Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Neutrophil granulocytes and...
Neutrophils in PDAC
Neutrophil elastase (NE)
Proteinase 3 (PR3)
Cathepsin G
Collagenase 2 (MMP-8)
Gelatinase B (MMP-9) and...
ADAMs
ADAMTS
Azurocidin
Therapeutic strategies
Concluding remarks
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):302-313. doi:10.7150/ijbs.14996 This issue Cite
Review
Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression
Klaus Felix1 ![]() , Matthias M. Gaida2
, Matthias M. Gaida2
1. Department of General Surgery, University of Heidelberg, INF 110, Heidelberg, Germany
2. Institute of Pathology, University of Heidelberg, INF 224, Heidelberg, Germany
Published 2016-1-28
Abstract
A hallmark of pancreatic ductal adenocarcinoma (PDAC) is the fibro-inflammatory microenvironment, consisting of activated pancreatic stellate cells, extracellular matrix proteins, and a variety of inflammatory cells, such as T cells, macrophages, or neutrophils. Tumor-infiltrating immune cells, which are found in nearly all cancers, including PDAC, often fail to eliminate the tumor, but conversely can promote its progression by altering the tumor microenvironment. Pancreatic cancer cells are able to attract polymorphonuclear neutrophils (PMN) via tumor secreted chemokines and in human PDAC, PMN infiltrates can be observed in the vicinity of tumor cells and in the desmoplastic tumor stroma, which correlate with undifferentiated tumor growth and poor prognosis. The behavior of tumor-infiltrating neutrophils in the tumor micromilieu is not yet understood at a mechanistic level. It has been shown that PMN have the potential to kill tumor cells, either directly or by antibody-dependent cell-mediated cytotoxicity, but on the other side various adverse effects of PMN, such as promotion of aggressive tumor growth with epithelial-to-mesenchymal transition and increased metastatic potential, have been described. Recent therapeutic approaches for PDAC focus not only the tumor cell itself, but also elements of the tumor microenvironment. Therefore, the role of PMN and their derived products (e.g. cytokines, proteases) as a new vein for a therapeutic target should be critically evaluated in this context. This review summarizes the current understanding of the interplay between proteases of tumor-infiltrating neutrophils and pancreatic tumor cells and elements of the desmoplastic stroma.
Keywords: inflammation, pancreatic cancer, microenvironment, neutrophils proteinases
Introduction
That tumorigenesis is associated with chronic inflammation, was first described by the German pathologist Rudolf Virchow nearly 150 years ago [1]. His conclusion was based on observations that different leukocytes were frequently found in neoplastic tissues, which Virchow described as “lymphoreticular infiltration”. This observation is still valid today, however a more detailed concept, how the specific inflammatory cells interact with tumor cells and how the tumor cells influence the inflammatory micromilieu was developed over the last decades.
In general, all tumors are composed of heterogeneous cell populations including genetically altered tumor cells, non-neoplastic cells (such as fibroblasts, vascular endothelial cells, residual tissue cells), and cells from the innate and adaptive immune system, which are recruited to the tumor microenvironment and hence are activated, creating a milieu with features of chronic inflammation [2]. The interaction between the multiple lineages of inflammatory cells with the tumor and non-neoplastic cells is a highly dynamic process, driving pathways towards proliferation, differentiation, extracellular matrix- remodeling, chemotaxis, angiogenesis, and chemoresistance. Therefore, the understanding of the molecular mechanisms of this interplay represents one of the major challenges to create new therapeutic strategies.
The cellular composition of the tumor microenvironment of pancreatic ductal adenocarcinoma (PDAC) consists like other tumors malignant and non-malignant cells, but the uniqueness is its enormous desmoplastic stroma component, consisting up to 90% of the whole tumor mass, seeded with different types of inflammatory cells such as neutrophils, macrophages, myeloid-derived suppressor cells, dendritic cells, lymphocytes, mast cells, and other non-neoplastic cells, such as endothelial cells, stellate cells, fibroblasts, and hematopoietic progenitor cells [3, 4]. In the tumor microenvironment, the different inflammatory cell types synthetize a huge variety of cytokines, chemokines, reactive oxygen and nitrogen species, cytotoxic and cell proliferative mediators, as well as different classes of proteinases (e.g. cysteine-, serine- metallo-). Upon activation, the inflammatory cells release these mediators into the extracellular compartment and aggravate the “smoldering” intra-tumor inflammation. In an orchestrated manner, these mediators create an environment conducting intracellular programs for proliferation, genomic instability, malignant transformation, tumor growth, and progression. Pivotal players of the immune cell released mediators are different types of proteases. Once activated, the members of the different protease families promote amongst others the turnover of the extracellular matrix (ECM), thus facilitating especially angiogenesis and tumor cell migration.
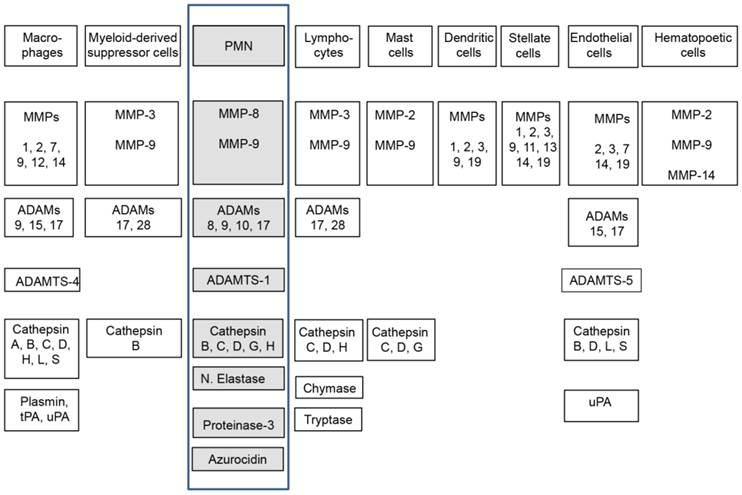
The expression of different types of proteases, in particularly (matrix metalloproteinases) MMPs and (a disintegrin and metalloproteinase) ADAMs, in the tumor microenvironment is very heterogeneous and even different types of tumor cells are able to express them, however, the major source of these proteinases are infiltrating stromal (inflammatory) cells into the tumor [9]. The different types of stromal cells found in the cancer micromilieu produce a specific cluster of proteinases (Figure 1), as well as the corresponding proteinase inhibitors, which are released into the extracellular space and specifically regulate themselves prior to their action in the tumor microenvironment [6]. Therefore, the cellular source of the proteases has important consequences on their function and activity in vivo and depends on the local balance between them and their physiological inhibitors. The proteolytic activity of proteases, such as MMPs can be regulated at different levels: gene expression, compartmentalization, conversion from zymogen to an active enzyme, and by specific inhibitors.
Stroma cell composition and their proteases. Highlighted (boxed) are here reviewed neutrophils, their expressed and secreted proteases [5-8].
Neutrophil granulocytes and granulocytic myeloid-derived suppressor cells
Neutrophils are the most prominent leukocyte type found in the blood circulation (up to 70% of peripheral white blood cells) [10], and well-studied in their basic biology and in terms of bacterial host defense [11]. In infections, chronic inflammation, and malignancies neutrophil granulocytes are systematically expanded in the bone marrow and released towards the site of disease or tumor in response to chemotactic factors (such as granulocyte-macrophage CSF, macrophage CSF granulocyte CSF, or neutrophil-attracting CXC-chemokines). The chemoattractants are released from the micromilieu of the pathologic site (reviewed in: [12-15]). PMN are frequently observed in the microenvironment of different types of tumors - however their pathophysiological roles in the context to tumors are “understudied”. The modest interest is probably based on the old dogma of their short life time. But this statement no longer holds true, due to observations that neutrophils in inflammatory environments can survive up to 5-6 days [16].
