Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Histopathology of PanIN
Molecular Biology of PanIN
Molecular Biomarkers of...
Molecular Biomarkers of...
Misregulation of MIcroRNA...
Strategies for Therapeutic...
Conclusions and Future Directions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):292-301. doi:10.7150/ijbs.14995 This issue Cite
Review
Molecular Biomarkers of Pancreatic Intraepithelial Neoplasia and Their Implications in Early Diagnosis and Therapeutic Intervention of Pancreatic Cancer
Junli Guo1,2, Keping Xie2 ![]() , Shaojiang Zheng1,2
, Shaojiang Zheng1,2 ![]()
1. Department of Pathology, Affiliated Hospital of Hainan Medical College, Hainan Cancer Hospital, Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, Haikou 571199, People's Republic of China
2. Department of Gastroenterology, Hepatology and Nutrition, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Published 2016-1-28
Abstract
Lack of early detection and effective interventions is a major reason for the poor prognosis and dismal survival rates for pancreatic cancer. Pancreatic intraepithelial neoplasia (PanIN) is the most common precursor of invasive pancreatic ductal adenocarcinoma (PDAC). Each stage in the progression from PanIN to PDAC is well characterized by multiple significant genetic alterations affecting signaling pathways. Understanding the biological behavior and molecular alterations in the progression from PanIN to PDAC is crucial to the identification of noninvasive biomarkers for early detection and diagnosis and the development of preventive and therapeutic strategies for control of pancreatic cancer progression. This review focuses on molecular biomarkers of PanIN and their important roles in early detection and treatment of pancreatic cancer.
Keywords: PanINs, Biomarker, Early Diagnosis, Therapeutic Intervention
Introduction
The overall 5-year survival rate in patients with pancreatic cancer is about 5%, and this disease is the fourth leading cause of cancer deaths in the United States [1]. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. Its high mortality rate is partly a result of difficulty in establishing an effective strategy for early detection and diagnosis and a lack of effective treatments of advanced-stage disease. The asymptomatic nature of early-stage pancreatic cancer and remote anatomic location of the pancreas increase the difficulty in early detection.
Main diagnostic procedures for PanINs include histopathological diagnosis, genetic diagnosis (detection of gene mutation) [2], imaging diagnosis, such as B-ultrasound, CT and MRI, detection of serum tumor markers, biopsy of circulation tumor DNA (ctDNA) and circulation tumor cell (CTC) in a noninvasive manner [3,4]. At present, histopathological diagnosis remains the most accurate and reliable gold standard of the diagnosis of PanINs. The accuracy of different diagnostic methods greatly varies. The sensitivity, specificity and accuracy of CA19-9 detection in the diagnosis of pancreatic cancer were 83.1%, 73% and 75%, respectively. Approximately 99% of KRAS gene mutation is intimately correlated with the incidence and progression of early lesions induced by pancreatic cancer. A substantial quantity of early pancreatic cancer can be diagnosed in a noninvasive manner by biopsy detection of ctDNA. Melo and colleagues have recently demonstrated that glypican-1 (GPC1) gene encoding protein, distributed in the cancer exosomes, can serve as a potential marker for noninvasive diagnosis and screening tool [4].
Authors reported that progression from preinvasive precursor lesions to invasive pancreatic cancer occurs over many years or decades, and the time required for parental pancreatic cancer to gain the capacity to invade and metastasize is typically more than 5 years [5]. Pancreatic intraepithelial neoplasias (PanINs) are well established as the most common precursors of PDAC [6]. PanINs are microscopic lesions (<5 mm in diameter) that are too small to be identified using current imaging techniques. Multiple PanINs are usually found in individuals with inherited susceptibility to pancreatic cancer [7, 8]. Diverse molecular changes take place in the sequential progression from precursor lesions to PDAC. Researchers have coupled improved understanding of the sequence of molecular changes in the progression from PanIN to PDAC with the establishment of genetically modified animal models that recapitulate these changes [9]. Such molecular alterations provide a road map for understanding PDAC development and progression. Improved diagnostic tools for the detection of preneoplastic lesions and PanINs are urgently needed to effectively control pancreatic cancer. One of the major goals in the biomarker field is to develop noninvasive diagnostic strategies based on these genetic alterations [10]. Thus, molecular biomarkers that improve the efficiency in detection of pancreatic cancer would greatly improve the treatment of PDAC.
Histopathology of PanIN
Morphological and molecular transformation of mutated cells results in the formation of precursor lesions [6]. Precursors of PDAC include PanIN, intraductal papillary mucinous neoplasia, mucinous cystic neoplasia, and intraductal tubular papillary neoplasia, of which PanIN is the most common [11]. Briefly, PanINs are microscopic lesions (<5 mm in diameter) generated in the small pancreatic ducts. The lesions are papillary or flat and composed of columnar and cuboidal cells with various amounts of mucin. According to their progression, PanINs are classified as low-grade (PanIN-1A and PanIN-1B), intermediate-grade (PanIN-2), or high-grade (PanIN-3) PanINs, indicating tissue deterioration into invasive neoplasia. The low-grade PanINs can be flat (PanIN-1A) or papillary (PanIN-1B), characterized by the absence of nuclear atypia and presence of nuclear polarity. PanIN-2 is more complicated than PanIN-1, having more nuclear changes such as loss of nuclear polarity, nuclear crowding, variation in nuclear size (pleomorphism), nuclear hyperchromasia, and nuclear pseudostratification; however, mitosis is rarely observed. PanIN-3, also referred to as pancreatic carcinoma in situ, exhibits extensive loss of polarity, nuclear atypia, and frequent mitoses. However, as a preinvasive lesion, PanIN-3 still exists in the basement membrane [3, 8]. The incidence of PanIN-1 is associated with age, and it is commonly detected, whereas PanIN-3 is uncommon in individuals with invasive pancreatic cancer.
