x108 per 35-mm culture dish or 2.25x108 per well of the 24-well plate) were added to infect the cells for 24 hours. The infected cells were cultured in fresh medium for another 24 hours. Cells in 35-mm dishes were harvested for preparation of protein and RNA samples for biochemical analysis, while cells in 24-well plate were fixed using 4% PFA for immunocytochemistry (ICC).
BrdU labeling
BrdU (B5002, Sigma-Aldrich, St. Louis, MO) in saline (10 mg/ml) was injected into pregnant mice at E12.5 and E16.5 (10 μl/g body mass, i.p.). Injected mice were sacrificed 1 hour later. Their hearts were collected, fixed in 4% PFA for 2 hours and embedded in paraffin. The incorporated BrdU were detected on the 5-μm thick coronal heart sections by using the BrdU In-Situ Detection Kit (#550803, BD biosciences, San Jose, CA). Three samples were examined for each genotype. Images were taken from six randomly selected fields for each sample. For cells in culture, BrdU (1 mM) in PBS was added to the medium at 10 μM. After one hour incubation, cells were rinsed in PBS and fixed in 4% PFA. BrdU signals were detected as described above. Experiments were performed with three repeats. Images were taken under microscope from three randomly selected fields for each experiment. BrdU-positive cells and total number of cells in the fields were counted. Student's t-test was used for statistical analysis of the data collected.
Cell culture
The H9C2 myoblastic cells were from a subclone of the initially cloned cells derived from an embryonic heart of BD1X rat. These cells were cultured in DMEM/F12 medium with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml of penicillin, 100 μg/ml of streptomycin and 2 mM of L-glutamine. These cells were only allowed to grow to 70% confluence. Mouse embryonic fibroblasts (MEFs) were cultured in high glucose DMEM medium with 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 μg of streptomycin.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as described previously [33]. E2F1, SRC-1 and SRC-3 antibodies were used in ChIP assays for the mouse cyclin E2 promoter. MEF cells, neonatal and adult mouse hearts were examined in these assays. The E2F1-binding region located 770 bp upstream of the transcriptional starting site and the negative control region in exon 6 of the cyclin E2 gene were measured by qPCR with specific primer pairs in ChIP assays. SRC-1 and SRC-3 antibodies were also used in ChIP assays for the enhancer regions of the mouse and rat myocardin genes. P0 mouse hearts, H9C2 cells and MEFs were examined in these ChIP assays. The enhancer region located 20-30 kb upstream of the myocardin's translational starting site [34] and the negative control region in exon 10 of the myocardin gene were measured by qPCR with specific primer pairs in ChIP assays.
Image analysis
Tissue sections and attached cells stained by H&E, ICC or IHC were imaged under a microscope by an RT Color Spot camera (Diagnostic Instruments, Sterling Heights, MI). The cells were analyzed and counted on the electronic images using the ImageTool software (UTHSCSA, San Antonio, TX). The thickness of ventricular wall and trabeculae and the areas of septum were quantitatively measured using the i-Solution Software (IMT Inc., Korea).
RESULTS
The spatiotemporal expression profiles of SRC-1 and SRC-3 during heart development
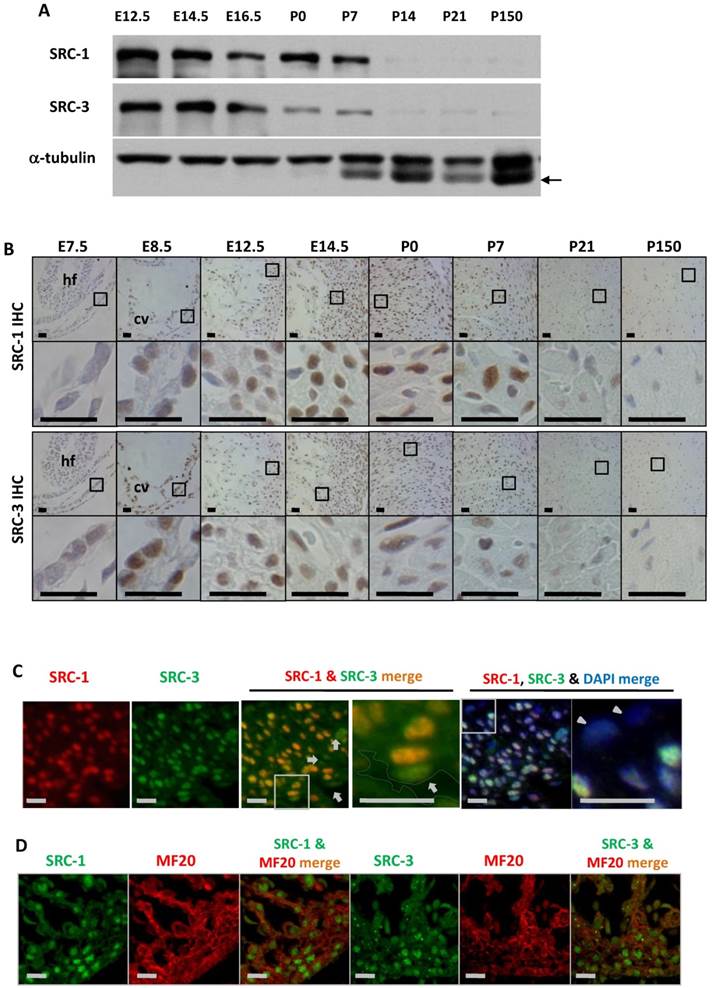
To define the cellular locations for SRC-1 and SRC-3 function during heart development, we examined the temporal and spatial expression patterns of SRC-1 and SRC-3 in the mouse hearts at different developmental stages. Both SRC-1 and SRC-3 proteins were detected from the heart tissue lysates at E12.5, E14.5, E16.5, P0 and P7 developmental stages, but both proteins were barely detectable from the heart tissue lysates of P14, P21 and adult (P150) mice. SRC-3 protein is significantly reduced in the mouse heart at P0 and P7 when compared with earlier stages (Fig. 1A). IHC analysis revealed that both proteins were extremely low in the myocardial cells at E7.5, but were expressed at high levels in the myocardial cells at E8.5, E12.5 and E14.5. In agreement with Western blotting results, SRC-1 protein levels kept high in the mouse hearts at P0 and P7 in the cardiomyocytes, while SRC-3 protein levels were reduced at these stages. Both SRC-1 and SRC-3 proteins became barely detectable in the myocardial cells in P21 and adult mice (Fig. 1B). Double immunofluorescence staining revealed that SRC-1 and SRC-3 were co-expressed in most heart cells at E12.5 (Fig. 1C). Double immunofluorescence staining for MF20, a myocardial cell marker, and SRC-1 or SRC-3 confirmed that both SRC-1 and SRC-3 were expressed in the cardiomyocytes at E12.5 (Fig. 1D). These results demonstrate that SRC-1 and SRC-3 are co-expressed at high levels in the cardiomyocytes during middle to late embryonic and neonatal stages, but at extremely low levels in the cardiomyocytes after P7.
