Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Overview of Pancreatic Cancer
Prelude
Non-cellular mechanisms...
Future directions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):273-282. doi:10.7150/ijbs.14951 This issue Cite
Review
The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms
Saswati Chand1, Kevin O'Hayer1,2,3, Fernando F. Blanco1,3, Jordan M. Winter1, Jonathan R. Brody1, ![]()
1. Department of Surgery, The Jefferson Pancreas, Biliary, and Related Cancer Center;
2. Department of Medical Oncology, and the
3. Department of Pharmacology & Experimental Therapeutics, Division of Clinical Pharmacology, Thomas Jefferson University, Philadelphia PA.
Published 2016-1-27
Abstract
Pancreatic cancer (pancreatic ductal adenocarcinoma, PDA) is infamously moving to the top of the list as one of the most lethal cancers with an overall 5 year survival rate of 7%. Multiple genomic-based and molecular characterization studies of PDA specimens and established animal models have provided the field with multiple targets and a progression model of this disease. Still, to date, the best therapeutic options are surgery and combination cytotoxic therapies. In general, even in the best case scenario (i.e., an early stage diagnosis and a response to a specific therapy), most of these fortunate patients' PDA cells acquire or exert resistance mechanisms and eventually kill the patient. Herein, we touch on a growing field of investigation that focuses on PDA cell therapeutic resistance mechanisms. We examine extrinsic elements (i.e., the tumor microenvironment, hypoxia) to the intrinsic processes within the cell (i.e., post-transcriptional gene regulation and somatic mutations) that are important for therapeutic efficacy and resistance. Even as better targeted and personalized approaches move through the clinical trial pipeline the discussed resistance mechanisms will most likely play a role in the management of this deadly disease.
Keywords: Pancreatic Ductal Adenocarcinoma, Chemotherapeutic Resistance, Hypoxia, HuR, DNA Damage
Overview of Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDA) is a highly lethal malignancy with a 5-year survival rate of 7%[1]. It is expected that by 2020 PDA will surpass breast and colorectal cancer to become the second most common cause of cancer-related deaths[2]. Currently, there are no active screening methods to detect PDA at early stages, and patients with localized disease exhibit no overt symptoms. Thus, PDA is often diagnosed too late and the few available therapeutic options have little durable activity. In fact, if evidence of the disease is detected early, the only potentially curative option for pancreatic cancer is surgery, followed by adjuvant chemotherapy. Nevertheless, early recurrence and disease progression after surgery is evident in a large proportion of patients. Though the underlying cause(s) of recurrence or disease progression remain largely unknown, key culprits are undetected micrometastases and cellular drug resistance mechanisms.
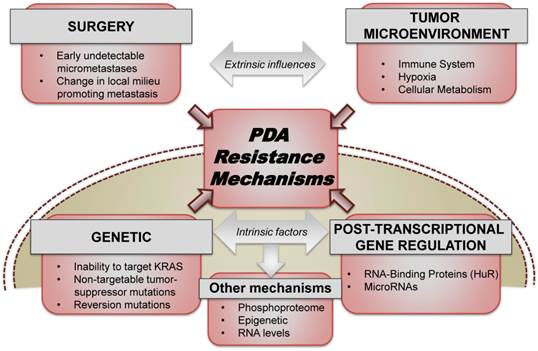
In the metastatic setting, two recent randomized-controlled trials demonstrated the advantage of combination therapies over single agent gemcitabine, a nucleoside analogue that had been the standard of care since 1997[3]. To this extent, combining Folinic acid, 5-Fluorouracil (5-FU), Irinotecan, and Oxaliplatin (FOLFIRINOX) provided patients with a 4.3 month increase in overall survival when compared to gemcitabine alone (hazard ratio for death, 0.57; 95% confidence interval [CI], 0.45 to 0.73; P<0.001)[4, 5]. Similarly, gemcitabine plus nab-paclitaxel (abraxane) increased overall survival by approximately 2 months when compared to single-agent gemcitabine (hazard ratio for death, 0.72; 95% confidence interval [CI], 0.62 to 0.83; P<0.001)[6]. Despite the recent success of these trials, patients who respond to these therapies inevitably recur and succumb to their disease. In this perspective, we survey current therapeutic resistance mechanisms both extrinsically including the tumor microenvironment and also intrinsically within the cellular machinery of PDA cells (Fig. 1).
PDA resistance mechanisms. Both extrinsic factors (which include the role of surgery, tumor microenvironment, stroma, etc.) and intrinsic cellular factors (ranging from somatic mutations, transcriptional and post-transcriptional reprogramming) contribute significantly to acute and acquired resistance to the existing therapeutic approaches used for treating PDA.
Prelude
We refer the reader to other outstanding reviews and publications on elements of the tumor microenvironment, drug availability and honing issues[7-10]. We will focus on the classical view of therapeutic resistance mechanisms that are most likely intact in the majority of PDAs. In brief, we are aware that there are distinct differences between innate/acute and acquired resistance therapeutic mechanisms. The somatic mutation section is clearly describing a putative acquired resistance event that would take at least months to develop, compared to other described mechanisms herein that may take only hours to disrupt the biology enough to affect drug efficacy. However, we disclose upfront that for the purpose of this review we do not label each aspect of resistance as acute or acquired.
1. An element of the microenvironment: Hypoxia-Induced Resistance: Tumor formation and the tumor microenvironment pose a unique set of challenges to the neoplastic cell including decreased glucose concentrations, oxidative stress, poor vascularization, low partial pressures of oxygen and low intratumoral perfusion (Fig. 1). This nutrient and oxygen poor environment imparts a selective pressure, favoring the growth of the most aggressive and fit PDA cells[11-13]. It is these cells which tend to be the most elusive to cytotoxic chemotherapeutic agents[11, 14, 15]. In order to overcome the harsh stress imposed by chronic hypoxia (e.g.: low oxygen pressure [pO2] and intratumoral perfusion) PDA cells orchestrate a multi-faceted response by activating hypoxia-inducible factors (HIFs; e.g.: HIF-1α, PIM1, CAIX, PDK1, CCND1)[16]. This potent acute cellular reprogramming activates pathways responsible for regulating cell motility, intracellular pH, mitochondrial function, angiogenesis, cellular metabolism, DNA repair, and cell survival[17-19].