Molecular Biology of PanIN
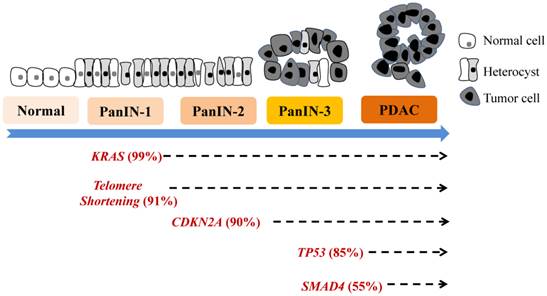
Various molecular alterations with major genetic irregularities affecting signaling pathways in each stage of progression from PanIN to PDAC are well characterized. These events destabilize multiple molecular processes, causing further aberrant cell-cycle progression, cell division, and cell growth. Early genetic events in PanIN-1A/B development include mutation of KRAS, epidermal growth factor receptor (EGFR), mucins (MUC1 and MUC6), TFF1, p16INK4A, S100A1, MUC5AC, S100A6, p21WAF1/C1P1, HER-2/neu, prostate stem cell antigen, fascin, matrix metalloproteinase (MMP)-7, and HOXB2 and telomere shortening [2-5, 8]. These mutations are followed by changes in Id-1/Id-2, cyclinD1, cyclooxygenase (COX)-2, 5-lipoxygenase, Hes1, Notch, pepsinigen C, KLF4, HOXA5, GATA5, gastrin, billin1/2, and CRABP1 in PanIN-2 and changes in Tp53, DPC4/Smad4, BRCA2, S100p, SHH, MUC4, TSLC-1, FAP, TopoIIa, 14-3-3s, Ki-67, and FOXM1 in PanIN-3 or PDAC [12, 13-21]. Oncogenic mutations of KRAS are detected in more than 90% of invasive pancreatic adenocarcinomas, whereas BRAF is mutated in a small subset of pancreatic cancers wild-type for KRAS [9, 12, 22]. When KRAS is activated by signaling partners such as EGFR, downstream signaling cascades, including Raf, mitogen-activated protein kinase, and phosphoinositide 3-kinase/AKT, are activated. Consequently, several signaling cascades are also deregulated, including Wnt, EGFR, mitogen-activated protein kinase, phosphoinositide 3-kinase/AKT, transforming growth factor-β, arachidonic acid metabolism, Notch, hedgehog, src, focal adhesion kinase, insulin-like growth factor, hepatocyte growth factor receptor, gastrin, cholescystokinin, and SMAD. Also, angiogenesis may be activated, cancer stem cells (CSCs) may emerge, and shortening of telomeres and other alterations may occur. A model of genetic progression of PanIN is shown in Figure 1, and the detailed molecular aberrations are described below. Molecular pathobiology is the phenotype of pancreaticlesions, often suggesting aberrant molecular changes at the genetic, transcriptomic, epigenetic, and proteomic levels that correlate with histological stages of PanIN development [9, 11-15, 23].
Genetic progression model of pancreatic carcinogenesis. The main molecular alterations that accumulate during pancreatic carcinogenesis can be classified as early (telomere shortening and activating KRAS mutations), intermediate (inactivating mutations or epigenetic silencing of CDKN2A), and late (inactivating mutations of TP53 and SMAD4) genetic events. More new molecular markers are emerging.
Molecular Biomarkers of Early-Stage PanINs
The distinct genetic changes generated in PanIN-1s are telomere shortening and gene mutations, including KRAS, cyclin-dependent kinase (CDK) inhibitor 2A, and p21WAF/CIP1 mutations [9, 12, 22].
Telomeres located at the ends of chromosomes are repeats of short DNA sequences that prevent the chromosomes from fusing during cell division. Researchers have detected remarkable shortening of telomeres in more than 90% of PanIN-1 cases as compared with that in normal ductal epithelium. [24]. Telomere shortening therefore is one of the earliest demonstrable genovariations in pancreatic cancer progression. Telomere shortening results in abnormal fusion of chromosome ends, chromosomal instability, and gene translocation followed by missegregation during mitosis. All of these variations eventually promote neoplastic progression [25]. Intact telomeres may serve as caretakers of the pancreatic ductal genome, and loss of telomere integrity in PanINs sets the stage for progressive accumulation of chromosomal abnormalities and, eventually, generation of frank neoplasia.
KRAS family members encode for small GTP-binding cytoplasmic proteins and regulate cell-cycle progression. KRAS mutation is one of the earliest genetic abnormalities in pancreatic carcinogenesis. The KRAS oncogene (chromosome 12p) is activated by a point mutation in more than 90% of pancreatic cancers, especially in codon 2 [10, 26-28]. Several signaling pathways downstream from KRAS including BRAF/mitogen-activated protein kinase and phosphoinositide 3-kinase/AKT, also may be activated by KRAS mutations [29-31]. However, RAS mutations are fairly common not only in patients with chronic pancreatitis but also in healthy individuals, causing concern about false-positivity in the diagnosis of pancreatic cancer [32, 33]. Inhibition of the KRAS signaling for treatment of pancreatic cancer using farnesyltransferase inhibitors has been disappointing thus far [34-36]. KRAS mutation is therefore a potentially interesting target for early detection and diagnosis of pancreatic cancer.
Progression of PanIN is related to loss of tumor suppressor function. The CDK inhibitor2A/p16 gene, which is located on chromosome arm 9p, encodes for a cell-cycle checkpoint protein and inhibits the cell cycle at the G1/S checkpoint by binding to CDKs, including CDK4 and CDK6 [37]. However, this gene is not expressed in PanIN-1 cells and is inactivated in more than 95% of pancreatic cancer cases [38, 39]. Their homozygous deletion and intragenic mutation in combination with loss of the remaining allele or hypermethylation of the promoter region are the mechanisms of gene silencing in pancreatic carcinogenesis [40, 41]. In some patients, loss of p16 causes familial atypical multiple mole melanoma syndrome, which is highly correlated with an increased risk of pancreatic cancer [24]. In genetically engineered mice, silencing of CDK inhibitor 2A induces the development of undifferentiated carcinoma [41].
p21WAF/CIP1 is a CDK inhibitor that inhibits the activity of cyclin E/cdk2 complexes and prevents phosphorylation of the Rb protein. Unlike proliferation and cell-cycle antigens, p21WAF/CIP1 is overexpressed in PanINs, especially the lower grade forms (PanIN-1A, 16%; PanIN-1B, 32%; PanIN-2, 56%; PanIN-3, 80%), leading to cyclin D1 abnormalities [39]. However, the mechanism of overexpression of p21WAF/CIP1 is not clear. A reasonable explanation is that overexpressed p21WAF/CIP1 sends negative feedback signals via the ductal epithelium as a result of activation of other mitogenic pathways, such as the KRAS-mediated signaling pathway.
MMP-7 is a member of the MMP family of zinc-dependent extracellular proteases. It takes part in cancer invasion and metastasis and endows resistance to apoptosis to cancer cells [43, 44]. MMP-7 is not expressed in a normal pancreas but is overexpressed in PanINs (>70% of PanIN-1s and the majority of invasive pancreatic adenocarcinomas), which is in line with its proposed tumorigenic characteristics [45]. That is an early event in pancreatic carcinogenesis.