The spatiotemporal expression patterns of SRC-1 and SRC-3 proteins during heart development. A. Western blot analysis of SRC-1 and SRC-3 in the hearts of E12.5, E14.5, E16.5, P0, P7, P14, P21 and P150 normal mice. Each tissue lysate was prepared from at least 3 mouse hearts. The α-tubulin served as a loading control. The arrow indicates a mouse IgG band detected by the secondary antibody. B. Detection of SRC-1 and SRC-3 positive cells by IHC in the E7.5, E8.5, E12.5, E14.5, P0, P7, P21 and P150 mouse hearts. The boxed areas in the first and third row panels are magnified in the second and fourth row panels, respectively. At least three mouse hearts at each stage were examined, and representative images were shown. hf, head fold; cv, common ventricular chamber of the primitive heart. C. Double immunofluorescent staining for SRC-1 and SRC-3 in the left ventricular wall of the E12.5 normal mouse heart. The nuclei were stained by DAPI. The boxed areas in the third and fifth panels are magnified in the fourth and sixth panels, respectively. The outlined area in the fourth panel indicates the heart chamber. The arrows indicate endothelial cells. The arrowheads indicate the epicardium. D. Double immunofluorescent staining for SRC-1 and MF20 and for SRC-3 and MF20 in the ventricular wall of the E12.5 normal mouse heart. Scale bars in panels B, C and D, 20 μm.
SRC-1 and SRC-3 coordinately regulate heart morphogenesis during embryonic development
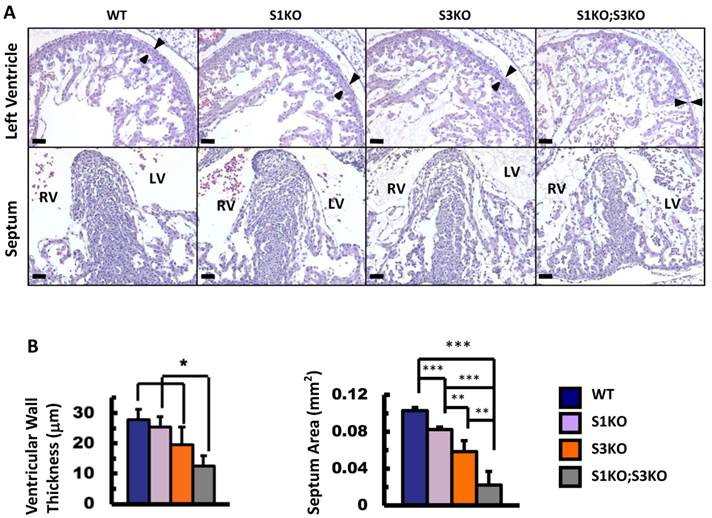
To define the roles of SRC-1 and SRC-3 in heart morphogenesis during embryonic development, we generated WT, SRC-1-/- (S1KO), SRC-3-/- (S3KO) and SRC-1-/-;SRC-3-/- (S1KO;S3KO) mice and examined the histology of hearts in these mouse embryos at E12.5. The heart morphology and tissue structures of S1KO embryos were very similar to those of WT embryos, although S1KO hearts showed slightly reduced septum area on the coronal sections (Fig. 2A and B). However, S3KO hearts exhibited moderately disorganized trabeculae, and both the ventricular wall thickness and the septum area of S3KO hearts were significantly reduced versus WT and S1KO hearts. Furthermore, S1KO;S3KO hearts had severely disorganized trabeculae network and the size of individual trabeculae was much smaller than that of WT and S1KO hearts. The thickness of the ventricular compact walls and the extent of the septum area in S1KO;S3KO hearts were severely reduced by 55% and 78% compared with the ventricular wall thickness and the septum area of WT hearts (Fig. 2A and B). These results demonstrate that SRC-3 and SRC-1 may coordinately regulate heart morphogenesis during heart development in embryos.
Knockout of SRC-1 and SRC-3 reduced ventricular wall and septum morphogenesis in the E12.5 mouse hearts. A. H&E-stained coronal sections of the E12.5 hearts from embryos with the indicated genotypes. The arrowheads indicate the thickness of the compact ventricular wall. RV, right ventricle; LV, left ventricle. Scale bars, 20 μm. B. The average ventricular wall thicknesses and septum areas measured from the mouse hearts with indicated genotypes. Each group had at least 4 independent samples. *, ** and ***, p<0.05, p<0.01 and p<0.001 by Student's t test.
Cardiomyocyte-specific knockout of both SRC-1 and SRC-3 in mice results in a phenotype simulating human NCC
Most S1KO;S3KO mouse embryos die before birth [25], which hampered our investigation of the redundant function for SRC-1 and SRC-3 in the heart at later stages. In order to investigate the myocardium-specific roles of SRC-1 and SRC-3 in heart development and function in all stages, we crossbred SRC-1-/-, SRC-3-/-, SRC-3f/f and Nkx2.5cre mouse lines, and generated the following combinatorial knockout (KO) mouse lines: SRC-3f/+ (S3F) normal control, SRC-1-/-;SRC-3f/+ for SRC-1 KO (S1KO;S3F), Nkx2.5cre;SRC-3f/- for myocardial SRC-3 KO (S3MKO) and Nkx2.5cre;SRC-1-/-;SRC-3f/- for myocardial SRC-1 and SRC-3 double KO (S1KO;S3MKO). As expected, SRC-1 mRNA and protein were detected in the hearts of newborn S3F and S3MKO mice, but not detected in the hearts of newborn S1KO;S3F and S1KO;S3MKO mice by qPCR, Western blotting and IHC (Additional File 1: Suppl. Fig. S1A-C). High levels of SRC-3 mRNA and protein were detected in the hearts of newborn S3F and S1KO;S3F mice. However, only trivial amount of SRC-3 mRNA and protein were detected in the hearts of newborn S3MKO and S1KO;S3MKO mice by qPCR and Western blotting (Additional File 1: Suppl. Fig. S1A and B), and no SRC-3 protein was detected in the majority of cardiomyocytes in these mice (Additional File 1: Suppl. Fig. S1C). These results demonstrate that SRC-1 and/or SRC-3 are selectively and efficiently knocked out in the cardiomyocytes of these mouse models.
Morphological defects observed in the hearts of S3MKO and S1KO;S3MKO mice. A. Photographs of the P0 mouse hearts with the indicated genotypes (upper panels) and H&E-stained coronal sections showing the right ventricle and the septum areas (lower panels). The red and green lines indicate the thicknesses of the compact ventricular wall and the noncompacted trabecular layer of S1KO;S3MKO heart, respectively. RV, right ventricle; SP, septum; Scale bars, 0.5 mm (upper panels) and 0.1 mm (lower panels). B. Quantitative analysis of the ratios of trabecular to ventricular wall thicknesses, septum areas and ventricular wall thicknesses of the P0 mouse hearts with the indicated genotypes. Each group has at least 4 independent samples. *, ** and ***, p<0.05, p<0.01 and p<0.001 by Student's t test. C. Other NCC-relevant phenotypes observed in S3MKO and S1KO;S3MKO P0 hearts. Arrows indicate blood clots among the trabeculae; the “#” signs indicate spongy-like areas of the ventricular wall tissue; arrowheads indicate myocardial infarction. Scale bar, 50 μm. D. Some histological defects observed in the septums of S1KO;S3MKO and S3MKO hearts at P0. The septum of S3F and S1KO;S3F hearts is normal. Some S1KO;S3MKO septums contain spongy-like tissue (*) and unsealed holes between the right and left ventricles (arrow). An unsealed hole between the right atrial and left ventricle (arrowhead) was observed in one of the S3MKO hearts. Scale bars, 0.1 mm.