Our mechanistic understanding of hypoxia-induced chemoresistance is limited, particularly in PDA models. Many previous studies focused on the role of HIF-1α and its downstream effectors in this process. In various tumor models, HIF-1α is implicated in the upregulation of VEGF, MDR/P-gp, CAIX, Glut1, WSB-1, Shh, LDH-A and Bcl2[20-25]. This transcriptional response is pleiotropic in nature and involved in apoptosis, cellular senescence, cellular metabolism, DNA repair, drug resistance, and oxidative stress[20-25]. HIF-1α has been directly linked to PDA hypoxic chemoresistance as well. In 2014, Cheng et.al. demonstrated under hypoxic conditions that siRNA inhibition of either HIF-1α or NFκB sensitized PDA cells to gemcitabine and increased apoptosis[26]. HIF-1α downstream targets have also been implicated in hypoxic chemoresistance. Maftouh et.al. examined the role of LDH-A, an enzyme involved in anaerobic glycolysis and known target of HIF-1α, in hypoxic chemoresistance[25]. They demonstrated increased gemcitabine chemosensitivity utilizing both siRNA as well as novel LDH-A inhibitors. This study provides rationale for targeting cellular metabolism in converting cells to a chemosensitive state under hypoxic stress.
HIF-1α independent pathways have also been implicated in hypoxic chemoresistance in PDA. Chen et.al. examined PIM1, a serine/threonine kinase with pleiotropic effects on cell survival, apoptosis, and metabolism[27]. They used a dominant negative PIM1 to rescue sensitivity to 5-FU, gemcitabine and cisplatin in hypoxia. They were further able to show that PIM1 acted to stabilize the mitochondrial membrane and inhibit apoptosis in a HIF-1α-independent manner. Our lab has delineated the mechanism by which PIM1 is upregulated in hypoxia and implicated the RNA binding protein HuR in hypoxic chemoresistance (Blanco et al, Oncogene in press). This work demonstrates upon hypoxic stimuli, HuR (ELAVL1) is translocated to the cytoplasm where it stabilizes the 3' UTR of the PIM1 transcript. Using siRNA as well as a novel pharmacologic inhibitor of HuR, the PIM1-HuR axis was demonstrated to be necessary for hypoxic chemoresistance to 5-FU and oxaliplatin. Finally, multiple studies by Onishi et.al have elucidated the importance of the sonic hedgehog (Shh) pathway in hypoxic chemoresistance[22, 23]. The Shh pathway is involved in pancreatic development and is upregulated under hypoxic conditions[23, 28]. Utilizing cyclopamine, an Shh pathway inhibitor, the investigators rescued 5-FU and gemcitabine resistance in hypoxic conditions. Accordingly, using siRNA towards HIF-1α, they showed that Shh mediated hypoxic chemoresistance was HIF-1α independent.
Translational researchers have recently attempted to use the hypoxic tumor environment as a therapeutic advantage. TH-302 (evofosfamide) is a 2-nitroimidazole prodrug of the cytotoxin bromo-isophosphoramide mustard. Under hypoxic conditions, the prodrug undergoes reduction and preferentially releases the active drug in the hypoxic tumor environment[29]. A recent phase II study reported that co-administering TH-302 with gemcitabine resulted in an increase in progression-free survival in patients with advanced PDA[30]. While current studies are being done in the backbone of gemcitabine-abraxane, these results indicate that targeting tumor hypoxia may be a viable approach to overcome resistance in PDA.
Although the studies outlined above have begun to elucidate the specific mechanisms as well as future avenues for targeting hypoxic chemoresistance, there is still much work to be done. Most studies have been performed using disparate model systems and cancer types. It is unknown how many of these mechanisms are tumor cell type specific or whether they represent more universal mechanisms of hypoxic chemoresistance. Furthermore, there has been no comprehensive evaluation using novel techniques to delve into this important mechanism of resistance. With the advent of gene editing, next generation sequencing, and proteomic advances over the past few years, unbiased screens may serve to answer these questions in the future.
2. Cancer Somatic mutations as drivers of resistance: Classical progression of pancreatic carcinogenesis has been classified into early (telomere shortening and activating mutations in KRAS), intermediate (inactivating mutations or epigenetic silencing of CDKN2A2) and late (inactivating mutations of TP53 and SMAD4) events[31]. High throughput sequencing and copy number studies of several PDA genomes have identified and validated genes such as KRAS, TP53, CDKN2A, SMAD4, etc. as well as identifying novel gene mutations that may be involved in cell growth, DNA repair, invasiveness and angiogenesis[15, 32-34] (Fig. 1). However, this extensive understanding of the somatic genetic landscape of PDA has yet to substantially contribute to an improvement in prognosis and treatment strategies. The unmet need can be potentially bridged by shifting focus to downstream events including, metabolic reprogramming, angiogenesis alterations, cell cycle abnormalities, overcoming stromal-microenvironment reaction, immune pathways and inflammation. While FOLFIRINOX and gemcitabine plus abraxane form the backbone of current frontline therapies for PDA, targeted therapeutics based on the above sequencing studies are being widely explored to combat and overcome acquired resistance in metastatic disease.