In addition, prostate stem cell antigen is overexpressed in 60% of invasive pancreatic cancers, including about 30% of PanIN-1s, 40% of PanIN-2s, and 60% of PanIN-3s, suggesting the potential for its overexpression as an early event in pancreatic cancer progression models [46]. Also, MUC1 expression in normal pancreatic ducts and acini is responsible for the formation of lumina. However, MUC1 is also often expressed in invasive PDAC cells [47, 48]. Authors reported that MUC1 was expressed in 6% of PanIN-1A and 5% of PanIN-1B cases compared with 43% of PanIN-2 and 85% of PanIN-3 cases [43]. Thus, MUC1 expression in normal intralobular and interlobular ducts appears to be decreased in the low-grade PanINs (PanIN-1A and -1B), but MUC1 appears to be highly expressed in the high-gradePanINs and even invasive adenocarcinomas [12]. In contrast with MUC1, MUC5, the gastric foveolar mucin, is not expressed in normal ductal epithelium, but it is overexpressed in PanINs at all stages [46, 49]. These mucins are also potentially detectable targets in cancer cells and may be developed as therapeutic agents for precursor pancreatic lesions [50].
Molecular Biomarkers of Intermediate- and Late-Stage PanINs
As with gene mutations in the early-stage PanINs, such as KRAS mutations, diverse downstream molecular aberrations, including cyclin D1, COX-2, TP53, and SMAD4 mutations, are identified in the intermediate- and late-stage PanINs,
As a co-factor involved in the phosphorylation and inactivation of Rb protein, cyclin D1 plays a key role in cell-cycle regulation [48]. It is overexpressed in more than 60% of PDAC cases [52, 53]. In addition, overexpression of cyclin D1 is reported to be an intermediate step in carcinogenesis along with its nuclear overexpression in 29% of PanIN-2 cases and 57% of PanIN-3 cases [46]. This has been associated with poor prognoses and decreased survival durations [54].
COX-2 mediates the metabolism of arachidonic acid into prostaglandins and other proinflammatory products. In tumorigenesis, COX-2 metabolites activate a series of signaling pathways, such as cancer cell proliferation, survival, invasion, and angiogenesis [55]. These processes may be secondary to activation of mitogen-activated protein kinase signaling and nuclear factor κB-mediated signaling [56]. The expression of COX-2 is higher in pancreatic tumors, including PanINs and PDAC, than in normal pancreatic ducts. Also, the expression of COX-2 is markedly higher in PanIN-2/3 than in PanIN-1A/1B [57]. Researchers suggested that selected COX-2 inhibitors are potential targets for chemoprevention of pancreatic cancer [58].
TP53 protein is a transcription factor that modulates molecules primarily involved in cell-cycle arrest and apoptosis [59]. The TP53 gene, located on chromosome 17p, is almost always inactivated by a combination of intragenic mutation and loss of the second wild-type allele in approximately 50-75% of pancreatic cancers [60]. Emerging evidence has demonstrated that loss of p53 function contributes to the genomic instability of pancreatic cancers [61]. Mutation of the TP53 gene is detected by immunolabeling for nuclear accumulation of the p53 protein [62], which can be exploited to assess lesions such as PanINs. Using immunohistochemistry, researchers observed obvious p53 accumulation in advanced PanIN-3s, which is consistent with TP53 gene mutation being a late genetic event in pancreatic cancer progression [46].
SMAD4 (DPC4) protein is critical to the signal transduction cascade that involves transforming growth factor-β and multiple targets in the transforming growth factor-β pathway. SMAD4/DPC4, which is located on chromosome arm 18q, is inactivated by its homozygous deletion and intragenic mutations accompanied by loss of the other allele in 55% of pancreatic cancer cases [63-65]. Recently, authors reported SMAD4 mutations to be associated with poor prognosis for and widespread metastasis of pancreatic cancer [66, 67]. Using nuclear labeling with immunohistochemistry, loss of SMAD4 is generally observed in pancreatic carcinogenesis, such as in PanIN-3s and infiltrating adenocarcinomas [68]. Loss of SMAD4 expression can be regarded as an important genetic event in the development of late PanINs, as well. The BRCA2 gene is part of a class of so-called caretaker genes that play important roles in DNA repair and maintenance of genome integrity. Furthermore, loss of BRCA2 gene function leads to pancreatic carcinogenesis [59]. Germ-line BRCA2 gene mutations occur in about 7-10% of patients with pancreatic cancer, including some with apparently sporadic disease. In one study, among three cases of pancreatic cancer with germ-line mutations of BRCA2, loss of the remaining wild-type allele occurred in only a single PanIN-3 and no low-grade PanINs, suggesting that inactivation of the BRCA2 gene is a late genetic event in pancreatic carcinogenesis [69].
Investigators found that expression of SOX17, a transcription factor that regulates biliary development and differentiation [70], increased throughout progression of PanINs of all stages using immunohistochemistry and tissue microarrays [71]. When combined with KrasG12D expression, SOX17 overexpression enhanced pancreatic tumorigenesis, suggesting that SOX17 expression and the ensuing biliary transdifferentiation promote tumor formation [71].
Furthermore, researchers have observed changes in the expression of many other proteins and apomucins, such as the proliferating antigen topoisomerase IIα, in PanIN and PDAC cases using immunohistochemistry and tissue microarrays. All of these genetic changes in PanINs and PDAC are summarized in Table 1 [16-21, 23, 61].
Molecular and genetic abnormalities in PanINs
| Abnormality | Normal pancreatic tissue | PanIN-1A | PanIN-1B | PanIN-2 | PanIN-3 | PDAC |
|---|---|---|---|---|---|---|
| Mucin 1 [112] | + | + | + | |||
| Kras mutation [113] | + | + | + | + | + | |
| Telomere shortening [21] | + | + | + | + | + | |
| PSCA [114] | + | + | + | + | + | |
| Mucin 5 [115] | + | + | + | + | + | |
| Fascin [116] | + | + | + | + | + | |
| MMP-7 [117] | + | + | + | + | + | |
| SOX17 [118] P16 [119] | + | + | + + | + + | + + | |
| Cyclin D1 [120] | + | + | + | |||
| KLF4 [121] | + | + | + | |||
| TP53 [122,123] | + | + | ||||
| SMAD4 [124] | + | + | ||||
| Topoisomerase IIα [115] | + | + | ||||
| Ki-67 [125] | + | + | ||||
| 14-3-3σ [115,126] | + | + | ||||
| Mucin 4 [127] | + | + | ||||
| FOXM1 [128,129] | + | + | ||||
| Mesothelin [130] | + | |||||
| Glypican-1 [4] | + |
PSCA, prostate stem cell antigen.