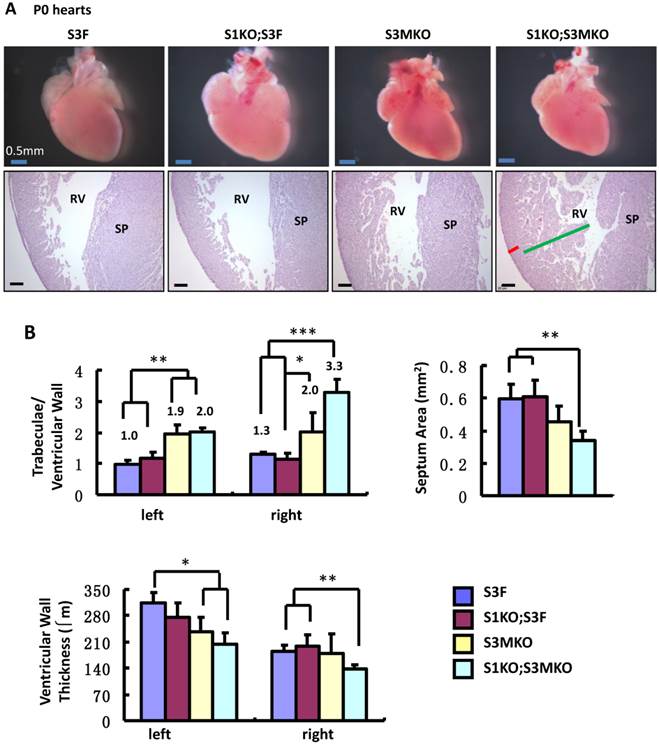
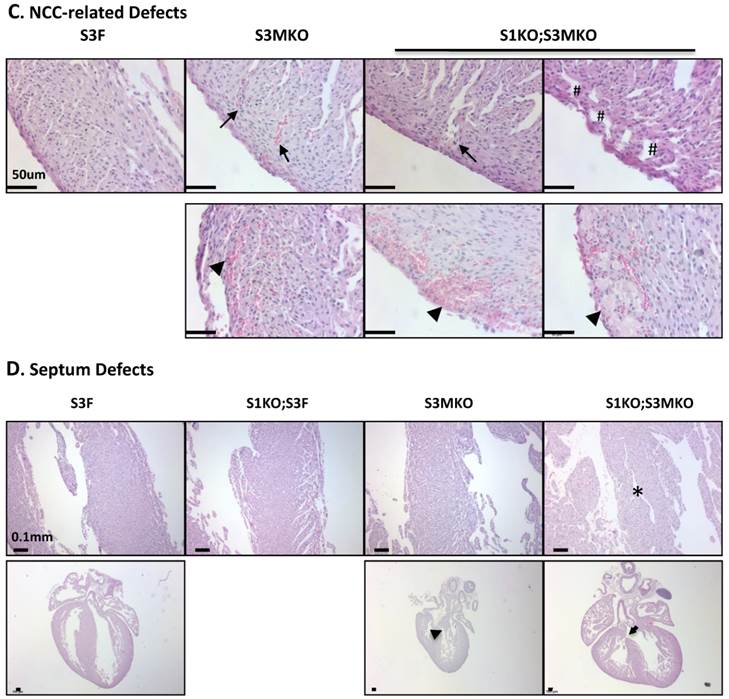
The superficial heart appearance was not significantly different among newborn S3F, S1KO;S3F, S3MKO and S1KO;S3MKO mice (Fig. 3A). The histological architectures of the cardiac compartments were also normal in S3F and S1KO;S3F mice (Fig. 3A). The thickness ratios of the trabeculae to compacted walls of both right and left ventricles of the newborn S3F and S1KO;S3F mice were maintained between 1.0 and 1.3 (Fig. 3B). In contrast, prominent trabeculae and deep intertrabecular recesses were detected in both right and left ventricles of all the examined newborn S1KO;S3MKO mice (Fig. 3A and B, and Table 1). The compacted ventricular walls in S1KO;S3MKO hearts were thinner than those in S3F hearts. The thickness ratios of the trabeculae to the compacted ventricular walls in S1KO;S3MKO hearts were 3.3 and 2.0 for the right and left ventricles (Fig. 3B), indicating that these hearts had developed a typical structural defect of NCC at the neonatal stage [4, 5]. On the coronal sections of S1KO;S3MKO hearts, the septum area was significantly smaller and the ventricular walls were much thinner compared with those in the normal S3F hearts (Fig. 3B). The septums in about 25% and the ventricular areas in about 50% of S1KO;S3MKO hearts also had many spongy spaces (Fig. 3A and C, and Table 1). These morphological defects could be a consequence of the failed trabecular compaction [5, 8, 9]. S1KO;S3MKO hearts also exhibited other pathological changes associated with NCC, such as blood clots among the trabeculae observed in all samples and myocardial infarction observed in about 50% of samples (Fig. 3C and Table 1). These NCC-related phenotypes became more prominent at later cardiac developmental stages (Additional File 1: Suppl. Fig. S2). Furthermore, 25% of S1KO;S3MKO hearts also had developmental defect in the inter-ventricular septum, displaying unsealed ventricular septal defect between ventricles (Fig. 3D and Table 1). Interestingly, some of the pathological lesions observed in S1KO;S3MKO hearts were also found in S3MKO hearts, although these lesions were less severe versus those in S1KO;S3MKO hearts. Specifically, the thickness ratios of their trabeculae to their right and left compacted ventricular walls were 2.2 and 1.9, respectively (Fig. 2B). The deep intertrabecular recesses and blood clots in these recesses were observed in most S3MKO hearts (Fig. 3C and Table 1). The myocardial infarction and interventricular septum defect were also observed in some S3MKO hearts (Fig. 3C and D, and Table 1). However, the spongy tissue, which was found in the septums and ventricular walls of S1KO;S3MKO hearts, was not observed in S3MKO hearts (Table 1). These results indicate that myocardial KO of both SRC-1 and SRC-3 in S1KO;S3MKO mice causes heart structural defects and pathological lesions similar to those observed in both isolated and nonisolated NCC human patients [4, 5, 35], while myocardial KO of only SRC-3 in S3MKO mice causes a partial phenotype of NCC. Since S1KO;S3F mice have normal hearts, SRC-3 may play a more dominant role over SRC-1 to support the morphogenesis of myocardial tissues or maintain the morphology of these myocardial tissues.