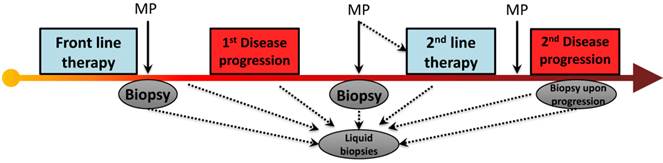
A review of recent modern chemotherapy trials reveal that front-line therapy may cause a RECIST response (or 30% shrinkage) in 1/3 of patients[4, 14] and stabilize disease in another 1/3, virtually all patients will ultimately progress, with a median time to progression of roughly 5 months. It is fascinating to consider that the mechanism of PDA drug resistance to chemotherapy is essentially unknown. Our group has focused on an understudied aspect molecular adaptation that occurs within hours of chemotherapy exposure, and is governed by regulatory proteins that rapidly change the expression of numerous pro-survival proteins by affecting their RNA stability (see section below)[35]. However, it is tempting to speculate that pro-survival somatic mutations are acquired in response to the intense selection pressure imposed by chemotherapy, and these heritable molecular events predominate in resistance clones. This molecular pattern has been well documented in numerous cancer genes, as 'secondary mutations' that occur in response to targeted therapies (e.g., KIT and imatinib [36], gefitinib and EGFR [37], crizotinib and ALK [38], vemurafanib and BRAF [39], vismodegib and SMO [40]). Interestingly, this theory has never been thoroughly investigated in pancreatic cancers that are resistant to standard chemotherapies. A clinical trial funded by the Pancreatic Cancer Action Network (PI Brody) will, in part, directly address this question if ex vivo modeling allows for sampling of pre- and post- treated specimens (Fig. 2).
PDA progression and treatment strategies. In the era of next generation sequencing and novel molecular diagnostics, the ability to use circulating tumor cells/DNA (and conventional biopsies) allows us to detect and map accumulated somatic mutations and molecular alterations which may be a result of disease progression or exposure to chemotherapy. (Abbreviations. MP- Molecular Profiling)
3. An alternative pathway to resistance: Post-transcriptional gene regulation (PTGR) is the modulation of RNA stability and expression, primarily mediated by RNA binding proteins (RBPs) and microRNAs (Fig. 1). Downstream effects of such modulation include alternative splicing, RNA processing and nuclear export and redirecting the RNA towards storage, translation or degradation. For instance, MUC4, a protein that plays a major role in pancreatic tumorigenesis, undergoes post-transcriptional regulation and alternative splicing to generate a variant MUC4/4, which has been linked to increased malignancy and resistance to apoptosis[41].
RNA-Binding proteins in cancer prognosis or treatment responses
Because of their ability to globally affect varied regulatory networks, RBPs regulate several cellular processes such as cancer initiation and progression, immunological responses, and neurological processes. Specifically in the context of tumorigenesis, post-transcriptional modulation contributes to changes in tumor cell growth and proliferation, angiogenesis, invasion and metastasis, drug responses and ultimately cancer prognosis. Of particular importance, RBPs such Human Antigen R (HuR), Tristetraproline (TTP), Sam68, eIF4E, La, AUF1, play a significant role in regulating tumor responses.
HuR and chemotherapeutic resistance
HuR's function is largely dependent on its overall expression and cellular localization in response to cancer-associated stimuli. Stress-induced cytoplasmic translocation of HuR and its subsequent stabilization of specific pro-survival transcripts have been linked to drug resistance in solid tumors. For instance, HuR-mediated up-regulation of proteins in the MAPK and JNK signaling pathways has been shown to cause tamoxifen resistance in breast cancer MCF-7 cells[42]. Several reports indicate the interplay between HuR and microRNA's resulting in chemotherapeutic resistance in ovarian[43], prostate[44], breast[45], colon[46, 47] and pancreatic cancer[48, 49]. A recent study indicated that cancer- associated stressors such as a highly oxidative environment, starvation and DNA-damaging agents correlate with a selective increase in expression of HuR, which in turn affects the phosphorylation of the initiation factor eIF4E and results in chemotherapeutic resistance[50]. In PDA, HuR expression regulates the stability and expression of enzyme dCK, which metabolizes the prodrug gemcitabine, thereby modulating response to the standard of care[51]. Another study pioneered the role of HuR in supporting a drug resistance phenotype through its stabilization of the mitotic kinase inhibitor, WEE1. In response to DNA damaging agents such as mitomycin C and platinum agents, HuR translocates to the cytoplasm wherein it stabilizes WEE1 mRNA, hence allowing cancer cells to pause at G2/M checkpoint, repair damaged DNA and evade apoptosis[35].
Role of MicroRNAs in cancer prognosis or treatment responses
MicroRNAs (mIRs) which can rapidly and effectively alter gene expression likely play a role in treatment efficacy[52-54]. For example, it has been reported that miR-22 regulates sensitivity of colorectal cancer cells to 5-FU by post-transcriptionally regulating crucial target genes which, in turn, facilitate autophagy[55]. Several studies have also indicated a link between deregulated miRNA expression profiles and chemoresistance in Ewing's sarcoma[56-59]. Similarly, microRNA signatures also influence resistance to TRAIL[60] and kinase inhibitors [61] in non-small cell lung and pancreatic cancer.
PARP Inhibition in PDA
Recently, Poly-ADP ribose polymerase (PARP) inhibitors (PARPi), which target the DNA damage repair (DDR) pathway, have delivered promising preclinical and clinical results. Mechanistically, PARPi work through the concept of synthetic lethality, by specifically targeting the Achilles' heel of cancer cells that are already carrying mutations in major DNA repair genes. Cells deficient in these enzymes encoded by tumor-suppressor genes, BRCA1 and BRCA2are therefore heavily dependent of PARP for DNA damage repair. PARP1, an enzyme with a native function to repair single-stranded breaks (SSBs) by base excision repair (BER) is now solely responsible for HR- function to repair DSBs arising from exposure to chemotherapy (alkylating agents, topoisomerase inhibitors, etc.). Therefore, inhibition of PARP1 via PARPi has proven to be effective towards treating patients with tumors that harbor mutations in DNA repair-related genes[62, 63]. This concept of synthetic lethality is a promising therapeutic strategy, which brings the medical oncology community closer to a 'personalized' approach towards treating a subset of PDA patients.