Misregulation of MIcroRNA Expression in PanINs
Besides the molecular changes that occur at the DNA and protein level during progression from PanIN to PDAC, molecular events at the RNA level, such as microRNA (miRNA) changes, are identified during pancreatic carcinogenesis.
MiRNAs are small noncoding RNAs of about 22 nucleotides that primarily bind to the 3' untranslated region of target mRNA and then negatively regulate protein expression at the posttranscriptional level by inhibiting translation and/or degrading target mRNA [69]. Researchers have identified more than 400 miRNAs in the human genome, many of which are implicated to have roles in regulation of cellular differentiation, proliferation, and apoptosis [37]. They are reported to be closely correlated with the progression of many diseases, including cancer [73]. Several miRNAs, including miR-21, miR-34, miR-146a, miR-155, miR-196a-2, and miR-200a/b, are overexpressed in PDAC cells [5, 16, 20, 21, 24-28], and some of them are aberrantly expressed in PanINs. For example, miR-155 overexpression is evident in PanIN-2s, and aberrant miR-21 expression is evident in PanIN-3s. Therefore, misregulation of miRNA expression is an early event in PanIN development [74]. With regard to alterations at the transcriptomic level, researchers recently exploited miRNAs as potential markers and therapeutic targets for pancreatic cancer.
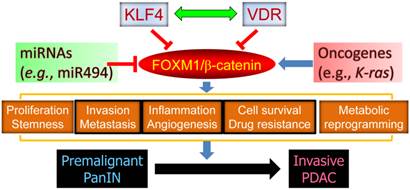
In addition, miR-494 is an important regulator of pancreatic cancer progression. In a recent study, we found that loss of SMAD4 expression in PDAC cells leads to reduced levels of miR-494 expression, increased levels of FOXM1 expression, and nuclear localization of β-catenin, meaning that miR-494 may be developed as a prognostic marker or therapeutic target for PDAC [17]. Extensive cross-talk between FOXM1 signaling and other major signaling pathways further substantiates a pivotal role for FOXM1 in pancreatic cancer development and progression (Figure 2).
The central role of FOXM1 signaling in pancreatic carcinogenesis. FOXM1 regulates various aspects of cancer cell biology via regulation of its downstream target genes and drives the progression of premalignant PanIN to invasive PDAC. Gain-of-function mutation of oncogenes and loss-of-function mutation of tumor suppressors may activate FOXM1 expression and signaling, and KLF4, VDR, and miR494 are known to repress the expression and function of FOXM1/β-catenin signaling.
Strategies for Therapeutic Intervention for PanINs
Early diagnosis of pancreatic cancer would enable the use of chemoprevention strategies to impede progression from PanIN to invasive carcinoma [22]. However, detection of pancreatic cancer at an early stage is difficult using the currently available modalities. The challenge in early diagnosis of pancreatic cancer is detection of early emerging symptoms. Extensive investigations of molecular signaling in pancreatic cancer cells have increased understanding of the molecular genetics of progression from PanIN to PDAC and resulted in progress in the diagnosis, staging, and treatment of localized pancreatic cancer. However, those studies did not result in any significant breakthroughs in early detection or significantly improve prognoses. Therefore, novel therapeutic strategies based on the molecular changes described above and on the concept of CSCs and stromal targeting should be explored to improve early diagnosis of and therapy for pancreatic cancer.
KRAS
KRAS is mutated in pancreatic cells early in the development of pancreatic cancer as evidenced by its identification in PanINs [75]. KRAS1 is a pseudogene, whereas KRAS2 is the proto-oncogene that results in a dominant-active form of the KRAS GTPase [26, 29]. KRAS2 is commonly mutated in many cancer types, including more than 90% of pancreatic cancer cases. The genetically modified model of pancreatic cancer established by Collins et al. [76] facilitated controllable activation and inactivation of oncogenic KRAS under different experimental conditions, including caerulein-induced pancreatitis and mutation of p53, which accelerated cancer onset. Of major importance to potential treatment of pancreatic cancer is that the oncoprotein KRAS is required for not only the initiation but also the maintenance of the disease in mice [76]. However, an important point is that KRAS mutations are fairly common, not only in patients with chronic pancreatitis, a recognized risk factor for pancreatic cancer [77], but also in healthy individuals [78]. These facts underline the fact that additional downstream genetic hits are urgently needed for accurate detection of pancreatic cancer based on KRAS mutations [79].
Interestingly, antroquinonol, a new Kras-targeting inhibitor, has had a marked anticancer effect on human pancreatic carcinoma cells, inducing cross-talk among apoptosis, autophagy, and senescence [80-82].
Tyrosine kinase receptors
Researchers have made considerable efforts in development of a number of tyrosine kinase receptors as potential therapeutic targets for pancreatic cancer, including EGFR, a member of the HER2 family of receptor tyrosine kinases, and insulin-like growth factor receptor. The National Cancer Institute of Canada Clinical Trials Group PA.3 phase III study (n = 569) was the first study to demonstrate a survival benefit of combination therapy with a tyrosine kinase inhibitor and gemcitabine in patients with pancreatic cancer [83]. Specifically, the combination of gemcitabine and the oral EGFR tyrosine kinase inhibitor erlotinib improved the median survival duration by 2 weeks over that with gemcitabine alone in patients with advanced pancreatic cancer (6.4 months versus 6.0 months). Overexpression of core genes in the EGFR pathway, such as AKT3 and PIK3CA, was predictive of response to treatment with EGFR inhibitors in an additional cohort of eight patients with implanted PDACs [84]. These findings demonstrated that treatment with tyrosine kinase inhibitors is promising in patients with PanINs and PDAC.
Investigators have successfully used two pharmacological approaches to inhibition of EGFR function in cancer treatment: delivery of neutralizing monoclonal antibodies and small molecule tyrosine inhibitors. The results of randomized trials of the addition of EGFR-targeted agents to gemcitabine compared with gemcitabine alone have been disappointing, although results of treatment with the EGFR tyrosine kinase inhibitor erlotinib were statistically significantly better but of marginal clinical benefit [85]. Recent studies demonstrated that targeting EGFR can be used for early diagnosis of orthotopic pancreatic tumors in vivo [86, 87].