In infections, PMN are triggered by chemotactic stimuli, thus migrating into the pathogen-infected area and phagocyte the microbes. For the elimination and degradation of microorganisms, reactive oxygen species (ROS), defensins, and serine-proteinases are rapidly shuttled to phagolysosomes, where they kill the invading microorganisms. In parallel, proteinases are released in huge amounts into the extracellular compartment to overwhelm their endogenous inhibitors and to accomplish focused proteolysis. The influx of PMN to the inflammatory microenvironment is followed by monocytes and dendritic cells which are recruited through chemoattractants, secreted by neutrophils, such as CCL2 [17], CCL3 [18] CCL19, and CCL20 [19]. PMN produce, store, and release a variety of serine-proteinases (e.g. neutrophil elastase, cathepsin-G, and proteinase-3), matrix metalloproteinases (e.g. MMP-8 and MMP-9) reactive oxygen species (ROS), different cytokines and chemokines, antimicrobial peptides, and the metal chelator proteins lactoferrin for Fe and calprotectin for Mn and Zn [20]. An intact neutrophil function is essential for survival of the host. The failure to resolve the inflammation due to malfunction or elimination of apoptotic neutrophils leads to accumulation of cytotoxic substances and is associated with severe pathologies. Disorders of PMN function in humans are characterized by recurrent invasive infections and early mortality. An example is chronic granulomatous disease, where PMN are unable to kill the microorganisms they had engulfed, due to the failure to produce reactive oxygen species. These patients suffer from severe infectious diseases, such as pneumonia, soft tissue and bone infections, and sepsis, resulting in a high mortality.
In the last years, PMN received attention in the context of inflammation driven tumorigenesis [21]. There is a concept that tumor associated neutrophils (TAN) might have different states of activation/polarization. Depending on the local concentration of TGF-β they can acquire a pro-tumor phenotype (N2 TAN) induced by high concentrations of this cytokine. In a TGF-β negative milieu in the presence of IFN-γ they resemble an anti-tumor phenotype (N1 TAN) [13, 21]. The N1 TAN counteract the tumor progression through tumor cell toxicity and enhancement of the antitumor immune memory response. On the other hand, the N2 TAN promote tumor growth, invasion, and metastasis through degradation of ECM, promotion of neovascularization, and immunosuppression [13]. Aside from migration into the tumor, it is discussed that TAN may also differentiate from myeloid-derived suppressor cells present in the microenvironment [22].
Therefore, one should mention that neutrophils are thought to have “direct relatives” in the population of the myeloid cells, the so called myeloid-derived suppressor cells (MDSC). MDSC are a very heterogeneous population of immune-suppressive cells, which characterization is still not finished and conflicting [23]. According to the current understanding MDSC are composed of cells in progenitor stage, in immature and more maturated forms [22]. According to their surface expression profile, murine studies subdivided MDSC into cells with more monocytic characteristics, so called mononuclear (Mo-MDSC) or correspondingly with granulocytic characteristics (G-MDSC). Most likely this two-sided separation does not hold true in humans, where more subsets are discussed [15]. Of importance is the fact that a significant overlap (e.g. in the expression of functional molecules or surface molecules) between neutrophils and G-MDSC exists, raised a couple of theories that both cell types might originate from a common progenitor cell and either enter the tumor micromilieu as finished cell due to lineage separation in the myelopoesis, or that a mature neutrophil transdifferentiate in the tumor micromilieu towards a tumor-associated neutrophil or a G-MDSC [15, 23]. At least one can state that G-MDSC not only have immunomodulating abilities but are also able to induce tumor cell migration or angiogenesis by the secretion of a variety of different proteases has been shown [23]. A comparison of peripheral neutrophils, tumor-associated neutrophils, and G-MDSC in a murine mesothelioma model showed that e.g. neutrophilic elastase was expressed to a similar level in all three groups, whereas the expression of several matrix metalloproteinases (e.g. MMP8, MMP9) varied [24]. Although a relationship between these cell types is overt, the present review will focus on neutrophils.
Neutrophils in PDAC
As mentioned above, PMN were originally studied in the context of host defense against invading bacteria, but now are also recognized as major protagonists of acute and chronic inflammation, and were shown to display immunomodulatory functions in the context of cancer [25].
Here, we review the recent understanding of the role of PMN in the pancreatic cancer microenvironment with special emphasis on their proteinases, which, depending on the circumstances, may either suppress or promote tumorigenesis, or even act independently of their proteolytic activity. Until today only a limited number of studies are available, however, to our knowledge all showed that presence of increased amounts of neutrophils in the tumor, often referred as tumor associated neutrophils (TAN), is associated with poor clinical outcome [26].
A link from infiltrating immune cells, such as neutrophils, in the micromilieu is directly tied to the role of Ras oncogenes, which are crucial for the promotion of malignant cellular transformation. Sparmann and Bar-Sagi demonstrated that the inflammatory chemokine CXCL-8 (synonym: interleukin-8 or IL-8) is a transcriptional target of Ras signaling. In a tumor xenograft model, they show that Ras-dependent CXCL-8 secretion is required for the initiation of tumor-associated inflammation and neovascularization and concluded that Ras oncogene can elicit a stromal response that fosters cancer progression [27]. Tumor cells for example from bronchial carcinoma or PDAC are able to attract PMN via tumor secreted chemokines such as CXCL-8 [28] or CXCL-16 [29]. The systemic inflammatory response from pancreatic cancer cells promotes the infiltration of neutrophils, which provide a favorable tumor environment for cancer progression, by the secretion interleukins IL-2, IL-6, IL-10, tumor necrosis factor-alpha (TNF-α), and vascular endothelial growth factor (VEGF) [30]. In human PDAC, PMN infiltrates can be observed in the vicinity of tumor cells or in the stroma, which correlate with undifferentiated tumor growth and poor prognosis [31, 32]. Similar findings have also been observed in animal breast cancer models [33], renal cancer [34], or lung cancer [35]. In addition increased peripheral blood neutrophils are described in cancer patients [25]. The correlation between TAN and tumor progression is not yet understood at a mechanistic level [25]. Some studies indicate that PMN have the potential to kill tumor cells, either directly [36] or by antibody-dependent cell-mediated cytotoxicity [37]. However, various adverse effects of PMN, such as promotion of aggressive tumor growth and increased metastatic potential, have been described [34, 38]. One hypothesis is that PMN-derived matrix-degrading proteases such as the metalloproteinases MMP-8, MMP-9, and MMP-17, or neutrophil elastase can modulate the composition of the extracellular matrix and facilitate metastasis [26]. Moreover, TAN are a major source of various pro-inflammatory cytokines such as IL-12, TNF-α, and GM-CSF, and chemokines such as CCL-3, CXCL-8, and CXCL-10, which can affect the migration of cancer cells and immune cells [21, 30]. Increased TNF-α and IL-10 levels lead to a decrease in the lymphocyte count as well as lymphocyte dysfunction [39, 40]. An imbalance of infiltrating immune cells may result in an inadequate immunologic reaction to cancer cells. The ratio of elevated neutrophils and decreased lymphocytes (neutrophil lymphocyte ratio; NLR) is associated with poor prognosis for cancer patients and this NLR could be a used as a marker for evaluating the systemic inflammatory response and outcome of individuals [41]. In addition, PMN are potent inductors of angiogenesis via VEGF or by the release of oncostatin-M, which induces cancer cells to secrete proangiogenic factors like VEGF [42, 43]. Reactive oxygen species, which were historically thought to be key factors in defense against bacteria and microbes, have been found to induce DNA damage of colonic epithelial cells and thus promote tumor induction and progression [44]. It was also reported that PMN-derived nitric oxygen species (NOS) can suppress T cell function [45]. Another means of host defense against bacteria are neutrophil extracellular traps (NETs), extruded DNA networks containing histones, granules, adhesion molecules, and proteases, which have been shown to promote circulating tumor cell adhesion and pulmonary metastasis [46]. A single murine study comparing only 4 mice with injected tumors, describes substantial differences in the expression of genes related for granule proteins, respiratory burst, cellular structure, apoptosis, and cytokine and chemokine production between TAN and peripheral blood PMN [24].