PDA stands as the third most common cancer associated with BRCA mutations[64]. Approximately 10% of PDA patients carry mutations in DNA repair genes such as BRCA1, BRCA2, Fanconi Anemia genes, PALB2, etc. which makes PARP inhibition the best personalized approach for treating this particular subset of PDA patients[65, 66]. Subsets of pancreatic cancer with specific somatic mutations correlate with in vitro chemosensitivity. A recent study found that the sensitivity of PARP1 inhibitor, additional to BRCA-deficiency, was influenced by expression profiles of DNA damage repair (DDR) pathway genes (ERCC3, RAD17, SUMO1, MUTYH, CRY1, HSP90B1, CDC37, RXRA, and USP5) [67]. Additionally, a screen identified the deubiquitylating enzyme USP11 as a participant in HR repair of DSBs. The loss of USP11 caused impaired recruitment of a subset of DSB repair proteins such as RAD51 and 53BP1 to the repair foci [68]. This suggests that, apart from BRCA and FA genes, PARP1 demonstrates synthetic lethality with other DDR genes. PARP1 is known to be hyperactive in BRCA1- or BRCA2-deficient cells and p53 also plays a role as a regulator of DNA repair pathways. PARP1 is shown to be hyperactive in BRCA1- or BRCA2-deficient cells. TP53 also plays a role as a regulator of DNA repair pathways. Therefore, patients carrying a gene defect in the DSB repair pathways together with p53 mutations are selected for PARP inhibitor therapy[69]. Currently, PARP inhibitors are in different phases of clinical trials, either as single agent (Olaparib) or in combination (Olaparib, Veliparib, Rucaparib, BMN673) with standard of care gemcitabine or other chemotherapeutic agents for treating patients with locally advanced, unresectable or metastatic pancreatic cancer.
However, de novo and acquired resistance to PARP inhibitors poses a significant clinical problem [70, 71]. To date, four PARPi resistant mechanisms have been highlighted in the literature [72]. A) Targeted mutation reversion: Successful targeted therapies in cancer can induce reversion mutations in a cell with a hypermutable state (e.g., Gleevec giving rise to BCR-Abl mutations) [73]. Secondary, reversion mutations that restore deleterious BRCA2 mutant function have been the predominantly described mechanism for PARP inhibitor resistance[74], and have been shown to be a result of acquired, not de novo mechanisms[75]. These findings were reproduced in cancer cells exposed to platinum-based therapies [76, 77]. A thorough study of independently derived PARPi- resistant BRCA2- mutant CAPAN1 cell lines indicated that BRCA2 function is not typically restored upon prolonged exposure to PARPi. Instead, deletion events in BRCA2 DNA either restored the ORF that encodes the C-terminal RAD51 binding domain or resulted in small tracts of DNA sequence homology arising from error-prone repair due to BRCA2 deficiency [77]. Perhaps the best evidence of a targeted mutation reversion came from a PARPi (olaparib) study that performed DNA sequencing on treatment-naive and post- treatment biopsies. However, only two patients harbored BRCA2 reversion mutations in olaparib-resistant metastases that restored BRCA2 function [78]. B) Hypermorphic/unclassified BRCA-alleles: Not all identified mutated BRCA1/2 patients respond to PARPi therapy and thus understanding all disease-related BRCA-alleles will be critical in predicting which patients will respond best to PARPi-based therapy. C) Ineffective PARPi uptake or drug export: Early pharmacodynamics studies have demonstrated that PARPi uptake is effective [79, 80], yet a P-glycoprotein-mediated drug resistance mechanism has been demonstrated in a mouse model [81]. Elevated expression of ABCB1, a P-glycoprotein efflux pump has been shown to cause Olaparib resistance; this can be abrogated by treatment with a PgP inhibitor, tariquidar[81]. However, it remains unknown whether these findings have any implications for patients. D) Rewiring of the DNA Damage Response: A more likely but complicated resistance mechanism is the compensation of DNA repair network, which will result in negating the 'synthetic lethal' setting in HR-deficient pancreatic cancer cells. Compensatory loss of another DNA repair factor, p53-binding protein 1 (53BP1) reduces non-homologous end joining (NHEJ) efficiency and is one of the best described DDR rewiring causing PARPi resistance [82, 83]. However, neither the validation of this DDR rewiring, nor the involvement of these molecules as biomarkers, have been fully established in human tumor samples from clinical trials.
An alternative post-transcriptional PARPi resistance mechanism (revisiting HuR): Recently, our group showed evidence that strongly suggests that PDA cells develop resistance to DNA damaging agents through post-transcriptional gene regulation[84] through the RNA-binding protein, HuR. Primarily localized in the nucleus, HuR translocates to the cytoplasm in response to cellular stress (e.g., DNA damage, nutrient depletion, and hypoxia)[85], where it becomes functionally active as a stress response protein. As an RBP, HuR binds to the AU-rich elements (AREs) typically in the 3'-untranslated regions (UTRs) of specific, survival target genes involved in cell proliferation, evading apoptosis [86], and mitotic inhibition (e.g., the mitotic kinase inhibitor WEE1) [84, 87-90]. Importantly, we have shown that chemical and genetic silencing of HuR results in modulation of key cell cycle regulator, WEE1 and efficacy of chemotherapy [84]. Indeed, a seminal study demonstrated that WEE1 inhibition enhances accumulation of lethal DNA damage and apoptosis, thereby enhancing sensitivity to PARP inhibitors in pancreatic cancer cells[91]. We have preliminary data demonstrating that PARPi: 1) induces HuR translocation from the nucleus to the cytoplasm and 2) HuR can regulate pro-survival transcripts that diminish the efficacy of PARPi and ultimately maybe a critical factor in causing PDA resistance to this targeted therapy (Chand et al., unpublished). Future studies will determine the significance that HuR biology has on the clinical effectiveness of PARPi-based therapies.