The Incidences of Heart Morphological Defects Observed in the Newborn S1KO;S3MKO and S3MKO Mice
| Heart Defects | S3F (n=4) | S1KO;S3F (n=4) | S3MKO (n=4) | S1KO;S3MKO (n=4) |
|---|---|---|---|---|
| Prominent trabecular noncompaction | 0 | 0 | 4 (100%) | 4 (100%) |
| Deep intertrabecular recession | 0 | 0 | 3 (75%) | 4 (100%) |
| Spongy septum | 0 | 0 | 0 | 1 (25%) |
| Spongy ventricular wall | 0 | 0 | 0 | 2 (50%) |
| Blood clots among trabeculae | 0 | 0 | 3 (75%) | 4 (100%) |
| Myocardial infarction | 0 | 0 | 2 (50%) | 2 (50%) |
| Interventricular septum defect | 0 | 0 | 1 (25%) | 1 (25%) |
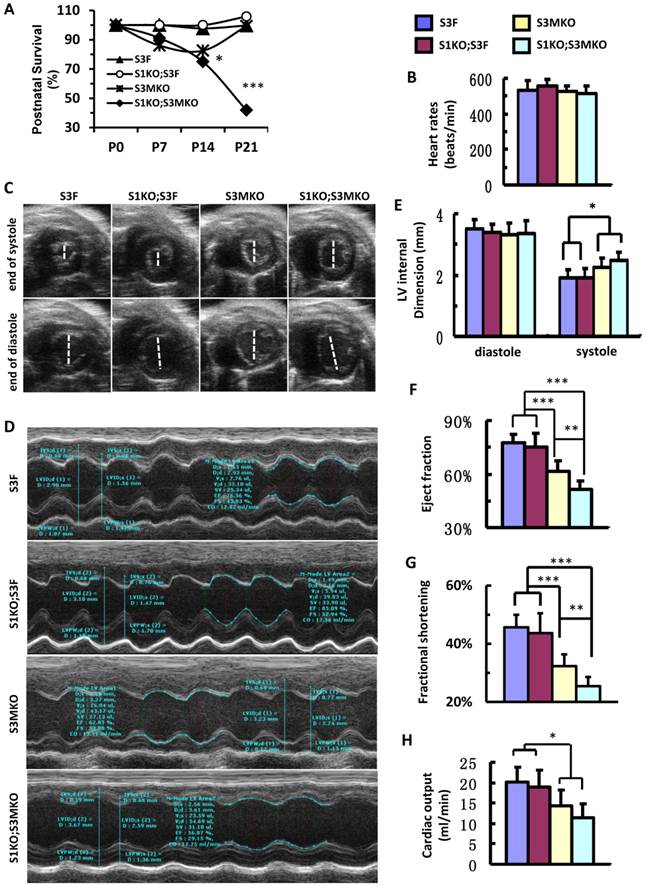
After birth, S1KO;S3F mice survived as well as control S3F mice. S3MKO mice also showed stable survival rate. However, similar to NCC patients [7], S1KO;S3MKO mice exhibited high mortality rates at young ages, about 70% at P14 and 45% at P21 after normalizing the data to P0 (Fig. 4A). Importantly, although the heart rates of the four groups of the survived adult mice were comparable (Fig. 4B), the heart function was totally dependent on specific genotypes of these age-matched adult mice. By looking at the real-time echocardiograph videos recorded from adult animals, the contractive extents of the hearts in S3F control mice and S1KO;S3F mice are comparable (Additional file 2: Suppl. Video 1 and Additional file 3: Suppl. Video 2). However, at the end of systole the ventricular chambers were larger in adult S3MKO mice and the largest in adult S1KO;S3MKO mice versus that in adult S3F mice (Additional File 4: Suppl. Video 3 and Additional File 5: Suppl. Video 4). Quantitative analysis of the real-time echocardiographs revealed that the diastolic dimensions of S3F, S1KO;S3F, S3MKO and S1KO;S3MKO hearts were similar. The systolic dimensions of S1KO;S3F hearts were also identical to that of S3F hearts. However, the systolic dimensions of S3MKO and S1KO;S3MKO hearts were significantly larger than the systolic dimensions of S3F and S1KO;S3F hearts (Fig. 4C-E). Accordingly, the cardiac ejection fractions of S3F and S1KO;S3F hearts were similar, about 75%. The cardiac ejection fraction of S3MKO hearts was reduced to 60%, which was significantly smaller than that of the control S3F hearts. The cardiac ejection fraction and fractional shortening of S1KO;S3MKO hearts were further respectively reduced to 51% and 25%, significantly smaller versus that of S3MKO hearts (Fig. 4F and G). These changes were also consistent with the largely reduced cardiac outputs of S3MKO and S1KO;S3MKO hearts (Fig. 4H). These results indicate that myocardial loss of SRC-3 or both SRC-1 and SRC-3 functions causes moderate or severe reductions in cardiac ejection fraction and blood output due to increased systolic dimensions of the heart chambers, a major cardiac functional failure of clinical NCC patients [5].
SRC-1 and SRC-3 regulate myocardial cell proliferation
During heart development, cardiomyocytes undergo mitosis at embryonic and early postnatal developmental stages, but become largely quiescent shortly after birth [36-39]. Indeed, our IHC analysis of the phosphor-S10-histone H3 (P-H3), a metaphase cell proliferation marker, and Ki67, a general cell proliferation marker, detected many proliferative cardiomyocytes in E9.5, E14.5. E16.5 and P0 mouse hearts, where high expression levels of SRC-1 and SRC-3 were detected. However, proliferative cardiomyocytes were not detected in the cardiomyocytes in the adult mouse hearts where SRC-1 and SRC-3 expression was extremely low (Additional File 1: Suppl. Fig. S3, and Fig. 1A and B). These results indicate that SRC-1 and SRC-3 expression positively correlates with cardiomyocyte proliferation during heart development, suggesting that these two proteins may be involved in the regulation of cardiomyocyte proliferation.
Postnatal survival and left ventricular dysfunction of SRC-1 and SRC-3 conditional knockout mice. A. The survival rates of the indicated genotypes after birth. The calculation was based on the observed numbers of S3F, S1KO;S3F, S3MKO and S1KO;S3MKO mice at P0 (n = 45, 47, 26 and 33), P7 (n = 36, 25, 18 and 16), P14 (n = 33, 19, 14 and 10) and P21 (n = 46, 72, 23 and 20) versus their predicted numbers at P0 (n = 40, 47, 46 and 47), P7 (n = 32, 25, 37 and 25), P14 (n = 30, 19, 30 and 19) and P21 (n = 41, 68, 41 and 68). Then, the survival rates for each genotype group were normalized to the survival rate at P0. * and ***, p < 0.05 and p < 0.001 by Chi-Square test. B. Heart rates measured by Doppler echocardiography (M-mode) from 5 to 7-month-old mice (n = 7-12 for each group) with the indicated genotypes. These mice were also used for collecting the data shown in panels C-G. C. Live heart images caught from Doppler echocardiograph videos (B-mode) at the ends of systole and diastole. The white lines indicate the internal dimension of ventricular chambers. D. M-mode echocardiographs of the left ventricles in mice with the indicated genotypes. E. The average internal dimensions of the left ventricular chambers of mice with the indicated genotypes. The data were calculated according to the Doppler echocardiographs (M-mode) recorded from 7-12 mice in each group. F. The average cardiac ejection fractions calculated from the Doppler echocardiographs (M-mode) of 7-12 mice in each group. G. The average cardiac fractional shortening calculated from the Doppler echocardiographs (M-mode) of 7-12 mice in each genotype group. H. The average cardiac outputs calculated from Doppler echocardiographs (M-mode) from 7-12 mice in each genotype group. *, ** and *** in panels E-H, p < 0.05, p < 0.01 and p < 0.001 by Student's t test, respectively.
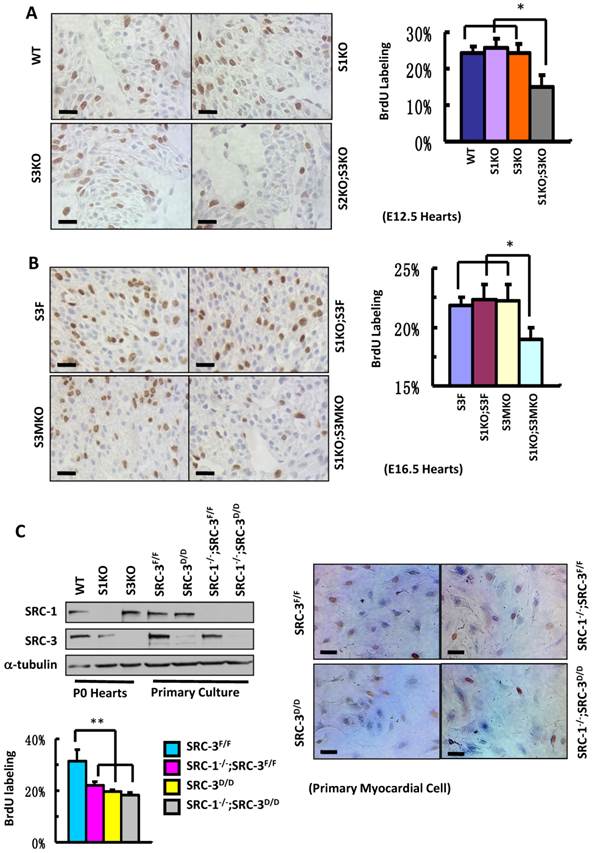
To define the roles for SRC-1 and SRC-3 in myocardial proliferation, we performed BrdU incorporation assays to compare the proliferation rates of myocardial cells with combinatorial SRC-1 and SRC-3 knockout genotypes in several mouse models. The ratios of BrdU-labeled versus total myocardial cells in the ventricular myocardium of WT, S1KO and S3KO hearts at E12.5 were comparable, about 25%, while the ratio of BrdU-labeled versus total myocardial cells in the same area of S1KO;S3KO hearts at E12.5 was markedly reduced to 15% (Fig. 5A). Furthermore, the percentages of BrdU-labeled myocardial cells in the ventricular areas of S3F, S1KO;S3F and S3MKO hearts at E16.5 were about 22%, but it was only 19% in the same area of S1KO;S3MKO hearts (Fig. 5B). These results indicate that both SRC-1 and SRC-3 support myocardial cell proliferation and the dysfunction of either one of these two genes is compensated from the other one during the embryonic heart development.