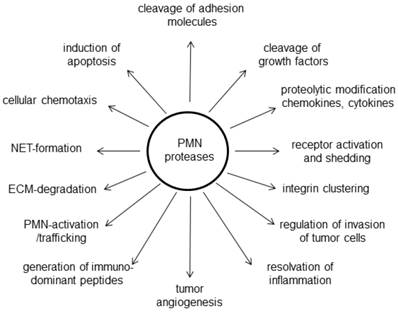
In a study by Reid et al. [32] the histological evaluation of approximately 500 pancreatic tumors revealed a preferential association of neutrophils to certain types of pancreatic neoplasms and that it occurred in a tumor-type- and cell-lineage-dependent manner. Among invasive pancreatic carcinomas, TAN were frequently observed in carcinomas with a micropapillary or undifferentiated growth pattern. In cystic pancreas tumors, neutrophils were preferentially associated with neoplasms of the pancreatobiliary-type lineage, detected in a significant proportion of the mucinous cystic neoplasms (MCN; in their carcinoma in-situ component) as well as in pancreatobiliary-type intraductal papillary mucinous neoplasms (IPMN) [32]. Whether the TAN were secondarily recruited by specific histologic groups of tumors or are primarily participating in their development is uncertain. At the moment, no detailed analyses of human TAN are available. However, a number of neutrophilic proteases were investigated in the context of pancreatic tumors (Figure 2). One note of caution has to be regarded in studying the role of neutrophils in (pancreatic) cancer. Although there are good and established mouse models for PDAC, they might not reflect the role of PMN in patients: Marked differences between human and murine PMN exist not only with regard to the percentage of circulating cells (70% in humans versus 30% in mice), but especially with regard to numerous granular proteins [11].
Diverse functions of PMN derived proteases in the microenvironment of pancreatic cancer
Neutrophil elastase (NE)
The enzymes human neutrophil elastase, as well as human cathepsin G and human proteinase 3 are the three most prominent serine proteases, stored in the azurophilic granula of neutrophil granulocytes [47]. A single neutrophil contains up to 1 pg neutrophil elastase, rising this enzyme to a most abundant enzyme [48]. The neutrophil elastase gene is named neutrophil elastase 2 with the gene symbol ELA2 to differentiate it from pancreatic elastase 1 and pancreatic elastase 3 (ELA1 and ELA3A and ELA3B). Originally thought to be a mediator of the host defense against bacteria [49], there is evidence that neutrophils release this enzyme in other chronic (abacterial) inflammatory settings such as psoriasis as well as in the cancer micromilieu [50, 51]. Neutrophil elastase degrades a variety of substrates including elastin, collagen, cadherins, proteoglycan, fibronectin, platelet receptors, complement receptors, thrombomodulin, lung surfactant, and growth factors (granulocyte-colony-stimulating factor, stromal cell-derived factor-1, and their respective receptors, G-CSFR and CXCR4). Neutrophil elastase mostly releases and acts locally but can be also detected in serum [52]. Among others, the major natural inhibitor of neutrophil elastase is α1-antitrypsin. Neutrophil elastase and α1-antitrypsin are a pair of protease and protease-inhibitor counterparts and an imbalance due to genetic aberrations in one of them is associated with for example with early onset of chronic liver disease, lung emphysema, bronchiectasis, asthma, rheumatoid arthritis, or aneurysma. Recently, it was reported that the imbalance of elastase and its inhibitors plays a significant role in cancer development and progression of malignant tumors, such as liver, lung, or colorectal cancer [53]. It is self-explaining that infiltrated neutrophils are the major source of neutrophil elastase in tumors and an increased concentration of neutrophil elastase in tumors is associated with late-stage disease and poor survival, in different tumors as breast cancer or lung cancer [53].
That neutrophil elastase has not only matrix degrading effects on the extracellular stroma [54, 55] was shown by its presence in neutrophil extracellular traps (NET) and in NET formation. [56, 57]. Additionally, we showed that neutrophil elastase actively promote pancreatic tumor cell migration and invasion [50]. Another group reported that the treatment of pancreatic cancer cells with a specific neutrophil elastase inhibitor ONO-5046.Na, reduced the proliferation activity as well as the migratory capacity of pancreatic cancer cells [58]. A rather explanation is that neutrophil elastase cleaves the tumor-cell adhesion molecule E-cadherin, allowing them to separate from the “main” tumor, followed by migration and invasion [50]. Moreover, the cleavage of E-cadherin on pancreatic (and liver) tumor cells induces an epithelial to mesenchymal transition of the tumor cell. Due to the loss of contact, the tumor cells lose their polarity and in line epithelial phenotype markers such as cytokeratins, and upregulate on the other hand mesenchymal transcription factors such as TWIST and Zeb1 and translocate β-catenin from the cytoplasm to the nucleus [51]. Of note, in a large cohort of human PDAC samples there was a negative correlation of the neutrophil infiltrate and the expression of E-cadherin on tumor cells [50] as well as a positive correlation of the neutrophilic infiltrate with nuclear accumulation of β-catenin and of ZEB1 expression in tumor cells. Another pro-metastatic mechanism might be that neutrophil elastase promotes the adhesion of pancreatic tumor cells on endothelial cells [59]. On the other hand, a study demonstrated a beneficial role of neutrophil elastase in a murine PDAC model, where a cleavage and thus activation of antithrombine inhibits tumor neoangiogenesis [60]. Based on the fact that neutrophil elastase is as a multifunctional enzyme with other abilities, (such as the cleavage of growth factors EGF or TGF-β from tumor cells [61], the regulation of crucial receptors such as EGFR and TLR4, or modulation of cytokine production [62]) it is to be assumed that also more biological functions in the pancreatic tumor microenvironment are likely.
Proteinase 3 (PR3)
Neutrophil serine proteinase 3 and the above described neutrophil elastase are homologous antimicrobial serine proteases of the neutrophils. They are key regulators of the inflammatory response and therefore considered as important cellular targets in a number of chronic inflammatory diseases. Both in common are the sequence identity of 56%, structural similarity, and preferential cleavage of peptide bonds after small hydrophobic amino acids. PR3 can be released extracellularly at sites of inflammation where it plays a crucial role in the pathogenesis by degrading elastin and other matrix proteins causing tissue destruction that may lead to chronic obstructive pulmonary disease, cystic fibrosis, acute lung injury, and inflammation of blood vessels [63, 64]. α-1-antitrypsin is the most important circulating inhibitor of PR3 and individuals with low serum levels of α-1-antitrypsin could develop disease in lung, liver and pancreas. Elevated levels of PR3 were recently reported in patients with type1 diabetes (T1D) and circulating NE and PR3 levels increased progressively with the increase of the positive numbers and titers of the autoantibodies against β-cell antigens [65]. Increased NE and PR3 in T1D patients are closely associated with elevated formation of neutrophil extracellular traps. By contrast, the circulating levels of α-1-antitrypsin were decreased in T1D patients. These findings support an early role of neutrophil activation and augmented neutrophil serine proteases activities in the pathogenesis of β-cell autoimmunity which is discussed to display a risk factor for PDAC [66].