Non-cellular mechanisms affecting therapeutic resistance
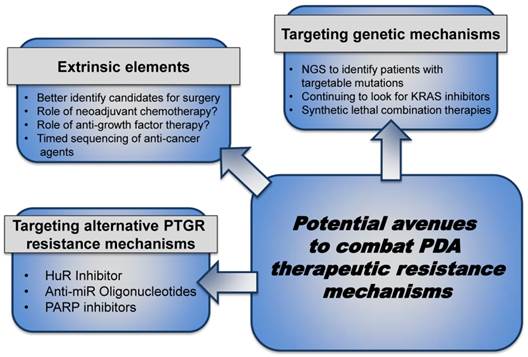
The above described cellular and molecular mechanisms either discuss how the milieu and/or the molecular machinery contribute to therapeutic resistance. Below we discuss how therapeutic pressures could either select for or strengthen metastatic clones. We also discuss possibilities in which to enhance outcomes (Fig. 3).
Combating PDA therapeutic resistance. Patients eligible for surgical resection can be further stratified to optimize treatment, gain maximal response after resection and improve prognosis by exploring neoadjuvant chemotherapy, anti-growth factor therapy, etc. to keep the tumor localized and prevent micrometastases. In the patients with metastatic disease, local or advanced, inhibiting the intrinsic pathways through combination 'targeted' strategies can improve response to current standard- of- care therapies. (Abbreviations. NGS- Next Generation Sequencing)
Surgery. Surgical resection provides the best chance of long term survival. The three-year survival for even the most effective modern chemotherapeutic regimens is 5% or less [4, 14]. In contrast, the 5-year survival in large surgical series is close to 20%, and roughly 1/3 of patients survive over 3 years [92]. Nevertheless, it is justifiable to question the role of surgery for the treatment of pancreatic cancer at all. The recurrence rate after resection, which is a pre-selected group of patients with a relatively favorable prognosis, is in excess of 90% [92]. Conversely, the cure rate is less than 10%. It stands to reason that occult residual disease (distant sites, regional metastases, or at the resection margin), are present in the majority of patients. Thus, why is resection a viable option, if the disease is so often systemic?
The strongest evidence in favor of surgery for localized disease is not biologic, but actually empiric. In Japan, forty-two patients with localized and resectable disease were randomized to resection and no adjuvant therapy vs. exploratory laparotomy and biopsy only, followed by chemoradiation. There were no long-term survivors in the group of patients who did not undergo resection, while 20% of patients in the resection group lived more than 3 years. Therefore, unlike most other cancers, the benefit of resection to treat pancreatic cancer is supported by randomized data. From a biologic standpoint, resection is able to functionally achieve a 'complete response' in one day's work (that is, render the patient with no radiographic evidence of disease). The patient typically has recovered completely by two months after surgery. In contrast, the most effective chemotherapy regimens are unable to achieve this result over the course of 6 months of treatment. If patients with pancreatic cancer ultimately die from a cancer syndrome when a critical volume of disease burden has been reached, then a complete resection can lengthen life by debulking 99% of the cancer cells. Assuming a cancer doubling time of 100 days, six doublings, or two years, are required to return to the pre-resection disease burden [93].
Rather than render surgery obsolete, it is more likely that improvements in chemotherapy in the future will expand the number of patients who are eligible or will likely benefit from resection. Simplistically, as treatments to control systemic disease improve, control of local disease by resection and/or radiation will likely become more important. Prospective, single-institution studies (albeit non-randomized) with modern multi-agent therapy reveal that the majority of patients with locally advanced disease may be converted from 'unresectable' to resectable, which far exceeds historical figures [94].
Despite the isolated, non-randomized studies surfacing now, the fact remains that roughly 20% of patients in the general population who develop pancreatic adenocarcinoma will have localized and resectable disease[95]. Pancreatic cancer is staged according to the AJCC TNM, 7th Ed. and based on the presence of regional lymph node metastases, distant metastases, and cancer involvement of major visceral vessels[96]. However, from a practical perspective, pancreatic cancer is surgically staged to determine resectability. Cancers localized to the pancreas are typically staged for resection using a multislice CT scan using three contrast phases (early arterial, late arterial, and venous) to carefully examine the relationship of the tumor to nearby vessels [97]. Localized cancers that do not invade and distort the superior mesenteric vein or portal vein, and do not abut the superior mesenteric or celiac arteries, are considered resectable and patients are typically offered resection (although neoadjuvant chemotherapy is an acceptable option). When the veins are distorted, or the arteries abutted by the tumor, the cancer is considered 'borderline resectable,' and most surgeons favor a neoadjuvant approach, using chemotherapy (with or without the addition or chemoradiation). Invasion of the veins with a technically reconstruction option, or encasement of the arteries indicate that the cancer is locally advanced, and unlikely to be treatable by resection. Modern chemotherapy regimens have increased the percentage of patients who are able to undergo an attempt at resection, after a course of neoadjuvant treatment for borderline or locally advanced pancreatic cancer.
Considerations: Although surgery is probably 'the best' option for a patient diagnosed with PDA and better adjuvant therapies need to be developed in order to improve outcomes, a few considerations remain about resectional therapy (Fig. 1): 1) Identification of biomarkers of early recurrence or progression, in order to spare patients with particularly aggressive cancers unnecessary surgery. 2) Examining the role of neoadjuvant treatment for resectable PDA. 3) Determining whether surgery, in some instances, cause a milieu in which growth factors or tissue dissection activate the spreading of micrometastateses. 4) Identifying whether neoadjuvant therapy, although relevant for down staging for resection, ultimately adds a selection process on distant metastatic cells and eventually strengthens the metastatic clones.
Immunotherapies: Tumor immunotherapy has become an effective tool in the treatment of many diseases, most notably melanoma[98]. A recent phase I study which evaluated the combination of an agonist CD40 monoclonal antibody in combination with gemcitabine in patients with metastatic chemotherapy-naive PDA indicated promise, though marginal [99]. Although it appears that like most promising therapeutics, PDA appears to be unresponsive to several new and established immunotherapeutic strategies. We do acknowledge the progress made and pursuit for a successful vaccine for use as a therapy [100].