Next, we isolated primary myocardial cells from the hearts of neonatal SRC-3f/f and SRC-1-/-;SRC-3f/f mice. When the primary cultures were examined at day 7 by immunostaining, we found more than 90% cells in the culture were actinin-positive myocardial cells (Additional File 1: Suppl. Fig. S4 and data not shown). We infected these cells with Ad5-CMV-Cre-GFP adenovirus that mediated Cre expression for deleting the floxed SRC-3 alleles or with Ad5-CMV-GFP adenovirus that served as a Cre-negative control. The efficient viral infection resulted in GFP expression in the majority of cells in all cultures (Additional File 1: Suppl. Fig. S5A). As expected, SRC-3 protein was detected by immunocytofluorescence in the control virus-infected SRC-3f/f and SRC-1-/-;SRC-3f/f primary myocardial cells, but not detected in most of the Cre-expressing virus-infected SRC-3f/f and SRC-1-/-;SRC-3f/f (hereafter designated as SRC-3d/d and SRC-1-/-;SRC-3d/d) primary myocardial cells (Additional File 1: Suppl. Fig. S5B). Western blotting further confirmed that SRC-3f/f cells express both SRC-1 and SRC-3. SRC-3d/d cells express SRC-1 and only residual SRC-3. SRC-1-/-;SRC-3f/f cells only express SRC-3. SRC-1-/-;SRC-3d/d cells only express residual SRC-3 (Fig. 5C). Interestingly, the proliferation rates of all SRC-1-/-;SRC-3f/f, SRC-3d/d and SRC-1-/-;SRC-3d/d primary myocardial cells were drastically reduced compared with SRC-3f/f control cells (Fig. 5C). In these cultures, myocardial cells were distinguished from cardiac fibroblasts based on their different morphologies. These results indicate that the loss of either SRC-1 or SRC-3 function in the myocardial cells can not be fully compensated by the other one's function under the in vitro culture condition, which is different from that observed under in vivo condition where SRC-1 and SRC-3 play redundant functions to support myocardial cell proliferation (Fig. 5A and B).
SRC-1 and SRC-3 regulate cyclin E2 expression in myocardial cells
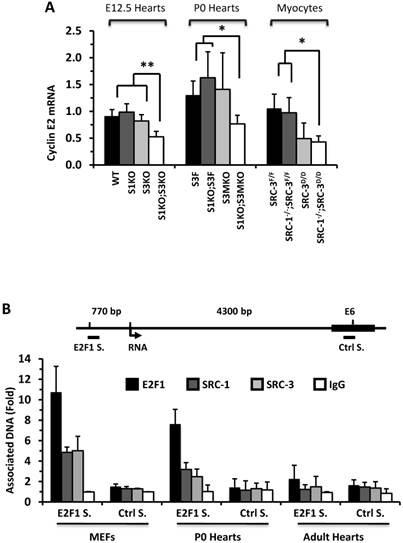
To understand why knockout of SRC-1 and SRC-3 severely attenuates myocardial cell proliferation, we measured the expression profiles of cyclins. The mRNA expression levels of cyclins D and A were not significantly different in the E12.5 and P0 mouse hearts and the primary cardiomyocytes developed from the P0 mouse hearts with different SRC-1 and SRC-3 genotypes (data not shown). At E12.5, expression levels of cyclin E2 mRNA were similar in WT, S1KO and S3KO hearts, but it was reduced more than 45% in S1KO;S3KO hearts. At P0, expression levels of cyclin E2 were also comparable in S3F, S1KO;S3F and S3MKO hearts, while it was reduced by 50% in S1KO;S3MKO hearts. Furthermore, expression levels of cyclin E2 were similar in SRC-3f/f and SRC-1-/-;SRC-3f/f primary myocardial cells, slightly reduced in SRC-3d/d primary myocardial cells and significantly reduced in SRC-1-/-;SRC-3d/d primary myocardial cells in culture (Fig. 6A). At E12.5, cyclin B1 expression was not significantly different in WT, S1KO and S3KO hearts, but it was reduced about 50% in S1KO;S3KO hearts. At P0, expression levels of cyclin B1 were also significantly reduced in S1KO;S3F, S3MKO and S1KO;S3MKO hearts versus S3F hearts. Intriguingly, cyclin B1 expression was not significantly different in all examined primary myocardial cells in culture, suggesting that SRC-1 and SRC-3 may not be the direct regulators of cyclin B expression (Additional File 1: Suppl. Fig. S6A). The consensus of these results suggests that SRC-1 and SRC-3 are involved in the regulation of cyclin E2 expression in myocardial cells, which should be responsible, at least in part, for SRC-1 and SRC-3-mediated myocardial cell proliferation during heart development.
E2F1 is a TF that drives cyclin E2 expression and SRC-3 is a coactivator for E2F1 [40, 41]. Therefore, we performed ChIP assays to examine whether SRC-1 and SRC-3 are associated with the functional E2F1 binding site proximate to the cyclin E2 promoter in MEFs and cardiac cells (Fig. 6B). Our assays revealed that E2F1, SRC-1 and SRC-3 were associated with the E2F1-binding region in both MEFs and the P0 mouse hearts (Fig. 6B), where both SRC-1 and SRC-3 were expressed (Additional File 1: Suppl. Fig. S6B). However, very little E2F1 and no SRC-1 and SRC-3 were associated with this E2F1-binding region in the adult mouse hearts (Fig. 6B), where little SRC-1 and SRC-3 are expressed (Additional File 1: Suppl. Fig. S6B) and the cardiomyocytes are not proliferative (Additional File 1: Suppl. Fig. S3). The specificity of these ChIP assays was confirmed by using non-immune IgG as a negative control for antibodies and a distant DNA region located in the 6th exon of the cyclin E2 gene that does not contain any E2F1-binding site (Fig. 6B). The recruitments of E2F1, SRC-1 and SRC-3 to the E2F1-binding site of cyclin E2 gene in the E12.5 and P0 hearts but not in the adult hearts were consistent with the high cyclin E2 expression in E12.5 and P0 hearts and the very low cyclin E2 expression in the adult hearts (Additional File 1: Suppl. Fig. S6C). These results support the notion that both SRC-1 and SRC-3 can serve as coactivators for E2F1 to upregulate cyclin E2 expression in the cardiomyocytes during heart development.