Cathepsin G
Another major neutrophilic serine protease is cathepsin G, a member of the large family of cathepsins [67]. Whereas in general tumor-promoting roles of cathepsin family members such as B, D, and others have been described for pancreatic cancer [68], as well as their particular inhibition was raised as potential therapeutic approach in PDAC [69], little is known about the role of cathepsin G in pancreatic cancer. Cathepsin G, which is stored in azurophilic granules of neutrophils as inactive preform, displays a variety of intra- and extracellular functions linked to tumorigenesis. The tumorigenic activities of cathepsin G include extracellular matrix degradation [67] cytokine/chemokine and receptor processing or shedding [70, 71], induction of chemotaxis of immune cells [72], increased tumor cell invasivity [73], but also induction of apoptosis when entering into the cell followed by the activation of caspases [74]. In addition, cathepsin G mediates tumor-cell adhesion in breast cancer cell by induction of E-cadherin [75]. Proteomic profiling of normal, chronic pancreatitis, and pancreatic cancer tissues revealed an upregulation of cathepsin G protein 1.9 fold in chronic pancreatitis, respectively 2.4 fold in PDAC tissue comparing to normal tissue samples [76]. However, this study does not separate the specific cell types producing cathepsin G. It is known that neutrophils are the most prominent source of cathepsin G, although pancreatic cancer cell lines are also able to express and secrete cathepsin G [77]. Interestingly, pancreatic cancer cells are also able to induce cathepsin G expression in myofibroblastic cells [78]. This data reflect that not only tumor-infiltrating neutrophils are a source of cathepsin G. In addition, SERPIN-A3, an inhibitor of cathepsin G, was shown to be overexpressed in PDAC [76], whereas TFPI2, another cathepsin G inhibitor is downregulated in PDAC [79]. Therefore one might speculate about complex networks regulating the serine protease cathepsin G in PDAC, due to its upregulation in a diversity of sources interacting with inhibitors. In spite of the described abilities of cathepsin G, the data about its role in PDAC are rare and needs further investigation.
Collagenase 2 (MMP-8)
MMP-8 is a Zn2+ metalloendopeptidase, predominantly expressed by neutrophils. MMP-8 is also expressed in few melanoma cell lines, chondrocytes, rheumatoid synovial fibroblasts, activated macrophages, smooth muscle cells, and endothelial cells [80]. In the tumor microenvironment MMP-8 reveals pro-host properties due to its potential to resolve an inflammation. This property is likely based on the fact that MMP-8 cleaves the neutrophil released chemokine CXCL-5 which exerts an enhanced attraction for neutrophils into the inflamed area. In murine models, it could be demonstrated that MMP-8 has a protective role in cancer through its ability to regulate the inflammatory response induced by carcinogens dimethylbenzanthracene and phorbolester (TPA) [81]. Furthermore, a decreased expression of MMP-8 was associated paradoxically with enhanced breast cancer metastasis [82]. For pancreatic cancer it has been described that an intratumor overexpression of MMP-8 facilitates the spread of oncolytic adenovirus, leading to a decrease of the tumor mass [83]. In a cohort of PDAC tissues, it was shown by immunohistochemistry that in approximately 75% of PDAC samples not only the tumor infiltrating neutrophils express MMP-8, but also the tumor cell itself [84].
Gelatinase B (MMP-9) and neutrophil gelatinase B-associated lipocalin (NGAL)
Sequestration of neutrophils within the pancreas is generally believed to be an early and important event in the evolution of pancreatitis. MMP-9 is present in considerable amounts in the granules of neutrophils and has been associated with pancreatitis [85, 86]. The massive infiltrate of neutrophils followed by their degranulation results in an extensive liberation of host defense proteins including MMP-8 (neutrophil collagenase), MMP-9 (gelatinase B), and NGAL (neutrophil gelatinase B-associated lipocalin). These last two proteins are forming a covalent linkage called NGAL-MMP9-complex. NGAL was previously reported to be overexpressed in pancreatic cancer [87]. Terris et al identified NGAL as a marker for intraductal papillary mucinous neoplasm (IPMN) a benign cystic tumor which can transfer to an invasive carcinoma [88]. The expression of NGAL was also found in various grades of the precursor lesions pancreatic intraepithelial neoplasia (PanIN) including the PanIN-1 stage lesion and therefore was suggested as a marker of the earliest premalignant changes in the pancreas [89]. The function of the NGAL-MMP-9 complex is not yet fully elucidated. It was proposed that MMP-9 covalently linked to NGAL protects MMP-9 from autodigestion [90]. An alternative hypothesis is that NGAL supports an allosteric activation of pro-MMP-9 [91]. Matrix metalloproteases are the major factors that degrade the extracellular matrix. The gelatinases as subfamily composed of MMP-2 (gelatinase A) and MMP-9 (gelatinase B) so called type IV collagenases were originally named for the ability to digest type IV collagen of the basement membrane [92]. In addition they were also found to possess a potent gelatinolytic activity that degrades collagen types V, VII, IX, X, fibronectin, and elastin [93]. The degradation of basement membranes and extracellular matrix plays a key role in the severity of inflammation and tumor progression. MMP-9 converts also other substrates as well and in particular cases can have a regulatory function. For instance, the neutrophil chemokine CXCL-8 is potentiated by amino-terminal truncation of six amino acids [94]. Furthermore, MMP-9 cleaves the big endothelin ET-1(AA 1-38) into ET-1(AA 1-32) that activates neutrophils and promotes leukocyte-endothelial cell adhesion and consequently, neutrophil trafficking into inflamed tissues areas [95]. Aside from these functions as a regulator and effector molecule in inflammation [96], MMP-9 increased proteolytic activity may further mediate pathological conditions leading to autoimmune diseases by the generation of immunodominant peptides [94, 96]. Overexpression of MMP-2 and MMP-9 in pancreatic cancer was previously reported [97, 98], and the gelatinases were also investigated as differential markers for CP/PDAC [99-101]. MMP-9 was originally thought to be involved in tumor angiogenesis indirectly by remodeling of the extracellular matrix [102] and interacting with VEGF [103, 104]. Nozawa et al. demonstrated that inflammatory cell derived MMP-9 is able to mobilize VEGF sequestered within the extracellular matrix [105] and particularly MMP-9 is very efficient enzyme for this release [106]. Other group showed that MMP-9 derived from PMN is a direct and potent factor in pancreatic tumor angiogenesis. They demonstrated that MMP-9 and PMN alone induced a robust in vitro angiogenesis, even in the absence of VEGF and in the presence of antibodies to VEGF. Thus PMN derived MMP-9 has a direct proangiogenic effect in addition to its additive VEGF interaction and is essential for tumor-associated blood vessel formation and tumor growth [107].