Notch
The Notch signaling pathway makes cell-fate decisions during embryogenesis. Notch signaling is produced after cell-to-cell contact and initiates the engagement of Notch receptors by Notch ligands [88]. The binding of Notch receptors by Notch ligands causes extracellular proteolysis via the action of metalloproteinase, a tumor necrosis factor-converting enzyme, ultimately resulting in γ-secretase-dependent intracellular proteolysis and release of the active notch intracellular domain from the plasma membrane. This domain is subsequently translocated to the nucleus and initiates the transcriptional activation and repression of target genes. The activity of Notch varies in different kinds of cancer cells, having oncogenic activity in some but tumor-suppressive activity in others [88]. Notch also is an oncogene involved in the progression of pancreatic cancer. Notch is a downstream pathway of KRAS, EGFR, and transforming growth factor-α signaling in pancreatic carcinogenesis that promotes cancer vascularization. As reported previously, aberrant regulation of the Notch pathway in pancreatic cancer cells contributes to tumor initiation, progression, and maintenance, and researchers have made efforts to target this pathway for cancer therapy [89]. Downregulation of Notch expression by small interfering RNA or curcumin is capable of inhibiting cell growth and inducing apoptosis in pancreatic cancer cells in vitro [90]. Therefore, regulation of the Notch pathway is a potential method of pancreatic cancer therapy. However, more investigations using in vitro and in vivo pancreatic cancer models should be carried out.
Authors reported that the Notch-targeting inhibitor RO4929097, an oral inhibitor of the γ-secretase enzyme, is safe when given as a single agent in patients with advanced solid pancreatic tumors [91]. Notch-1 and Notch-4 are novel transcriptional targets of PEA3 in breast cancer cells, and targeting of PEA3 and/or Notch pathways may be a new therapeutic strategy for triple-negative and possibly other subtypes of breast cancer [92, 93].
BRCA2 poly(ADP-ribose) polymerase inhibition
As a tumor suppressor gene, BRCA2 takes part in DNA damage repair, and germ-line mutations of the BRCA2 gene increase the risk of pancreatic cancer [90]. These findings demonstrated that inactivation of the BRCA2 gene is responsible for approximately 10% of familial pancreatic cancer cases [90, 94]. Therefore, activation of the BRCA2 gene can be expected to prevent the development of pancreatic cancer. Patients with pancreatic cancer have received treatment with poly(ADP-ribose) polymerase inhibitors, which target BRCA-deficient cancers and have exhibited exciting effectiveness [95]. Several clinical-stage poly(ADP-ribose) polymerase inhibitors, including veliparib, rucaparib, olaparib, niraparib, and talazoparib, have undergone evaluation of their poly(ADP-ribose) polymerase-trapping activity and use in pancreatic cancer therapy [93-95].
CSCs
Much interest in pancreatic cancer research has focused on CSCs. Nguyen et al. [99] defined CSCs as cells in a malignant clonal population that can propagate cancer. CSCs are believed to be responsible for self-renewal, maintenance, and metastasis of tumors that must be eradicated for cancer therapy. Although traditional therapies are capable of eliminating rapidly proliferating cells within the tumor bulk, CSCs are thought to be somewhat quiescent and may evade treatment. Thus, strategies for targeting these cells are expected to result in successful cancer treatment. Li et al. [100] identified c-Met as a stem cell marker in pancreatic cancer cases and reported that expression of c-Met was necessary for tumor growth and metastasis. Therefore, targeting pancreatic CSCs via c-Met and related pathways is a promising platform for stopping the development of tumor metastases [100]. In a recent study, investigators found that DCLK1 marked a morphologically distinct subpopulation of cells with CSC properties in preinvasive pancreatic cancer cases [101]. Kure et al. [102] examined the correlation between development of PanIN and expression of CSC markers, including CD24, CD44, CD133, CXCR4, ESA, and nestin, using immunohistochemical analysis and detected CD24-, CD44-, CXCR4-, ESA-, and nestin-positive cells in the following tissues, which are listed in order of increasing percentage: normal duct < low-grade PanIN < high-grade PanIN < PDAC.
Stromal targeting therapy
Many anticancer drugs have been highly effective in cell models and xenograft and/or allograft mice models of pancreatic cancer but have failed in patients with pancreatic cancer. An emerging explanation for the discrepancy between their effectiveness in preclinical and clinical trials is the absence of stromal microenvironments in pancreatic cancer cells and mouse models. A pancreatic tumor is characterized by a dense fibrotic stromal matrix [103] composed of activated fibroblasts/stellate cells, inflammatory cells, and other cell types, such as endothelial cells. These cells form a microenvironment that plays a crucial role in cancer initiation, progression, and chemoresistance [104-106]. The delivery of chemotherapeutic drugs to cancer cells in the stroma in vivo is the first step in cancer cell internalization and targeting. Investigations using genetically engineered mice with pancreatic cancer bearing well-developed stromal components similar to those of human tumors have indicated that drugs do not penetrate the stromal mass and hence are not able to effectively approach cancer cells [103, 106]. Therefore, development of approaches to modifying the pancreatic tumor stroma, which facilitates the access of chemotherapeutic drugs to cancer cells, should attract extensive attention. Marimastat is a broad-spectrum synthetic MMP inhibitor that researchers first tested in a large randomized phase 3 trial in patients with advanced pancreatic cancer [107-109]. In vivo studies demonstrated that human pancreatic stellate cells in conditioned medium increased pancreatic tumor cell proliferation, migration, invasion, and colony formation and that treatment with human pancreatic stellate cells in conditioned medium rendered pancreatic cancer cells more resistant to gemcitabine and radiation therapy [110, 111].
Conclusions and Future Directions
A series of molecular genetic alterations, such as KRAS mutations, occur in PanIN, the most common precursor of PDAC. These molecular alterations are closely correlated with each stage in the progression from PanINs to PDAC by affecting downstream signaling pathways. A lack of early detection of and effective interventions for these genetic irregularities is a major contributor to the poor prognosis and dismal survival rates for pancreatic cancer. The availability of noninvasive molecular biomarkers of PanIN provides the basis for designing rational early-detection strategies and therapeutic intervention trials for pancreatic cancer. Use of these markers can resolve the diagnostic conundrum for early detection of PanIN progression to invasive carcinoma and serves as a guideline for the design of novel therapeutic intervention strategies. Chemical entities and therapeutic genes such as small interfering RNA and miRNA may be employed to regulate signal pathways, including KRAS, tyrosine kinase receptors, Notch, and BRCA2, for pancreatic cancer therapy. In addition, targeting inhibition of CSCs and regulation of tumor stroma are emerging promising routes. Further understanding of the detailed downstream cascades of molecular genetic alterations and development of corresponding targeting drugs and therapeutic strategies in to block the stepwise progression of PanIN are needed. Finally, it is important to use some animal models other than clinical patient cohorts for discovering early diagnostic markers, e.g., GEMMs, since most of the clinical patients already develop pancreatic cancer thus some of the early events may be missed. Moreover, it will be convenient to collect precancerous tissue and blood specimens during the monitoring of the animal models.