Knockout of SRC-1 and SRC-3 compromises cardiomyocyte proliferation. A. IHC for detecting BrdU-labeled proliferating cells (brown) and the average percentages of BrdU-labeled cardiomyocytes in the hearts of WT, S1KO, S3KO and S1KO;S3KO mouse embryos at E12.5 (n = 3). B. IHC for detecting BrdU-labeled cells (brown) and the average percentages of proliferating cells in the hearts of S3F, S1KO;S3F, S3MKO and S1KO;S3MKO mouse embryos at E16.5 (n = 3). C. Knockout of SRC-1 and SRC-3 in primary myocardial cells in culture decreases their proliferation. In Western blot analysis, samples prepared from WT, S1KO and S3KO hearts at P0 were used as positive, SRC-1 knockout and SRC-3 knockout controls, respectively. Samples prepared from SRC-3f/f, SRC-3d/d, S1KO;SRC-3f/f and S1KO;SRC-3d/d primary cardiomyocytes were used to assay SRC-1 and SRC-3 protein levels. Each pool of the primary cardiomyocytes was prepared from multiple hearts with the same genotype. α-tubulin served as a loading control. The lower band detected by SRC-3 antibody is a SRC-3 splicing isoform. The proliferating cardiomyocytes were labeled by BrdU incorporation and detected by immunocytochemistry (brown). The BrdU-labeled cells were counted versus the total cell numbers for each genotype group. Scale bars in all images, 20 μm. Each group in all panels contained at least three independent samples and at least three randomly selected regions of each independent sample were examined for quantitative data analysis. * and **, p<0.05 and p<0.01 by Student's t test.
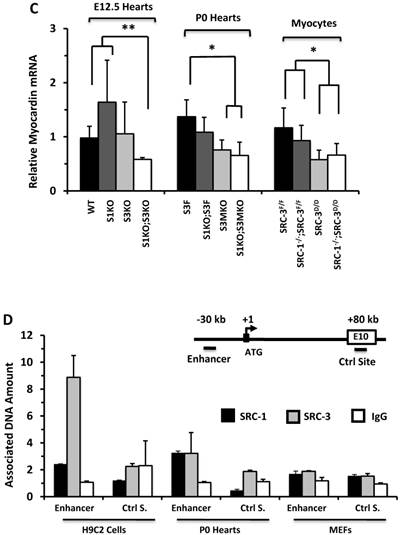
SRC-1 and SRC-3 regulate cyclin E2 and myocardin expression. A. Relative expression levels of cyclin E2 mRNA in the hearts (n = 4-6 for each group) of mice with the indicated genotypes at E12.5 and P0 and in the cultured primary cardiomyocytes (n = 3-4 for each group) with the indicated genotypes. B. E2F1, SRC-1 and SRC-3 are associated with the E2F1-binding site proximate to the promoter of the cyclin E2 gene. The E2F1-binding site (E2F1 S.), the RNA transcriptional initiation site and the distant negative control site (Ctrl S.) in extron 6 (E6) of the mouse cyclin E2 gene are sketched. ChIP assays were performed with MEFs, P0 heart tissues and adult heart tissues using antibodies against E2F1, SRC-1 and SRC-3, and non-immune IgG as a negative control. Relative amounts of DNA associated with E2F1, SRC-1 and SRC-3 were quantitatively assayed by real time PCR. The experiments were repeated three times. C. Relative mRNA levels of myocardin in E12.5 (n = 4-6 for each group), P0 hearts (n = 4-6 for each group) and the primary myocardial cells in culture with the indicated genotypes. * and ** in panels A and C, p < 0.05 and p < 0.01 by Student's t test. D. SRC-1 and SRC-3 are associated with the enhancer site of the mouse myocardin gene. The known functional enhancer region and the distant negative control site in E10 of the mouse myocardin gene are sketched. ChIP assays were carried out with H9C2 cells, P0 hearts and MEFs using SRC-1 and SRC-3 antibodies, and the non-immune IgG as a negative control. The relative amounts of precipitated DNA were quantitatively measured by real time PCR. The experiments were repeated three times.
SRC-1 and SRC-3 work with myocardin to regulate myocardial cell differentiation
Multiple genes including ACTC1, TNNT2, MYH7, TAZ, LDB3, DTNA, FKBP12 and RXRα play important roles in myocardial cell differentiation and function during heart development. These gene mutations are usually associated with NCC development in human or mouse [42-48]. We measured the expression of these genes in the normal control and SRC-1 and/or SRC-3 knockout hearts and found a significant decrease in ACTC1 expression in the E12.5 S1KO;S3KO mouse hearts versus the E12.5 WT, S1KO and S3KO hearts as well as in the P0 S1KO;S3MKO mouse hearts versus the P0 S3F, S1KO;S3F and S3MKO hearts (Additional File 1: Suppl. Fig. S7A), while the expression of other genes did not change significantly in SRC-1 and/or SRC-3 mutant mouse hearts (Additional File 1: Suppl. Fig. S7B). Since ACTC1 encodes an essential protein for differentiated myocardial cells, its decreased expression suggests a contribution of SRC-1 and SRC-3 to myocardial differentiation during heart development.
The expression of ACTC1 is regulated by myocardin, a master regulator of smooth muscle gene expression required for myocardial differentiation [34, 49]. qPCR measurements revealed that myocardin expression was significantly reduced in the E12.5 S1KO;S3KO hearts versus WT and S1KO hearts, in the P0 S1KO;S3MKO and S3MKO hearts versus S3F hearts, and in SRC-1-/-;SRC-3d/d and SRC-3d/d primary myocytes versus SRC-3f/f and SRC-1-/-;SRC-3f/f primary myocytes (Fig. 6C). To understand the molecular mechanism responsible for SRC-1 and SRC-3-regulated myocardin expression, we performed ChIP assays for the enhancer element that controls myocardin transcription during heart development [34] (Fig. 6D). These ChIP assays revealed that both SRC-1 and SRC-3 were recruited to this enhancer region in H9C2 myoblastic cells and P0 mouse heart cells (Fig. 6D) that express SRC-1, SRC-3 (Additional File 1: Suppl. Fig. S6B) and myocardin (Additional File 1: Suppl. Fig. S7C), while both SRC-1 and SRC-3 were not associated with this region of the myocardin gene in MEFs (Fig. 6D) that express SRC-1 and SRC-3 (Additional File 1: Suppl. Fig. S6B) but not myocardin (Additional File 1: Suppl. Fig. S7C). The specificity of these ChIP assays was further confirmed by including negative controls using the non-immune IgG substitute for SRC-1 or SRC-3 antibodies and a distant DNA region in exon 10 of the myocardin gene that does not contain any known enhancers (Fig. 6D). These results suggest that SRC-1 and SRC-3 may promote myocardin expression through working as transcriptional coactivators for a TF associated with the major functional enhancer of the myocardin gene.
DISCUSSION
NCC is a recently defined genetic cardiomyopathy that causes frequent mortality among children suffering this disease. The incidence of NCC is estimated as 0.05% to 0.24% [50]. Because the responsible genetic mutations for NCC are still unknown, genetic diagnosis and prognosis are currently unavailable, and NCC is mainly diagnosed by clinical imaging. Furthermore, the molecular mechanisms responsible for the pathogenesis of NCC are unclear. In this study, we demonstrated that knockout of SRC-1 and SRC-3 in the mouse myocardial lineage resulted in prominent trabeculae, deep intertrabecular recesses, thin ventricular wall and septum, failed trabecular compaction, decreased internal ventricular dimension, decreased cardiac ejection fraction and output, as well as a high rate of postnatal mortality. Similar but lesser morphological and functional abnormalities were also observed in the hearts of S3MKO mice with myocardial knockout of SRC-3. These phenotypes very closely simulate the morphological and functional cardiac abnormalities of NCC patients, suggesting that dysfunction of SRC-1 and SRC-3 or their regulatory gene networks may develop NCC in human.