Considerations: The mutational and cellular heterogeneity of PDA makes this a difficult disease to bolster the immune system to fight against. Thus, although tumor immunotherapy is an extremely promising strategy to combat cancer, 1) it remains to be determined if it is an effective treatment option for PDA cells. 2) Further studies are required to define if there are too few tumor infiltrating B lymphocytes that can circulate and find their way to PDA tumor cells in order for a T-cell based therapeutic strategy to work. 3) Future investigation should identify whether depleting the cellular elements of the tumor microenvironment will allow for proper honing and targeting of immuno- and chemotherapy. 4) Recognizing if PDA cells produce factors or enzymes, such as IDO2, that need to be targeted in order to truly combat the immune blockage against PDA.[101]
Targeted and personalized (and combination therapies): Combination therapies and even cytotoxic therapies along with new targeted approaches are being evaluated in clinical trials. Even in the best 'personalized approach' scenario, these targeted or combinatory approaches will most likely yield resistant PDA cells. As the therapies become better and we establish a better understanding of a molecularly tailored approach to treating PDA patients, we may need to think about other aspects of combination therapy and personalized approaches to treating this disease.
Considerations: 1) Determining the importance of the 'timed sequencing' of drug administration in combination treatment strategies in an effort to slow down and overcome resistance mechanisms. For instance, if one drug induces the subcellular localization of HuR (see above), that induces a survival network for another drug, it is crucial to determine if these drugs should be given separately or spaced out over days versus hours. 2) Additional elements of the cell machinery (not discussed here) include the adaptable phosphoproteome. As technologies advance, following the phosphoproteomic signature may be very relevant to drug resistance and optimal drug selection for a patient who recurs for 2nd and 3rd line therapies(Fig. 2) [102]. 3) Evaluate if targeting a specific disrupted pathway could either be trumped or further enhanced by the addition of a cytotoxic agent (e.g., a generic DNA damaging agent could potentially set the stage for a targeted approach against a DNA repair pathway such as PARP inhibition).
Future directions
We are hopeful that an era of better drug selections and therapeutic options are around the corner for PDA patients. Yet, until 'personalized medicine' and better targeted therapies are ready for the clinic, optimizing current therapeutic strategies that have activity in patients will be critical. Similarly, even if 'personalized therapy' becomes a reality, we will need to better understand how PDA cells become resistant to the best matched, available therapies (Fig. 3). Monitoring every patient's tumor in real time (i.e., biopsying and sequencing the tumor as a moving target) is what we may need to do to manage this disease effectively (Fig. 2).
Abbreviations
PDA: pancreatic ductal adenocarcinoma; 5-FU: 5 fluoruracil; HIF: hypoxia inducible factor; PIM1: proviral integration site for Moloney murine leukemia virus 1; CAIX: carbonic anhydrase IX; PDK1: pyruvate dehydrogenase kinase, isoenzyme 1; CCND1: cyclin D1; VEGF: vascular endothelial growth factor; MDR (ABCB1,pgp): multidrug resistance; Glut-1: glucose transporter 1; WSB1: WD repeat and SOCS box containing 1; Shh: sonic hedgehog; LDH-A: lactate dehydrogenase A; Bcl-2: B-cell CLL/lymphoma 2; NFκB: nuclear factor kappa light chain enhancer of activated B cells; HuR (ELAVL1): human antigen R; KRAS: Kirsten rat sarcoma viral oncogene homolog; CDKN2A: cyclin dependent kinase 2A; TP53: tumor protein p53; SMAD4: mothers against decapentaplegic homolog 4; RECIST: response evaluation criteria in solid tumors; KIT: v-kit Hardy Zuckerman 4 feline sarcoma viral oncogene homolog; EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma receptor tyrosine kinase; BRAF: Raf 1 proto-oncogene; SMO: smoothened; MUC4: mucin 4; TTP: tristetraproline; eIF4E: eukaryotic initiation factor 4E; La: La protein; MAPK: mitogen activated protein kinase; JNK: jun kinase; AUF1: AU-rich element RNA binding protein 1; WEE1: G2 checkpoint kinase; BRCA: breast cancer succeptibility gene; PARP: poly-ADP ribose polymerase; SSB: single strand break; BER: base excision repair; PALB2: partner and localizer of BRCA2; ERCC3: excision repair cross-complementation group 3; RAD: cell cycle checkpoint protein RAD; SUMO1: small ubiquitin-like modifier 1; MUTYH: mutY DNA glycosylase; CRY1: cryptochrome circadian clock 1; HSP90: heat shock protein 90B1; CDC37: cell division cycle 37; RXRA: retinoid x receptor alpha; USP: ubiquitin specific peptidase; BCR-Abl: breakpoint cluster region-Abelson murine leukemia viral oncogene; ARE: AU rich element; UTR: untranslated region; CD40: cluster of differentiation 40; IDO2: indoleamine 2,3-dioxygenase 2; RBP's: RNA binding proteins; DDR: DNA damage response; NGS: next generation sequencing.