ADAMs
The ADAMs (a disintegrin and metalloproteinase) are a family of transmembrane and secreted proteins involved in cell adhesion and proteolytic cleavage of the ectodomains of diverse cell surface receptors and cell signaling molecules. Most ADAMs are membrane-anchored and function in the pericellular space. Thirteen of the currently known 21 ADAMs have proteolytic properties and influence the cell signaling within the tumor microenvironment. ADAMs fulfill a broad spectrum of functions and have been implicated in neurological and cardiovascular diseases, asthma, infection, inflammation and cancer [7, 8]. In pancreatic cancer it was shown that ADAM8 expression is associated with increased invasiveness and reduced patient survival [108] and that ADAM10 is required for pancreatic cancer cell migration and invasion [109]. Silencing of ADAM10 in pancreatic cancer cells, decreases their CXCL-16 liberation, and thus inhibits the migration of neutrophils [29]. Furthermore, neutrophilic ADAM10 is required for normal neutrophil migration towards sites of inflammation [110]. ADAM9 is known to be expressed by monocytes and macrophages and it was recently reported that ADAM9 is also a product of human and murine PMN [111]. ADAM9 is likely transcribed by PMN precursors in the bone marrow and stored as a preformed proteinase within PMN granules and vesicles. ADAM9 is redistributed to the PMN surface when PMN are activated by proinflammatory mediators as is the case for PMN-derived serine proteinases and MMPs [111]. ADAM9 degrades several extracellular matrix proteins, including fibronectin, entactin, laminin, and insoluble elastin, as potently as MMP-9. Furthermore, ADAM9 was described to activate neutrophils [112]. Aside of ADAM9, ADAM8 [113], and ADAM17 [114] are further members of the ADAM family known to be expressed by primary PMN. In mice ADAM9, 10, and 17 are potential mediators in the biology of the pancreas with distinct dynamic expression patterns during pancreas development and maturation [115]. ADAM17 has been discovered to be an important proteolytic sheddase of ALCAM and by this cleavage enhancing tumor cell motility and migratory capacity and therefore the metastatic potential [116]. Kahlert et al. could show that ADAM17 is significantly overexpressed in pancreatic cancer compared to normal pancreatic tissue and that the disruption of ALCAM-mediated cell adhesion is a relevant step in pancreatic cancer progression and early relapse of disease [117].
ADAMTS
In close relation to ADAMs and MMPs are the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) families, members of the superfamily of zinc-endopeptidases so called metzincins [118]. Activated neutrophils were shown to produce ADAMTS-family members [119]. The ADAMTS have a protease domain, an adjacent disintegrin domain, one or more thrombospondin domains and are generally secreted and soluble. They play roles in ECM assembly, ovulation, and cancer. The role of these metzincin proteinases in cancer has been extensively reviewed by Murphy [8]. Masui et al. [120] investigated the expression of ADAMTS-1(METH-1) in human pancreatic cell lines and pancreatic cancer tissue and in surrounding noncancerous pancreatic tissue specimens. They observed that pancreatic cancer samples with higher expression of ADAMTS-1 showed significantly frequent lymph node metastasis or retroperitoneal invasion, most likely due to the cleavage of substrates in the surrounding ECM such as heparan-sulfate proteoglycans syndecan-1, and glypican-1, all shown to be overexpressed in pancreatic cancer.
Azurocidin
Azurocidin (also known as CAP37 or heparin-binding protein) is a member of the serine protease family, but without protease activity [121]. It is an abundant protein of neutrophils, mediating especially the extravasation of PMN and antimicrobial activity towards bacteria or candida species [122]. The role in the tumor micromilieu of PDAC is not known. The poor stromal reaction in PDAC areas of neutrophils might not only result from digestion of the extracellular matrix, but also to other regulatory effects of proteases, such as induction of apopotosis. Whereas a decrease of apoptosis in endothelial cells mediated by azurocidin was described [123], another study shows in human umbilical vein endothelial cells an interference of azurocidin to the mitochondrial metabolism followed by the induction of apopotosis [124]. Our data indicate that PMN are activated by pancreatic stellate cells and secrete upon activation azurocidin, which in turn causes a “quiescent” status of pancreatic stellate cells (Gaida MM unpublished data).
Therapeutic strategies
Pancreatic cancer is characterized to be a therapy-refractory cancer. Indeed, novel therapeutic approaches in PDAC are urgently needed, because it is a highly aggressive neoplasm with a 5-year survival rate of approximately 5%, and the limited therapeutic options are only effective in early stages of disease [125]. There is a rising body of interest in the development of so called “stroma targeted therapies”, not only for the cancer cell itself, but also for the cells of the micromilieu, which reveal a more stable and predictable behavior, compared to the tumor cell. Considering this facts, immune cells may become a primary target, not simply for the prediction of the prognosis, but more for the direct treatment and intervention of cancers including, PDAC. Depending on the activation state and their released mediators they may either act tumoricidal or conversely enhance tumor growth. Considering neutrophils as a potent source of protumorigenic mediators favoring cancer development and progression, there are several reasons to target PMN, respectively their released products: PMN possess a very efficient migration machinery and can be quickly recruited to areas of early cancer development and interact with tumor cells in the very early steps of malignant transformation. They can migrate in and out of the tumor microenvironment, transferring important signals, and can also be detected in the peripheral circulation. Primed by cancer cells, they have a prolonged life cycle and can reside in the tumor microenvironment for several days. Additionally they are equipped with a variety of matrix-remodeling proteinases which can modulate the tumor microenvironment.
Thus the identification of PMN derived and released tumor promoting substances may lead to novel therapeutic approaches. Most of their proteins are premade in the bone marrow, and stored in their granules. Several proteases, in particular MMPs, were described to be upregulated in different cell types in malignant tissues thus showing an enhanced capability to digest various proteins of the extracellular matrix. Therefore, since several decades, proteases are in the focus as targets also for (pancreatic) cancer therapy [126, 127]. Unfortunately, the development of synthetic MMP-inhibitors and their tests in preclinical trials were not promising [128] but at the same time shed light on the biological functions of MMPsin the context of tumors. Additionally to the components of the ECM also other non-matrix substrates of MMPs were identified as targets including other proteinases, proteinase inhibitors, clotting factors, chemotactic molecules, growth factors, growth factor binding proteins, cell surface receptors, and cell-cell and cell-matrix adhesion molecules (reviewed in [129]). MMP cleavage events are associated with both stimulation and repression of cellular proliferation, with enhancement and abrogation of apoptosis, and increased and decreased chemotaxis and migration. Consequently, certain MMP activities may aggravate a pathological condition, whereas others may be beneficial to the host [130]. An understanding of the biological outcome of MMPs cleavage products is required for the therapeutic application of MMP inhibitors to the treatment and prevention of complex diseases. Additionally a new focus of research is the non-proteolytic function of MMPs. Dufour et al. demonstrated that the hemopexin domain of MMP-9 is essential for the MMP-9 mediated epithelial cell migration [131, 132]. In CLL the hemopexin domain of MMP-9 prolongs the B cell survival through binding to B cell surface receptors and prevents cell apoptosis [133]. A plethora of synthetic substrates have been postulated mainly based on in vitro studies [134], few have been identified as a true substrate in an in vivo context [135]. The therapeutic implications for neutrophil elastase inhibitors in malignomas were tested, mainly in vitro but also in animal models, and a variety of substances have been shown to inhibit neutrophil elastase, such as designed natural occurring peptide-inhibitors binding to the active site of the enzyme e.g. α-1-antitrypsin or human monocyte/neutrophil elastase inhibitor (M/NEI), the natural products heparin or epigallocatechin-3-gallate from green tea, different fatty acids, or synthetic neutrophil-elastase inhibitors such as coumarin or the antibiotic-family members beta-lactame or cephalosporins [136]. In an in vivo mouse study for lung cancer, it was shown that the treatment of genetically-modified LSL-K-ras mice with the specific neutrophil elastase inhibitor ONO-5046, significantly reduced the tumor growth [137]. Furthermore the treatment of mice with the neutrophil elastase inhibitor GW311616A, prevented the formation of metastasis in a lung cancer model [46]. A couple of other studies showed that a specific inhibition of neutrophil elastase can inhibit the development, growth and the progression of a variety of tumors, including lung and colonic cancer [53]. Data for PDAC are rare. An in vitro study with the neutrophil elastase inhibitor ONO-5046, described an inhibition of proliferation and chemotaxis of the tumor cells [58]. Regarding the clinical implication of cathepsin G treatment in PDAC, to our knowledge, no studies are available. But, the inhibition of cathepsins is discussed as therapeutic approach for PDAC [69]. Due to the fact that a variety of neutrophil elastase and cathepsin G inhibitors are available as well as neutrophils display a significant population in the pancreatic cancer microenvironment [29, 50, 109], studying their role as additional treatment options in PDAC might be useful.