At the current time, it remains challenging to define the responsible genes and molecular regulations for NCC development due to limited availability of appropriate animal models. Mouse lines with RXRα or FKBP12 knockout have been reported to develop prominent myocardial trabecularization during embryonic development, but both lines are lethal before birth [47, 48, 51], making it impossible to study the roles of these genes in neonatal and postnatal development of NCC. Furthermore, since this NCC-like cardiac phenotype observed in RXRα and FKBP12 knockout mouse embryos is always associated with other congenital heart problems [47, 48, 51], these mouse lines can not model the isolated NCC disease. Moreover, myocardium-specific knockout of either RXRα or FKBP12 does not cause a NCC-like phenotype in mouse hearts [52, 53], suggesting that the NCC-like phenotype observed in their global knockout embryos is a secondary defect. Thus, RXRα and FKBP12 knockout mouse lines may not be ideal models for studying NCC. Instead, the S1KO;S3MKO mouse line is a more appropriate animal model of human NCC disease (Table 2).
S1KO;S3MKO and S3MKO mice are better noncompaction cardiomyopathy (NCC) models versus other knockout mouse models.
| NCC-related Abnormalities | S1KO;S3MKO | S3MKO | RXRα KO | FKBP12 KO | |
|---|---|---|---|---|---|
| Thin ventricular wall, small septum, & disorganized trabecular network | Yes | Yes | Yes | Yes | |
Major Structural Defects | Prominent myocardial trabecularization | Yes | Yes | Yes | Yes |
| Deep intertrabecular recesses | Yes | Yes | Yes | Yes | |
Diagnosis | The ratio of noncompacted to compacted myocardial structure (>2) | Yes | Yes | Yes | Yes |
Sub-types | Isolated form | Yes | Yes | No | No |
| Non-isolated form | Yes | Yes | Yes | Yes | |
Symptoms | High mortality rate from infant to young adulthood | Yes | No | No | No |
| Left ventricular systolic dysfunction | Yes | Yes | N.A. | N.A. | |
| Blood clots in the recesses | Yes | Yes | N.A. | N.A. | |
| Myocardial infarction | Yes | Yes | N.A. | N.A. | |
N.A., not available due to lethality.
Firstly, this model links myocardial proliferation and differentiation problems to NCC development, providing an opportunity to study the relationship between myocardial development at early heart developmental stages and NCC initiation and progression at later stages. Secondly, the NCC phenotype observed in the majority of S1KO;S3MKO mice is not associated with other congenital heart defects and thereby can serve as a model for isolated NCC. Thirdly, S1KO;S3MKO mice develop all NCC-related cardiac structural abnormalities and common symptoms, including prominent myocardial trabecularization, deep intertrabecular recesses, intramyocardial thrombosis and myocardial infarction at perinatal stages, high rate mortality at young ages and ventricular dysfunction in survived adult mice (Table 2). In addition, S3MKO mice also exhibit a lesser severe NCC-like phenotype versus S1KO;S3MKO mice, suggesting that the S3MKO mouse line may be used as a model of NCC with less severe symptoms (Table 2). Since S1KO mice do not have any NCC-like abnormalities and S1KO;S3MKO mice have much more severe NCC-like phenotype versus S3MKO mice, SRC-3 may play a more prominent role than SRC-1 does to support heart development and function. Finally, given the similarities between the cardiac phenotypes of S1KO;S3MKO and S3MKO mice and the human NCC disease, it should be interesting to investigate whether SRC-1 and SRC-3 genes have any genetic mutations and/or loss of functions in human patients with NCC in future studies.
Interestingly, SRC-1 and SRC-3 are mainly expressed in the cardiomyocytes of the mouse heart from E7.5 to P14. Since their temporal expression profiles positively correlate with the time window of cardiomyocyte proliferation and differentiation during cardiac development, we proposed that SRC-1 and SRC-3 could play important roles in myocardial proliferation and differentiation. Indeed, our data demonstrated that myocardial double knockouts of SRC-1 and SRC-3 drastically reduced cardiomyocyte proliferation in the developing hearts at both E12.5 and E16.5 and in the primary myocardial cells in culture. Interestingly, although knockout of either SRC-1 or SRC-3 did not significantly change cardiomyocyte proliferation in the hearts at E12.5 and E16.5, knockout of either SRC-1 or SRC-3 in the primary cardiomyocyte significantly decreased their proliferation in culture. These results suggest that both SRC-1 and SRC-3 are involved in the regulation of cardiomyocyte proliferation in vivo and in vitro, while the loss of SRC-1 or SRC-3 function in the heart is compensated by each other or by other unknown factors under in vivo conditions.
SRC family members serve as transcriptional coactivators for supporting gene expression mediated by multiple TFs, including nuclear hormone receptors, E2F1, AP-1, PEA3 and Mef2c [12, 15, 33, 54-57]. In this study, we found double knockouts of SRC-1 and SRC-3 markedly reduced cyclin E2 expression in E12.5 and P0 hearts and primary cardiomyocytes and also reduced cyclin B1 expression in E12.5 and P0 hearts. These results suggest that SRC-1 and SRC-3 may control myocardial proliferation through regulating the expression of cyclins. ChIP assays confirmed that E2F1, SRC-1 and SRC-3 are associated with a chromatin region that contains a known E2F1-binding site important for cyclin E2 expression [40]. These results indicate that cyclin E2 is a direct target gene of E2F1, SRC-1 and SRC-3. These observations are consistent with previous studies showing that SRC-3 interacts with E2F1 to promote E2F1-mediated gene expression [41, 58] and that SRC-3 accelerates cell proliferation in breast and prostate tumor cells [32, 59]. Additionally, we also found that SRC-1 and SRC-3 are recruited to a major enhancer region of the myocardin gene and knockout of SRC-1 and/or SRC-3 reduced myocardin expression levels in E12.5 and P0 hearts and in the primary myocytes in culture. Although TF working with SRC-1 and SRC-3 at this enhancer is still unknown, these results indicate that SRC-1 and SRC-3 may also regulate myocardin expression to regulate cardiomyocyte differentiation during heart development. Together, these results suggest SRC-1 and SRC-3 work with multiple TFs to regulate both myocardial cell proliferation and differentiation during heart development and their dysfunction would decrease the myocardial cell number and the ventricular contrasting force.
It has been proposed that NCC may be caused by arrested cardiac embryogenesis [5]. During early heart development, the trabeculae, a loose interwoven fiber network in the ventricular chambers, is formed from cardiomyocytes to increase myocardial oxygenation. These trabeculae process compaction to form the compacted ventricular wall at the end of systole. It is believed that the failure of trabecula-processed compaction will lead to NCC [5]. Since the thin myocardial tissues have been observed in the mouse hearts with perturbed epicardial development, insufficient coronary circulation has also been proposed to influence myocardial compaction [60]. However, no study has investigated how NCC development is related to the disturbed cardiomyocyte proliferation and differentiation and their underlying molecular mechanisms. In this study, we found that SRC-1 and SRC-3 are mainly expressed in the cardiomyocytes during heart development in the embryonic and early postnatal stages to regulate their proliferation and differentiation, while the NCC-related heart dysfunction characterized by the reduced eject fraction and cardiac output is observed in the adult S1KO;S3MKO and S3MKO mice where both SCR-1 and SRC-3 proteins are barely detectable. These findings indicate that the NCC-like phenotype in these adult knockout mice is a consequence of myocardium-developmental problems experienced in the embryonic and early postnatal developmental stages. Thus, this study provides a first direct link between the abnormal development of cardiomyocytes at early developmental stages and the NCC development at later stages of the entire lifespan. These initial findings also suggest that studies to develop therapeutic strategies for treating NCC should be focused on early heart developmental stages.
Supplementary Material
Additional File 1Suppl. Figures S1-S7.
Suppl. Video 1.
Suppl. Video 2.
Suppl. Video 3.
Suppl. Video 4.