Acknowledgements
This work is supported by research grants from the National Natural Scientific Foundation of China (81460461, 81372465, 81260350, and 81060184), Hainan Provincial Science & Technology Cooperation Program (KJHZ2014-23), and Hnky2015ZD-13, and Haikou Key Program of Science & Technology (2014059; to S. Zheng), and Hnkyzx2014-07 (to J. Guo), and grants R01-CA129956, R01-CA148954, R01CA152309, and R01CA172233 from the National Cancer Institute, National Institutes of Health (to K. Xie).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2013;63:11-30
2. Childs EJ, Mocci E, Campa D, Bracci PM. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47(8):911-6
3. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, O'Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA Jr, Von Hoff DD, Johansen JS, Drebin JA, Velculescu VE. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;7:7686
4. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, Reissfelder C, Pilarsky C, Fraga MF, Piwnica-Worms D, Kalluri R. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015Jul9;523(7559):177-82
5. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2011;467:1114-1117
6. Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, Lüttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions, Am. J. Surg. Pathol. 2001;25:579-586
7. Koorstra JB, Feldmann G, Habbe N, Maitra A. Morphogenesis of pancreatic cancer: role of pancreatic intraepithelial neoplasia (PanINs), Langenbecks Arch. Surg. 2008;393:561-570
8. Cooper CL, O'Toole SA, Kench JG. Classification, morphology and molecular pathology of premalignant lesions of the pancreas. Pathology. 2013;45:286-304
9. Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007;14:224-232
10. Singh M, Maitra A. Precursor lesions of pancreatic cancer: molecular pathology and clinical implications. Pancreatology. 2007;7:9-19
11. Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 1998;22:163-169
12. Shen R, Wang Q, Cheng S, Liu T, Jiang H, Zhu J, Wu Y, Wang L. The biological features of PanIN initiated from oncogenic Kras mutation in genetically engineered mouse models. Cancer Lett. 2013;339:135-143
13. Singh M, Maitra A. Precursor lesions of pancreatic cancer: molecular pathology and clinical implications. Pancreatology. 2007;7:9-19
14. Turrini O, Cano C, Legoffic A, Delpero JR, Dagorn JC, Iovanna J. Genetic alterations in precancerous pancreatic lesions and their clinical implications. Gastroenterol. Clin. Biol. 2009;33:1028-1035
15. Ottenhof NA, Milne AN, Morsink FH, Drillenburg P, Ten Kate FJ, Maitra A, Offerhaus GJ. Pancreatic intraepithelial neoplasia and pancreatic tumorigenesis: of mice and men. Arch. Pathol. Lab Med. 2009;133:375-381
16. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y, Xie K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res. 2014;20:4370-4380
17. Li L. et al. Down-regulation of microRNA-494 via loss of SMAD4 increases FOXM1 and β-Catenin signaling in pancreatic ductal adenocarcinoma cells. Gastroenterology. 2014;147:485-497
18. Quan M, Wang P, Cui J, Gao Y, Xie K. The roles of FOXM1 in pancreatic stem cells and carcinogenesis. Mol Cancer. 2013;12:159-167
19. Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD, Le X, Wei D, Huang S, Xie K. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. 2012;72:655-665
20. Gao Y, Jia Z, Kong X, Li Q, Chang DZ, Wei D, Le X, Suyun H, Huang S, Wang L, Xie K. Combining betulinic acid and mithramycin a effectively suppresses pancreatic cancer by inhibiting proliferation, invasion, and angiogenesis. Cancer Res. 2011;71:5182-5193
21. Wei D, Wang L, Kanai M, Jia Z, Le X, Li Q, Wang H, Xie K. KLF4α up-regulation promotes cell cycle progression and reduces survival time of patients with pancreatic cancer. Gastroenterology. 2010;139:2135-2145
22. Costello E, Greenhalf W, Neoptolemos JP. New biomarkers and targets in pancreatic cancer and their application to treatment. Nat. Rev. Gastroenterol Hepatol. 2012;9:435-444
23. Haugk B. Pancreatic intraepithelial neoplasia-can we detect early pancreatic cancer? Histopathology. 2010;57:503-514
24. van Heek NT. et al. Maitra, Telomere shortening is nearly universal in pan-creatic intraepithelial neoplasia. Am. J. Pathol. 2002;161:1541-1547
25. Koorstra JB, Hustinx SR, Offerhaus GJ, Maitra A. Pancreatic carcinogenesis. Pancreatology. 2008;8:110-125
26. Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol. 1995;18:1-6
27. Jimeno A, Hidalgo M. Molecular biomarkers: their increasing role in the diagnosis, characterization, and therapy guidance in pancreatic cancer. Mol. Cancer Ther. 2006;5:787-796
28. Goggins M. Identifying molecular markers for the early detection of pancre-atic neoplasia. Semin. Oncol. 2007;34:303-310
29. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-1806
30. Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, Kern SE. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am. J. Pathol. 2003;163:1255-1260
31. Schneider G, Schmid RM. Genetic alterations in pancreatic carcinoma. Mol. Cancer. 2003;2:15
32. van Heek NT, Rauws EA, Caspers E, Drillenburg P, Gouma DJ, Offerhaus GJ. Long-term follow-up of patients with a clinically benign extrahepatic biliary stenosis and K-ras mutation in endobiliary brush cytology. Gastrointest. Endosc. 2002;55:883-888
33. Yan L, McFaul C, Howes N, Leslie J, Lancaster G, Wong T, Threadgold J, Evans J, Gilmore I, Smart H, Lombard M, Neoptolemos J, Greenhalf W. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology. 2005;128:2124-2130
34. Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: astrategic target for anticancer therapeutic development. J. Clin. Oncol. 1999;17:3631-3652
35. Porzner M, Seufferlein T. Novel approaches to target pancreatic cancer. Curr. Cancer Drug Targets. 2011;11:698-713
36. Bendell J, Goldberg RM. Targeted agents in the treatment of pancreatic cancer: history and lessons learned. Curr. Opin. Oncol. 2007;19:390-395
37. Maitra A. Hruban RH, Pancreatic cancer. Annu. Rev. Pathol. 2008;3:157-188
38. Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140-2143
39. Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740-4744
40. Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994;8:27-32
41. Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126-3130
42. Biankin AV, Kench JG, Morey AL, Lee CS, Biankin SA, Head DR, Hugh TB, Henshall SM, Sutherland RL. Overexpression of p21(WAF1/CIP1) is an early event in the development of pancreatic intraepithelial neoplasia. Cancer Res. 2001;61:8830-8837
43. Shiomi T, Okada Y. MT1-MMP and MMP-7 in invasion and metastasis of human cancers. Cancer Metastasis Rev. 2003;22:145-152
44. Vargo-Gogola T, Fingleton B, Crawford HC, Matrisian LM. Matrilysin (matrix metalloproteinase-7) selects for apoptosis-resistant mammary cells in vivo. Cancer Res. 2002;62:5559-5563
45. Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J. Clin. Invest. 2002;109:1437-1444
46. Maitra A, NAdsay V, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neo-plasia tissue microarray, Mod. Pathol. 2003;16:902-912
47. Terada T, Ohta T, Sasaki M, Nakanuma Y, Kim YS. Expression of MUC apomucins in normal pancreas and pancreatic tumours. J. Pathol. 1996;180:160-165
48. Nagata K, Horinouchi M, Saitou M, Higashi M, Nomoto M, Goto M, Yonezawa S. Mucin expression profile in pancreatic cancer and the precursor lesions. J. Hepatobiliary Pancreat. Surg. 2007;14:243-254
49. Kim GE, Bae HI, Park HU, Kuan SF, Crawley SC, Ho JJ, Kim YS. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. 2002;123:1052-1060
50. Mohammed A, Janakiram NB, Lightfoot S, Gali H, Vibhudutta A, Rao CV. Early detection and prevention of pancreatic cancer: use of genetically engineered mouse models and advanced imaging technologies. Curr. Med. Chem. 2012;19:3701-3713
51. Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73-92
52. Poch B, Gansauge F, Schwarz A, Seufferlein T, Schnelldorfer T, Ramadani M, Beger HG, Gansauge S. Epidermal growth factor induces cyclin D1 in human pancreatic carcinoma: evidence for a cyclin D1-dependent cell cycle progression. Pancreas. 2001;23:280-287
53. Gansauge S, Gansauge F, Ramadani M, Stobbe H, Rau B, Harada N, Beger HG. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997;57:1634-1637
54. Kornmann M, Ishiwata T, Itakura J, Tangvoranuntakul P, Beger HG, Korc M. Increased cyclin D1 in human pancreatic cancer is associated with de-creased postoperative survival. Oncology. 1998;55:363-369
55. Sarkar FH, Adsule S, Li Y, Padhye S. Back to the future: COX-2 inhibitors for chemoprevention and cancer therapy. Mini Rev. Med. Chem. 2007;7:599-608
56. Wu KK. Control of cyclooxygenase-2 transcriptional activation by pro-inflammatory mediators. Prostaglandins Leukot. Essent. Fatty Acids. 2005;72:89-93
57. Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA, Cameron JL, Hruban RH, Wilentz RE. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am. J. Clin. Pathol. 2002;118:194-201
58. Sclabas GM, Uwagawa T, Schmidt C, Hess KR, Evans DB, Abbruzzese JL, Chiao PJ. Nuclear factor kappa B activation is a potential target for preventing pancreatic carcinoma by aspirin. Cancer. 2005;103:2485-2490
59. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789-799
60. Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025-3033
61. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469-483
62. Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR. An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol. 1994;172:5-12
63. Iacobuzio-Donahue CA, Song J, Parmiagiani G, Yeo CJ, Hruban RH, Kern SE. Missense mutations of MADH4: characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res. 2004;10:1597-1604
64. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350-353
65. Iacobuzio-Donahue CA, Wilentz RE, Argani P, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Dpc4 protein in mucinous cystic neoplasms of the pancreas: frequent loss of expression in invasive carcinomas suggests a role in genetic progression. Am J Surg Pathol. 2000;24:1544-1548
66. Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, Goggins M, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Cameron JL, Olino K, Schulick R, Winter J, Herman JM, Laheru D, Klein AP, Vogelstein B, Kinzler KW, Velculescu VE, Hruban RH. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674-4679
67. Iacobuzio-Donahue CA. et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806-1813
68. Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002-2006
69. Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the devel-opment of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767-1771
70. Spence JR, Lange AW, Lin SC. et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell. 2009;17:62-74
71. Delgiorno KE, Hall JC, Takeuchi KK, Pan FC, Halbrook CJ, Washington MK, Olive KP, Spence JR, Sipos B, Wright CV, Wells JM, Crawford H. Identification C and Manipulation of Biliary Metaplasia in Pancreatic Tumors. Gastroenterology. 2014;146:233-244
72. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297
73. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259-269
74. du Rieu MC, Torrisani J, Selves J, Al Saati T, Souque A, Dufresne M, Tsongalis GJ, Suriawinata AA, Carrere N, Buscail L, Cordelier P. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin Chem. 2010;56:603-612
75. Klimstra DS, Longnecker DS. K ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547-1550
76. Collins MA. et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639-653
77. Grocock CJ. et al. The variable phenotype of the p.A16V mutation of cationic trypsinogen (PRSS1) in pancreatitis families. Gut. 2010;59:357-363
78. Yan L, McFaul C, Howes N, Leslie J, Lancaster G, Wong T, Threadgold J, Evans J, Gilmore I, Smart H, Lombard M, Neoptolemos J, Greenhalf W. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology. 2005;128:2124-2130
79. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-361
80. Yu CC, Chiang PC, Lu PH, Kuo MT, Wen WC, Chen P, Guh J. Antroquinonol H, natural ubiquinone derivative, induces a cross talk between apoptosis, autophagy andsenescence in human pancreatic carcinoma cells. J Nutr Biochem. 2012;23:900-907
81. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548-551
82. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272-281
83. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-1966
84. Jimeno A. et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 2008;68:2841-2849
85. Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, Ciardiello F. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13:802-810
86. Ardito CM, Grüner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, Pal D, Briel T, Herner A, Trajkovic-Arsic M, Sipos B, Liou GY, Storz P, Murray NR, Threadgill DW, Sibilia M, Washington MK, Wilson CL, Schmid RM, Raines EW, Crawford HC, Siveke JT. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304-317
87. Hudson SV, Huang JS, Yin W, Albeituni S, Rush J, Khanal A, Yan J, Ceresa BP, Frieboes HB, McNally LR. Targeted noninvasive imaging of EGFR-expressing orthotopic pancreatic cancer using multispectral optoacoustic tomography. Cancer Res. 2014;74:6271-6279
88. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338-351
89. Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011;31:1105-1113
90. Iovanna J, Mallmann MC. et al. Current knowledge on pancreatic cancer. Front Oncol. 2012;2:6
91. De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, Blatchford PJ, Quackenbush K, Messersmith W. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs. 2014;32:739-745
92. Egloff AM, Grandis JR. Molecular pathways: context-dependent approaches to Notch targeting as cancer therapy. Clin Cancer Res. 2012;18:5188-95