Therapeutic approaches for inhibition of ADAMs are still preliminary. Similar to the MMPs they exert proteolytic and non-protease functions and most attempts to develop therapies against cancer were made for the targeting of proteolytic ADAMs. Learned from the experiences and pitfalls with early generation of synthetic MMP-inhibitors more caution was put into the development of highly selective active site directed inhibitors for individual ADAMs [138]. Recent interest in selective inhibition of ADAM17 is its modulation of ErbB/EGFR signaling in the focus of immune diseases, inflammation and tumorigenesis [139]. ADAM17 is produced by neutrophils and also found overexpressed in pancreatic cancer [117]. Recently Ardito et al. applying a murine pancreatic tumor model showed that genetic ablation or pharmacological inhibition of ADAM17 effectively eliminates KRAS-driven tumorigenesis in vivo [140].
Concluding remarks
Excessive release of proteases mediates tissue remodeling and damage, and therefore a marked neutrophil accumulation, as source of several important proteases, has an important role in the pathogenesis of many diseases, in particular inflammation and cancer. In this short review, we discuss the influx and role of neutrophils into the pancreatic cancer microenvironment, with special emphasis on their proteases. There is increasing evidence that proteases released from neutrophils are pivotal players in tumor initiation and progression, also in pancreatic cancer, which is especially characterized by distinct inflammatory reaction in its microenvironment. Those neutrophils are not only “bystanders” in the tumor micromilieu but “active players”, releasing a variety and high amounts of biological active proteases. Although found in the majority of PDAC tissues, the data about the role of PMN are limited. It is widely accepted that pancreatic cancer progression is highly dependent on factors released by the tumor micromilieu, and when progressed, this tumor is basically refractory to standard chemo- and radiotherapies. Therefore, the understanding and interaction with the inflammatory micromilieu of PDAC it seems to be an additional vein to establish new therapeutic options.
Competing Interests
The authors have declared that no competing interest exists.
References
1. R V. Die krankhaften Geschwülste. Berlin: August Hirschwald. 1864 -1865
2. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286-291
3. Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Lohr JM, Friess H. Pancreatic cancer microenvironment. Int J Cancer. 2007;121:699-705
4. Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, Nakanishi S, Usui Y, Higuchi K, Takahashi M. et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015;75:2629-2640
5. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21:228-237
6. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
7. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258-289
8. Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8:929-941
9. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-174
10. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381-1390
11. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459-489
12. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253-268
13. Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol. 2012;82:296-309
14. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: mechanisms and consequences for tumor progression. Semin Cancer Biol. 2013;23:141-148
15. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. 2012;61:1155-1167
16. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012-2020
17. Yoshimura T, Takahashi M. IFN-gamma-mediated survival enables human neutrophils to produce MCP-1/CCL2 in response to activation by TLR ligands. J Immunol. 2007;179:1942-1949
18. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970-974
19. Scapini P, Laudanna C, Pinardi C, Allavena P, Mantovani A, Sozzani S, Cassatella MA. Neutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP-3alpha)/CCL20 and MIP-3beta/CCL19. Eur J Immunol. 2001;31:1981-1988
20. Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R. et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962-965
21. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell. 2009;16:183-194
22. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238-244
23. Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol. 2013;23:171-182
24. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, Horng W, Fridlender G, Bayuh R, Worthen GS, Albelda SM. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One. 2012;7:e31524
25. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33:949-955
26. Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71:2411-2416
27. Sparmann A, Bar-Sagi D. Ras oncogene and inflammation: partners in crime. Cell Cycle. 2005;4:735-736
28. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, Mayaud C, Milleron B, Baud L, Cadranel J. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol. 1998;152:83-92
29. Gaida MM, Gunther F, Wagner C, Friess H, Giese NA, Schmidt J, Hansch GM, Wente MN. Expression of the CXCR6 on polymorphonuclear neutrophils in pancreatic carcinoma and in acute, localized bacterial infections. Clin Exp Immunol. 2008;154:216-223
30. Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195-203
31. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914-923
32. Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, Altinel D, Adsay V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. 2011;24:1612-1619
33. Welch DR, Schissel DJ, Howrey RP, Aeed PA. Tumor-elicited polymorphonuclear cells, in contrast to "normal" circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc Natl Acad Sci U S A. 1989;86:5859-5863
34. Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol. 2009;27:4709-4717
35. Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B, Mayaud C, Antoine M, Soler P, Cadranel J. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res. 2003;63:1405-1412
36. Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97:339-345
37. Kindzelskii AL, Petty HR. Early membrane rupture events during neutrophil-mediated antibody-dependent tumor cell cytolysis. J Immunol. 1999;162:3188-3192
38. Aeed PA, Nakajima M, Welch DR. The role of polymorphonuclear leukocytes (PMN) on the growth and metastatic potential of 13762NF mammary adenocarcinoma cells. Int J Cancer. 1988;42:748-759
39. Salazar-Onfray F, Lopez MN, Mendoza-Naranjo A. Paradoxical effects of cytokines in tumor immune surveillance and tumor immune escape. Cytokine Growth Factor Rev. 2007;18:171-182
40. Bellone G, Turletti A, Artusio E, Mareschi K, Carbone A, Tibaudi D, Robecchi A, Emanuelli G, Rodeck U. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537-547
41. Yang JJ, Hu ZG, Shi WX, Deng T, He SQ, Yuan SG. Prognostic significance of neutrophil to lymphocyte ratio in pancreatic cancer: A meta-analysis. World J Gastroenterol. 2015;21:2807-2815
42. Mueller MD, Lebovic DI, Garrett E, Taylor RN. Neutrophils infiltrating the endometrium express vascular endothelial growth factor: potential role in endometrial angiogenesis. Fertil Steril. 2000;74:107-112
43. Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res. 2005;65:8896-8904
44. Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z. et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A. 2012;109:E1820-1829
45. Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155-1166
46. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013
47. Hajjar E, Broemstrup T, Kantari C, Witko-Sarsat V, Reuter N. Structures of human proteinase 3 and neutrophil elastase-so similar yet so different. FEBS J. 2010;277:2238-2254