Acknowledgements
This work was supported by National Institutes of Health grants CA112403, DK058242, DK59820 and HL093484. We thank the Genetically Engineered Mouse (GEM) Core of Baylor College of Medicine for generating genetically manipulated mouse models. The GEM Core is partially supported by the NIH grant P30 CA125123.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890-900
2. Marelli AJ, Mackie AS, Ionescu-Ittu R. et al. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation. 2007;115:163-72
3. Bruneau BG. The developmental genetics of congenital heart disease. nature. 2008;451:943-8
4. Jenni R, Oechslin E, Schneider J. et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666-71
5. Weiford BC, Subbarao VD, Mulhern KM. Noncompaction of the ventricular myocardium. Circulation. 2004;109:2965-71
6. Maron BJ, Towbin JA, Thiene G. et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-16
7. Markiewicz-Loskot G, Moric-Janiszewska E, Loskot M. et al. Isolated ventricular non-compaction: clinical study and genetic review. Europace. 2006;8:1064-7
8. Miranda EP, Albuquerque LA, Menezes RA Jr. et al. Isolated noncompacted myocardium in an elderly patient. Clinics (Sao Paulo). 2008;63:567-70
9. Swinkels BM, Boersma LV, Rensing BJ. et al. Isolated left ventricular noncompaction in a patient presenting with a subacute myocardial infarction. Neth Heart J. 2007;15:109-11
10. Martinez-Baca Lopez F, Alonso Bravo RM, Rodriguez Huerta DA. Echocardiographic features of non-compaction cardiomyopathy: missed and misdiagnosed disease. Arq Bras Cardiol. 2009;93:e33-5
11. Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681-92
12. Xu J, Wu RC, O'Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615-30
13. Xu J, Qiu Y, DeMayo FJ. et al. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science. 1998;279:1922-5
14. Nishihara E, Yoshida-Komiya H, Chan CS. et al. SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J Neurosci. 2003;23:213-22
15. Walsh CA, Qin L, Tien JC. et al. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int J Biol Sci. 2012;8:470-85
16. Gehin M, Mark M, Dennefeld C. et al. The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol Cell Biol. 2002;22:5923-37
17. Mukherjee A, Soyal SM, Fernandez-Valdivia R. et al. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol Cell Biol. 2006;26:6571-83
18. Xu J, Liao L, Ning G. et al. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379-84
19. Onate SA, Tsai SY, Tsai MJ. et al. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354-7
20. Chen H, Lin RJ, Schiltz RL. et al. Nuclear Receptor Coactivator ACTR Is a Novel Histone Acetyltransferase and Forms a Multimeric Activation Complex with P/CAF and CBP/p300. Cell. 1997;90:569-80
21. Torchia J, Rose DW, Inostroza J. et al. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677-84
22. Anzick SL, Kononen J, Walker RL. et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965-8
23. Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci U S A. 1997;94:8479-84
24. Mark M, Yoshida-Komiya H, Gehin M. et al. Partially redundant functions of SRC-1 and TIF2 in postnatal survival and male reproduction. Proc Natl Acad Sci U S A. 2004;101:4453-8
25. Chen X, Liu Z, Xu J. The cooperative function of nuclear receptor coactivator 1 (NCOA1) and NCOA3 in placental development and embryo survival. Mol Endocrinol. 2010;24:1917-34
26. Liu Z, Liao L, Zhou S. et al. Generation and validation of a mouse line with a floxed SRC-3/AIB1 allele for conditional knockout. Int J Biol Sci. 2008;4:202-7
27. Tien JC, Liao L, Liu Y. et al. The steroid receptor coactivator-3 is required for developing neuroendocrine tumor in the mouse prostate. Int J Biol Sci. 2014;10:1116-27
28. Moses KA, DeMayo F, Braun RM. et al. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176-80
29. Lee DK, Liu Y, Liao L. et al. The prostate basal cell (BC) heterogeneity and the p63-positive BC differentiation spectrum in mice. Int J Biol Sci. 2014;10:1007-17
30. Liu Z, Chen X, Zhou S. et al. The histone H3K9 demethylase Kdm3b is required for somatic growth and female reproductive function. Int J Biol Sci. 2015;11:494-507
31. Li Q, Chu MJ, Xu J. Tissue- and nuclear receptor-specific function of the C-terminal LXXLL motif of coactivator NCoA6/AIB3 in mice. Mol Cell Biol. 2007;27:8073-86
32. Kuang SQ, Liao L, Zhang H. et al. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875-85
33. Qin L, Chen X, Wu Y. et al. Steroid receptor coactivator-1 upregulates integrin alpha(5) expression to promote breast cancer cell adhesion and migration. Cancer Res. 2011;71:1742-51
34. Creemers EE, Sutherland LB, McAnally J. et al. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development. 2006;133:4245-56
35. Rapezzi C, Leone O, Ferlito M. et al. Isolated ventricular non-compaction with restrictive cardiomyopathy. Eur Heart J. 2006;27:1927
36. Zak R. Cell proliferation during cardiac growth. Am J Cardiol. 1973;31:211-9
37. Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res. 1974;35(suppl II):17-26
38. Li JM, Poolman RA, Brooks G. Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol. 1998;275:H814-22
39. Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15-26
40. Caldon CE, Sergio CM, Schutte J. et al. Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc. Mol Cell Biol. 2009;29:4623-39
41. Louie MC, Zou JX, Rabinovich A. et al. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157-71
42. Klaassen S, Probst S, Oechslin E. et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893-901
43. Monserrat L, Hermida-Prieto M, Fernandez X. et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28:1953-61
44. Bleyl SB, Mumford BR, Thompson V. et al. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet. 1997;61:868-72
45. Ichida F, Tsubata S, Bowles KR. et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103:1256-63
46. Xing Y, Ichida F, Matsuoka T. et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88:71-7
47. Shou W, Aghdasi B, Armstrong DL. et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. nature. 1998;391:489-92
48. Gruber PJ, Kubalak SW, Pexieder T. et al. RXR alpha deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J Clin Invest. 1996;98:1332-43
49. Imamura M, Long X, Nanda V. et al. Expression and functional activity of four myocardin isoforms. Gene. 2010;464:1-10
50. Madan S, Mandal S, Bost JE. et al. Noncompaction cardiomyopathy in children with congenital heart disease: evaluation using cardiovascular magnetic resonance imaging. Pediatr Cardiol. 2012;33:215-21
51. Mascrez B, Ghyselinck NB, Chambon P. et al. A transcriptionally silent RXRalpha supports early embryonic morphogenesis and heart development. Proc Natl Acad Sci U S A. 2009;106:4272-7
52. Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943-9
53. Maruyama M, Li BY, Chen H. et al. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ Res. 2011;108:1042-52
54. Chen SL, Dowhan DH, Hosking BM. et al. The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev. 2000;14:1209-28
55. Qin L, Liao L, Redmond A. et al. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937-50
56. Qin L, Liu Z, Chen H. et al. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009;69:3819-27
57. Tien JC, Xu J. Steroid receptor coactivator-3 as a potential molecular target for cancer therapy. Expert Opin Ther Targets. 2012;16:1085-96
58. Yuan Y, Qin L, Liu D. et al. Genetic screening reveals an essential role of p27kip1 in restriction of breast cancer progression. Cancer Res. 2007;67:8032-42
59. Chung AC, Zhou S, Liao L. et al. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Res. 2007;67:5965-75
60. Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15:165-76
Author contact
![]() Corresponding author: Jianming Xu, PhD, One Baylor Plaza, Houston, TX 77030, USA. E-mail: jxuedu
Corresponding author: Jianming Xu, PhD, One Baylor Plaza, Houston, TX 77030, USA. E-mail: jxuedu
Received 2015-4-14
Accepted 2015-6-6
Published 2015-7-15