Acknowledgements
This work was supported by NIH-NIGMS T32 GM008562 20; the Mary Halinski Pancreatic Cancer Research Fund; Fund for a Cure, Nancy Kay Engel Pancreatic Cancer Research Fund, and the Elkan Katz Memorial Fund for Pancreatic Cancer Research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Society AC. American Cancer Society: Cancer Facts and Figures. http: //wwwcancerorg/research/cancerfactsfigures/index. 2013
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
3. Burris HA3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403-13
4. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817-25
5. Conroy T. et al. Randomized phase III trial comparing FOLFIRINOX (F: 5FU/leucovorin [LV], irinotecan [I], and oxaliplatin [O]) versus gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA): Preplanned interim analysis results of the PRODIGE 4/ACCORD 11 trial. J Clin Oncol. 2010:28
6. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE. et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548-54
7. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, NY. 2011;331:1612-6
8. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266-76
9. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY. 2009;324:1457-61
10. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer cell. 2012;21:418-29
11. Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010;31:1703-9
12. Anderson AR, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905-15
13. Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM. et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48:919-22
14. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691-703
15. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6
16. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967-75
17. Buchler P, Reber HA, Buchler MW, Friess H, Lavey RS, Hines OJ. Antiangiogenic activity of genistein in pancreatic carcinoma cells is mediated by the inhibition of hypoxia-inducible factor-1 and the down-regulation of VEGF gene expression. Cancer. 2004;100:201-10
18. Chang Q, Jurisica I, Do T, Hedley DW. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71:3110-20
19. Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771-80
20. Liu L, Ning X, Sun L, Zhang H, Shi Y, Guo C. et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008;99:121-8
21. Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A. et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075-83
22. Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T. et al. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci. 2011;102:1144-50
23. Onishi H, Morifuji Y, Kai M, Suyama K, Iwasaki H, Katano M. Hedgehog inhibitor decreases chemosensitivity to 5-fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci. 2012;103:1272-9
24. Tong Y, Li QG, Xing TY, Zhang M, Zhang JJ, Xia Q. HIF1 regulates WSB-1 expression to promote hypoxia-induced chemoresistance in hepatocellular carcinoma cells. FEBS Lett. 2013;587:2530-5
25. Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R. et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer. 2014;110:172-82
26. Cheng ZX, Wang DW, Liu T, Liu WX, Xia WB, Xu J. et al. Effects of the HIF-1alpha and NF-kappaB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol Rep. 2014;31:1891-8
27. Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R. et al. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene. 2009;28:2581-92
28. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059-87
29. Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L. et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem. 2008;51:2412-20
30. Borad MJ, Reddy SG, Bahary N, Uronis HE, Sigal D, Cohn AL. et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. J Clin Oncol. 2015;33:1475-81
31. Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer journal (Sudbury, Mass. 2012;18:492-501
32. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications. 2015;6:6744
33. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW. et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science (New York, NY. 2009;324:217
34. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
35. Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA. et al. HuR Posttranscriptionally Regulates WEE1: Implications for the DNA Damage Response in Pancreatic Cancer Cells. Cancer research. 2014;74:1128-40
36. Wang CM, Huang K, Zhou Y, Du CY, Ye YW, Fu H. et al. Molecular mechanisms of secondary imatinib resistance in patients with gastrointestinal stromal tumors. Journal of cancer research and clinical oncology. 2010;136:1065-71
37. Nurwidya F, Takahashi F, Murakami A, Kobayashi I, Kato M, Shukuya T. et al. Acquired resistance of non-small cell lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Respiratory investigation. 2014;52:82-91
38. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W. et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011;71:6051-60
39. Hoogstraat M, Gadellaa-van Hooijdonk CG, Ubink I, Besselink NJ, Pieterse M, Veldhuis W. et al. Detailed imaging and genetic analysis reveal a secondary BRAF(L) (505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment cell & melanoma research. 2015;28:318-23
40. Pricl S, Cortelazzi B, Dal Col V, Marson D, Laurini E, Fermeglia M. et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Molecular oncology. 2015;9:389-97
41. Zhu Y, Zhang JJ, Xie KL, Tang J, Liang WB, Zhu R. et al. Specific-detection of clinical samples, systematic functional investigations, and transcriptome analysis reveals that splice variant MUC4/Y contributes to the malignant progression of pancreatic cancer by triggering malignancy-related positive feedback loops signaling. J Transl Med. 2014;12:309
42. Hostetter C, Licata LA, Witkiewicz A, Costantino CL, Yeo CJ, Brody JR. et al. Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol Ther. 2008;7:1496-506
43. Prislei S, Martinelli E, Mariani M, Raspaglio G, Sieber S, Ferrandina G. et al. MiR-200c and HuR in ovarian cancer. BMC Cancer. 2013;13:72
44. Kojima K, Fujita Y, Nozawa Y, Deguchi T, Ito M. MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate. 2010;70:1501-12
45. Srikantan S, Abdelmohsen K, Lee EK, Tominaga K, Subaran SS, Kuwano Y. et al. Translational control of TOP2A influences doxorubicin efficacy. Mol Cell Biol. 2011;31:3790-801
46. Yamakuchi M. MicroRNA Regulation of SIRT1. Front Physiol. 2012;3:68
47. Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167-80
48. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH. et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745-52
49. Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L. et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816
50. Martinez A, Sese M, Losa JH, Robichaud N, Sonenberg N, Aasen T. et al. Phosphorylation of eIF4E Confers Resistance to Cellular Stress and DNA-Damaging Agents through an Interaction with 4E-T: A Rationale for Novel Therapeutic Approaches. PLoS One. 2015;10:e0123352
51. Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A. et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer research. 2009;69:4567-72
52. Qu J, Zhao L, Zhang P, Wang J, Xu N, Mi W. et al. MicroRNA-195 chemosensitizes colon cancer cells to the chemotherapeutic drug doxorubicin by targeting the first binding site of BCL2L2 mRNA. Journal of cellular physiology. 2015;230:535-45