48. Wiedow O, Muhle K, Streit V, Kameyoshi Y. Human eosinophils lack human leukocyte elastase. Biochim Biophys Acta. 1996;1315:185-187
49. Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91-94
50. Gaida MM, Steffen TG, Gunther F, Tschaharganeh DF, Felix K, Bergmann F, Schirmacher P, Hansch GM. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur J Immunol. 2012;42:3369-3380
51. Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hansch GM, Gaida MM. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768
52. Yamanouchi H, Fujita J, Hojo S, Yoshinouchi T, Kamei T, Yamadori I, Ohtsuki Y, Ueda N, Takahara J. Neutrophil elastase: alpha-1-proteinase inhibitor complex in serum and bronchoalveolar lavage fluid in patients with pulmonary fibrosis. Eur Respir J. 1998;11:120-125
53. Sun Z, Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol. 2004;5:182-190
54. Mainardi CL, Hasty DL, Seyer JM, Kang AH. Specific cleavage of human type III collagen by human polymorphonuclear leukocyte elastase. J Biol Chem. 1980;255:12006-12010
55. Mainardi CL, Dixit SN, Kang AH. Degradation of type IV (basement membrane) collagen by a proteinase isolated from human polymorphonuclear leukocyte granules. J Biol Chem. 1980;255:5435-5441
56. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-1535
57. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677-691
58. Kamohara H, Sakamoto K, Mita S, An XY, Ogawa M. Neutrophil elastase inhibitor (ONO-5046.Na) suppresses the proliferation, motility and chemotaxis of a pancreatic carcinoma cell line, Capan-1. Res Commun Mol Pathol Pharmacol. 1997;98:103-108
59. Nozawa F, Hirota M, Okabe A, Shibata M, Iwamura T, Haga Y, Ogawa M. Elastase activity enhances the adhesion of neutrophil and cancer cells to vascular endothelial cells. J Surg Res. 2000;94:153-158
60. Kisker O, Onizuka S, Banyard J, Komiyama T, Becker CM, Achilles EG, Barnes CM, O'Reilly MS, Folkman J, Pirie-Shepherd SR. Generation of multiple angiogenesis inhibitors by human pancreatic cancer. Cancer Res. 2001;61:7298-7304
61. Sato T, Takahashi S, Mizumoto T, Harao M, Akizuki M, Takasugi M, Fukutomi T, Yamashita J. Neutrophil elastase and cancer. Surg Oncol. 2006;15:217-222
62. Bergin DA, Greene CM, Sterchi EE, Kenna C, Geraghty P, Belaaouaj A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J Biol Chem. 2008;283:31736-31744
63. Voynow JA, Fischer BM, Zheng S. Proteases and cystic fibrosis. Int J Biochem Cell Biol. 2008;40:1238-1245
64. Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, Noel LH, Guillevin L, Ravaud P, Sermet-Gaudelus I. et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. 1999;10:1224-1233
65. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, Bornstein SR, Zhou Z, Lam KS, Xu A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes. 2014;63:4239-4248
66. Ganss R, Hanahan D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998;58:4673-4681
67. Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins mediate tumor metastasis. World J Biol Chem. 2013;4:91-101
68. Sulpizio S, Franceschini N, Piattelli A, Di Sebastiano P, Innocenti P, Selvaggi F. Cathepsins and pancreatic cancer: the 2012 update. Pancreatology. 2012;12:395-401
69. Michl P. Targeting cathepsins: a new glimmer of hope for pancreatic cancer therapy? Gut. 2012;61:790-791
70. Wiedow O, Meyer-Hoffert U. Neutrophil serine proteases: potential key regulators of cell signalling during inflammation. J Intern Med. 2005;257:319-328
71. Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994;352:231-235
72. Chertov O, Ueda H, Xu LL, Tani K, Murphy WJ, Wang JM, Howard OM, Sayers TJ, Oppenheim JJ. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med. 1997;186:739-747
73. Shamamian P, Pocock BJ, Schwartz JD, Monea S, Chuang N, Whiting D, Marcus SG, Galloway AC, Mignatti P. Neutrophil-derived serine proteinases enhance membrane type-1 matrix metalloproteinase-dependent tumor cell invasion. Surgery. 2000;127:142-147
74. Bird PI. Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins. Immunol Cell Biol. 1999;77:47-57
75. Kudo T, Kigoshi H, Hagiwara T, Takino T, Yamazaki M, Yui S. Cathepsin G, a neutrophil protease, induces compact cell-cell adhesion in MCF-7 human breast cancer cells. Mediators Inflamm. 2009;2009:850940
76. Pan S, Chen R, Stevens T, Bronner MP, May D, Tamura Y, McIntosh MW, Brentnall TA. Proteomics portrait of archival lesions of chronic pancreatitis. PLoS One. 2011;6:e27574
77. Yamaguchi N, Yamamura Y, Koyama K, Ohtsuji E, Imanishi J, Ashihara T. Characterization of new human pancreatic cancer cell lines which propagate in a protein-free chemically defined medium. Cancer Res. 1990;50:7008-7014
78. Basso D, Millino C, Greco E, Romualdi C, Fogar P, Valerio A, Bellin M, Zambon CF, Navaglia F, Dussini N. et al. Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genes. Gut. 2004;53:1159-1166
79. Dawelbait G, Winter C, Zhang Y, Pilarsky C, Grutzmann R, Heinrich JC, Schroeder M. Structural templates predict novel protein interactions and targets from pancreas tumour gene expression data. Bioinformatics. 2007;23:i115-124
80. Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M, Schonbeck U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899-1904
81. Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252-257
82. Agarwal D, Goodison S, Nicholson B, Tarin D, Urquidi V. Expression of matrix metalloproteinase 8 (MMP-8) and tyrosinase-related protein-1 (TYRP-1) correlates with the absence of metastasis in an isogenic human breast cancer model. Differentiation. 2003;71:114-125
83. Cheng J, Sauthoff H, Huang Y, Kutler DI, Bajwa S, Rom WN, Hay JG. Human matrix metalloproteinase-8 gene delivery increases the oncolytic activity of a replicating adenovirus. Mol Ther. 2007;15:1982-1990
84. Jones LE, Humphreys MJ, Campbell F, Neoptolemos JP, Boyd MT. Comprehensive analysis of matrix metalloproteinase and tissue inhibitor expression in pancreatic cancer: increased expression of matrix metalloproteinase-7 predicts poor survival. Clin Cancer Res. 2004;10:2832-2845
85. Muhs BE, Patel S, Yee H, Marcus S, Shamamian P. Increased matrix metalloproteinase expression and activation following experimental acute pancreatitis. J Surg Res. 2001;101:21-28
86. Yokota T, Denham W, Murayama K, Pelham C, Joehl R, Bell RH Jr. Pancreatic stellate cell activation and MMP production in experimental pancreatic fibrosis. J Surg Res. 2002;104:106-111
87. Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845-867
88. Terris B, Blaveri E, Crnogorac-Jurcevic T, Jones M, Missiaglia E, Ruszniewski P, Sauvanet A, Lemoine NR. Characterization of gene expression profiles in intraductal papillary-mucinous tumors of the pancreas. Am J Pathol. 2002;160:1745-1754
89. Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, Lele SM, Ouellette M, Pour PM, Sasson AR. et al. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540-1547
90. Kubben FJ, Sier CF, Hawinkels LJ, Tschesche H, van Duijn W, Zuidwijk K, van der Reijden JJ, Hanemaaijer R, Griffioen G, Lamers CB, Verspaget HW. Clinical evidence for a protective role of lipocalin-2 against MMP-9 autodegradation and the impact for gastric cancer. Eur J Cancer. 2007;43:1869-1876
91. Tschesche H, Zolzer V, Triebel S, Bartsch S. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. Eur J Biochem. 2001;268:1918-1928
92. Nethery A, O'Grady RL. Identification of a metalloproteinase co-purifying with rat tumour collagenase and the characteristics of fragments of both enzymes. Biochim Biophys Acta. 1989;994:149-160