93. Clementz AG, Rogowski A, Pandya K, Miele L. Osipo NOTCH-1 C and NOTCH-4 are novel gene targets of PEA3 in breast cancer: novel therapeutic implications. Breast Cancer Res. 2011;13:R63
94. Bartsch DK, Gress TM. Langer P, Familial pancreatic cancer-current knowledge. Nat Rev Gastroenterol Hepatol. 2012;9:445-453
95. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293-301
96. Vyas S, Chang P. New PARP targets for cancer therapy. Nat Rev Cancer. 2014;14:502-509
97. Shen Y, Aoyagi-Scharber M, Wang B. Trapping Poly(ADP-Ribose) Polymerase. J Pharmacol Exp Ther. 2015;353:446-457
98. Valsecchi ME, Díaz-Cantón E, de la Vega M, Littman SJ. Recent treatment advances and novel therapies in pancreas cancer: a review. J Gastrointest Cancer. 2014;45:190-201
99. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-143
100. Li C, Wu JJ, Hynes M. et al. c Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141:2218-2227
101. Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, Zhang H, Pasricha PJ, Bardeesy N, Matsui W, Maitra A, Leach SD. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells With Stem Cell Properties in Preinvasive Pancreatic Cancer. Gastroenterology. 2014;146:245-256
102. Kure S, Matsuda Y, Hagio M, Ueda J, Naito Z, Ishiwata T. Expression of cancer stem cell markers in pancreatic intraepithelial neoplasias and pancreatic ductal adenocarcinomas. Int J Oncol. 2012;41:1314-1324
103. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2010;60:861-868
104. Polyak K, Haviv I. Campbell IG, Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30-38
105. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918-926
106. Olive KP. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-1461
107. Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer Cell. 2014;25:711-712
108. Kong X, Li L, Li Z, Xie K. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: Molecular basis for therapeutic implications. Cytokine Growth Factor Rev. 2012;23:343-356
109. Xie D, Xie K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015;2(2):133-143
110. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918-26
111. Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS, Apte MV. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585-2596
112. Gold DV, Karanjawala Z, Modrak DE, Goldenberg DM, Hruban RH. PAM4-reactive MUC1 is a biomarker for early pancreatic adenocarcinoma. Clin Cancer Res. 2007Dec15;13(24):7380-7
113. Löhr M. et al. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17-23
114. Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, Ryu B, Skinner HG, Goggins M, Jaffee EM, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Discovery of new markers of cancer through serial analysis of gene expres-sion: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001Jun1;61(11):4320-4
115. Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A. Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003Sep;16(9):902-12
116. Li A, Morton JP, Ma Y, Karim SA, Zhou Y, Faller WJ, Woodham EF, Morris HT, Stevenson RP, Juin A, Jamieson NB, MacKay CJ, Carter CR, Leung HY, Yamashiro S, Blyth K, Sansom OJ, Machesky LM. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology. 2014;146(5):1386-96
117. Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J Clin Invest. 2002Jun;109(11):1437-44
118. Delgiorno KE, Hall JC, Takeuchi KK, Pan FC, Halbrook CJ, Washington MK, Olive KP, Spence JR, Sipos B, Wright CV, Wells JM, Crawford H. Identification C and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology. 2014Jan;146(1):233-44.e5
119. Wilentz RE, Geradts J, Maynard R. et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740-4744
120. Poch B, Gansauge F, Schwarz A. et al. Epidermal growth factor induces cyclin D1 in human pancreatic carcinoma: evidence for a cyclin D1-dependent cell cycle progression. Pancreas. 2001;23:280-287
121. Funel N, Morelli M, Giovannetti E, Del Chiaro M, Pollina LE, Mosca F, Boggi U, Cavazzana A, Campani D. Loss of heterozygosity status of D9S105 marker is associated with downregulation of Krüppel-like factor 4 expression in pancreatic ductal adenocarcinoma and pancreatic intraepithelial lesions. Pancreatology. 2011;11(1):30-42
122. Hingorani SR, Wang L, Multani AS. et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469-483
123. Hingorani SR, Petricoin EF, Maitra A. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437-450
124. Wilentz RE, Iacobuzio-Donahue CA, Argani P. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002-2006
125. Klein WM1, Hruban RH, Klein-Szanto AJ, Wilentz RE. Direct correlation between proliferative activity and dysplasia in pancreatic intraepithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod Pathol. 2002Apr;15(4):441-7
126. Sinha P, Hütter G, Köttgen E, Dietel M, Schadendorf D, Lage H. Increased expression of epidermal fatty acid binding protein, cofilin, and 14-3-3-sigma (stratifin) detected by two-dimensional gel electrophoresis, mass spectrometry and microsequencing of drug-resistant human adenocarcinoma of the pancreas. Electrophoresis. 1999Oct;20(14):2952-60
127. Swartz MJ, Batra SK, Varshney GC. et al. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am. J. Clin. Pathol. 2002;117:791-796
128. Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD, Le X, Wei D, Huang S, Xie K. A novel FoxM1-caveolin signaling pathway promotes pancreatic can-cer invasion and metastasis. Cancer Res. 2012Feb1;72(3):655-65
129. Quan M, Wang P, Cui J, Gao Y, Xie K. The roles of FOXM1 in pancreatic stem cells and carcinogenesis. Mol Cancer. 2013Dec10;12:159
130. Hassan R, Laszik ZG, Lerner M, Raffeld M, Postier R, Brackett D. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreasand chronic pancreatitis. Am J Clin Pathol. 2005Dec;124(6):838-45
Author contact
![]() Corresponding authors: Shaojiang Zheng, M.D., Ph.D., Department of Pathology, Affiliated Hospital of Hainan Medical College, Hainan Cancer Hospital, Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, 3 Xueyuan Road, Haikou, Hainan 571199, People's Republic of China. Tel and Fax: +86 898 667 233 33; E-mail: zhenghohocom; or Keping Xie, M.D., Ph.D., Department of Gastroenterology, Hepatology and Nutrition, Unit 426, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030, USA. Tel: 713-834-6685; Fax: 713-745 1163; E-mail: kepxieorg.
Corresponding authors: Shaojiang Zheng, M.D., Ph.D., Department of Pathology, Affiliated Hospital of Hainan Medical College, Hainan Cancer Hospital, Hainan Provincial Key Laboratory of Carcinogenesis and Intervention, 3 Xueyuan Road, Haikou, Hainan 571199, People's Republic of China. Tel and Fax: +86 898 667 233 33; E-mail: zhenghohocom; or Keping Xie, M.D., Ph.D., Department of Gastroenterology, Hepatology and Nutrition, Unit 426, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030, USA. Tel: 713-834-6685; Fax: 713-745 1163; E-mail: kepxieorg.