53. Zhang Y, Geng L, Talmon G, Wang J. MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. The Journal of biological chemistry. 2015;290:6215-25
54. Zhao L, Liu W, Xiao J, Cao B. The role of exosomes and "exosomal shuttle microRNA" in tumorigenesis and drug resistance. Cancer letters. 2015;356:339-46
55. Zhang H, Tang J, Li C, Kong J, Wang J, Wu Y. et al. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015;356:781-90
56. Ahmed AA, Zia H, Wagner L. Therapy resistance mechanisms in Ewing's sarcoma family tumors. Cancer Chemother Pharmacol. 2014;73:657-63
57. Misawa A, Katayama R, Koike S, Tomida A, Watanabe T, Fujita N. AP-1-Dependent miR-21 expression contributes to chemoresistance in cancer stem cell-like SP cells. Oncol Res. 2010;19:23-33
58. Radhakrishnan M, Hammond G, Jones PB, Watson A, McMillan-Shields F, Lafortune L. Cost of improving Access to Psychological Therapies (IAPT) programme: an analysis of cost of session, treatment and recovery in selected Primary Care Trusts in the East of England region. Behav Res Ther. 2013;51:37-45
59. Iida K, Fukushi J, Matsumoto Y, Oda Y, Takahashi Y, Fujiwara T. et al. miR-125b develops chemoresistance in Ewing sarcoma/primitive neuroectodermal tumor. Cancer Cell Int. 2013;13:21
60. Garofalo M, Quintavalle C, Di Leva G, Zanca C, Romano G, Taccioli C. et al. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene. 2008;27:3845-55
61. Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 2014;4:411-35
62. Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V. et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013;19:5485-93
63. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C. et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512-9
64. Brennan GT, Relias V, Saif MW. BRCA and pancreatic cancer. JOP. 2013;14:325-8
65. Bhalla A, Saif MW. PARP-inhibitors in BRCA-associated pancreatic cancer. JOP. 2014;15:340-3
66. Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S. et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111:1132-8
67. Cui Y, Brosnan JA, Blackford AL, Sur S, Hruban RH, Kinzler KW. et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6519-30
68. Wiltshire TD, Lovejoy CA, Wang T, Xia F, O'Connor MJ, Cortez D. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. The Journal of biological chemistry. 2010;285:14565-71
69. Nguyen D, Zajac-Kaye M, Rubinstein L, Voeller D, Tomaszewski JE, Kummar S. et al. Poly(ADP-ribose) polymerase inhibition enhances p53-dependent and -independent DNA damage responses induced by DNA damaging agent. Cell cycle. 2011;10:4074-82
70. Fojo T, Bates S. Mechanisms of resistance to PARP inhibitors-three and counting. Cancer discovery. 2013;3:20-3
71. Montoni A, Robu M, Pouliot E, Shah GM. Resistance to PARP-Inhibitors in Cancer Therapy. Frontiers in pharmacology. 2013;4:18
72. Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540-7
73. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN. et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science (New York, NY. 2001;293:876-80
74. Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP. et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer research. 2009;69:6381-6
75. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W. et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008-15
76. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer research. 2008;68:2581-6
77. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA. et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111-5
78. Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K. et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. The Journal of pathology. 2013;229:422-9
79. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009;361:123-34
80. Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T. et al. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nature communications. 2013;4:1504
81. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17079-84
82. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology. 2010;17:688-95
83. Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A. et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243-54
84. Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA. et al. HuR Posttranscriptionally Regulates WEE1: Implications for the DNA Damage Response in Pancreatic Cancer Cells. Cancer research. 2014;74:1128-40
85. Burkhart RA, Peng Y, Norris ZA, Tholey R, Talbott VA, Liang Q. et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol Cancer Res. 2013
86. Pineda DM. et al. HuR's post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer biology & therapy. 2012 TBD
87. Kurosu T, Ohga N, Hida Y, Maishi N, Akiyama K, Kakuguchi W. et al. HuR keeps an angiogenic switch on by stabilising mRNA of VEGF and COX-2 in tumour endothelium. Br J Cancer. 2011;104:819-29
88. Annabi B, Currie JC, Moghrabi A, Beliveau R. Inhibition of HuR and MMP-9 expression in macrophage-differentiated HL-60 myeloid leukemia cells by green tea polyphenol EGCg. Leukemia research. 2007;31:1277-84
89. Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. The EMBO journal. 2000;19:2340-50
90. Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley interdisciplinary reviews RNA. 2010;1:214-29
91. Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR. et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20:5085-96
92. Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J. et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J Gastrointest Surg. 2006;10:1199-210 discussion 210-1
93. Friberg S, Mattson S. On the growth rates of human malignant tumors: implications for medical decision making. Journal of surgical oncology. 1997;65:284-97
94. Sherman WH, Chu K, Chabot J, Allendorf J, Schrope BA, Hecht E. et al. Neoadjuvant gemcitabine, docetaxel, and capecitabine followed by gemcitabine and capecitabine/radiation therapy and surgery in locally advanced, unresectable pancreatic adenocarcinoma. Cancer. 2015;121:673-80
95. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5-29
96. Edge SE, Byrd DR. AJCC Cancer Staging Manual. 7th ed. New York: Springer. 2009
97. Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Annals of surgical oncology. 2010;17:2803-5
98. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (New York, NY. 2015;348:62-8
99. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A. et al. A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2013;19:6286-95
100. Salman B, Zhou D, Jaffee EM, Edil BH, Zheng L. Vaccine therapy for pancreatic cancer. Oncoimmunology. 2013;2:e26662
101. Witkiewicz AK, Costantino CL, Metz R, Muller AJ, Prendergast GC, Yeo CJ. et al. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. Journal of the American College of Surgeons. 2009;208:781-7 discussion 7-9
102. Pierobon M, Wulfkuhle J, Liotta L, Petricoin E. Application of molecular technologies for phosphoproteomic analysis of clinical samples. Oncogene. 2015;34:805-14
Author contact
![]() Corresponding author: Jonathan R. Brody, Ph.D., Department of Surgery, Thomas Jefferson University. 1015 Walnut Street Ste 620, Curtis Bldg, Philadelphia, PA 19107. Email: jonathan.brodyedu, Telephone: (215) 955-2693; Fax: (215) 923-6609
Corresponding author: Jonathan R. Brody, Ph.D., Department of Surgery, Thomas Jefferson University. 1015 Walnut Street Ste 620, Curtis Bldg, Philadelphia, PA 19107. Email: jonathan.brodyedu, Telephone: (215) 955-2693; Fax: (215) 923-6609