93. Senior RM, Griffin GL, Fliszar CJ, Shapiro SD, Goldberg GI, Welgus HG. Human 92- and 72-kilodalton type IV collagenases are elastases. J Biol Chem. 1991;266:7870-7875
94. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673-2681
95. Fernandez-Patron C, Zouki C, Whittal R, Chan JS, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of endothelin-1[1-32]. FASEB J. 2001;15:2230-2240
96. Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol. 2001;22:571-579
97. Gress TM, Muller-Pillasch F, Lerch MM, Friess H, Buchler M, Adler G. Expression and in-situ localization of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in pancreatic cancer. Int J Cancer. 1995;62:407-413
98. Koshiba T, Hosotani R, Wada M, Miyamoto Y, Fujimoto K, Lee JU, Doi R, Arii S, Imamura M. Involvement of matrix metalloproteinase-2 activity in invasion and metastasis of pancreatic carcinoma. Cancer. 1998;82:642-650
99. Bramhall SR, Stamp GW, Dunn J, Lemoine NR, Neoptolemos JP. Expression of collagenase (MMP2), stromelysin (MMP3) and tissue inhibitor of the metalloproteinases (TIMP1) in pancreatic and ampullary disease. Br J Cancer. 1996;73:972-978
100. Lekstan A, Lampe P, Lewin-Kowalik J, Olakowski M, Jablonska B, Labuzek K, Jedrzejowska-Szypulka H, Olakowska E, Gorka D, Filip I, Dranka-Bojarowska D. Concentrations and activities of metalloproteinases 2 and 9 and their inhibitors (TIMPS) in chronic pancreatitis and pancreatic adenocarcinoma. J Physiol Pharmacol. 2012;63:589-599
101. Mroczko B, Lukaszewicz-Zajac M, Wereszczynska-Siemiatkowska U, Groblewska M, Gryko M, Kedra B, Jurkowska G, Szmitkowski M. Clinical significance of the measurements of serum matrix metalloproteinase-9 and its inhibitor (tissue inhibitor of metalloproteinase-1) in patients with pancreatic cancer: metalloproteinase-9 as an independent prognostic factor. Pancreas. 2009;38:613-618
102. Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422-433
103. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-744
104. Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288-36295
105. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493-12498
106. Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681-691
107. Bausch D, Pausch T, Krauss T, Hopt UT, Fernandez-del-Castillo C, Warshaw AL, Thayer SP, Keck T. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis. 2011;14:235-243
108. Valkovskaya N, Kayed H, Felix K, Hartmann D, Giese NA, Osinsky SP, Friess H, Kleeff J. ADAM8 expression is associated with increased invasiveness and reduced patient survival in pancreatic cancer. J Cell Mol Med. 2007;11:1162-1174
109. Gaida MM, Haag N, Gunther F, Tschaharganeh DF, Schirmacher P, Friess H, Giese NA, Schmidt J, Wente MN. Expression of A disintegrin and metalloprotease 10 in pancreatic carcinoma. Int J Mol Med. 2010;26:281-288
110. Pruessmeyer J, Hess FM, Alert H, Groth E, Pasqualon T, Schwarz N, Nyamoya S, Kollert J, van der Vorst E, Donners M. et al. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood. 2014;123:4077-4088
111. Roychaudhuri R, Hergrueter AH, Polverino F, Laucho-Contreras ME, Gupta K, Borregaard N, Owen CA. ADAM9 is a novel product of polymorphonuclear neutrophils: regulation of expression and contributions to extracellular matrix protein degradation during acute lung injury. J Immunol. 2014;193:2469-2482
112. Amendola RS, Martin AC, Selistre-de-Araujo HS, Paula-Neto HA, Saldanha-Gama R, Barja-Fidalgo C. ADAM9 disintegrin domain activates human neutrophils through an autocrine circuit involving integrins and CXCR2. J Leukoc Biol. 2015
113. Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, Fourie A, Tugores A, Valenzuela-Fernandez A, Mollinedo F. et al. Expression and regulation of the metalloproteinase ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-selectin shedding. J Immunol. 2007;178:8053-8063
114. Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J Leukoc Biol. 2007;82:173-176
115. Asayesh A, Alanentalo T, Khoo NK, Ahlgren U. Developmental expression of metalloproteases ADAM 9, 10, and 17 becomes restricted to divergent pancreatic compartments. Dev Dyn. 2005;232:1105-1114
116. Rosso O, Piazza T, Bongarzone I, Rossello A, Mezzanzanica D, Canevari S, Orengo AM, Puppo A, Ferrini S, Fabbi M. The ALCAM shedding by the metalloprotease ADAM17/TACE is involved in motility of ovarian carcinoma cells. Mol Cancer Res. 2007;5:1246-1253
117. Kahlert C, Weber H, Mogler C, Bergmann F, Schirmacher P, Kenngott HG, Matterne U, Mollberg N, Rahbari NN, Hinz U. et al. Increased expression of ALCAM/CD166 in pancreatic cancer is an independent prognostic marker for poor survival and early tumour relapse. Br J Cancer. 2009;101:457-464
118. Stocker W, Bode W. Structural features of a superfamily of zinc-endopeptidases: the metzincins. Curr Opin Struct Biol. 1995;5:383-390
119. Krampert M, Kuenzle S, Thai SN, Lee N, Iruela-Arispe ML, Werner S. ADAMTS1 proteinase is up-regulated in wounded skin and regulates migration of fibroblasts and endothelial cells. J Biol Chem. 2005;280:23844-23852
120. Masui T, Hosotani R, Tsuji S, Miyamoto Y, Yasuda S, Ida J, Nakajima S, Kawaguchi M, Kobayashi H, Koizumi M. et al. Expression of METH-1 and METH-2 in pancreatic cancer. Clin Cancer Res. 2001;7:3437-3443
121. Iversen LF, Kastrup JS, Bjorn SE, Rasmussen PB, Wiberg FC, Flodgaard HJ, Larsen IK. Structure of HBP, a multifunctional protein with a serine proteinase fold. Nat Struct Biol. 1997;4:265-268
122. Linder A, Soehnlein O, Akesson P. Roles of heparin-binding protein in bacterial infections. J Innate Immun. 2010;2:431-438
123. Olofsson AM, Vestberg M, Herwald H, Rygaard J, David G, Arfors KE, Linde V, Flodgaard H, Dedio J, Muller-Esterl W, Lundgren-Akerlund E. Heparin-binding protein targeted to mitochondrial compartments protects endothelial cells from apoptosis. J Clin Invest. 1999;104:885-894
124. Wang L, Liu Z, Dong Z, Pan J, Ma X. Azurocidin-induced inhibition of oxygen metabolism in mitochondria is antagonized by heparin. Exp Ther Med. 2014;8:1473-1478
125. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
126. Hess KR, Abbruzzese JL. Matrix metalloproteinase inhibition of pancreatic cancer: matching mechanism of action to clinical trial design. J Clin Oncol. 2001;19:3445-3446
127. Saif MW. Anti-angiogenesis therapy in pancreatic carcinoma. JOP. 2006;7:163-173
128. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387-2392
129. McCawley LJ, Matrisian LM. Matrix metalloproteinases: they're not just for matrix anymore!. Curr Opin Cell Biol. 2001;13:534-540
130. Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589-601
131. Dufour A, Sampson NS, Zucker S, Cao J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J Cell Physiol. 2008;217:643-651
132. Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem. 2010;285:35944-35956
133. Redondo-Munoz J, Ugarte-Berzal E, Terol MJ, Van den Steen PE, Hernandez del Cerro M, Roderfeld M, Roeb E, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A. Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell. 2010;17:160-172
134. Funel N, Costa F, Pettinari L, Taddeo A, Sala A, Chiriva-Internati M, Cobos E, Colombo G, Milzani A, Campani D. et al. Ukrain affects pancreas cancer cell phenotype in vitro by targeting MMP-9 and intra-/extracellular SPARC expression. Pancreatology. 2010;10:545-552
135. Dufour A, Sampson NS, Li J, Kuscu C, Rizzo RC, Deleon JL, Zhi J, Jaber N, Liu E, Zucker S, Cao J. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011;71:4977-4988
136. Moroy G, Alix AJ, Sapi J, Hornebeck W, Bourguet E. Neutrophil elastase as a target in lung cancer. Anticancer Agents Med Chem. 2012;12:565-579
137. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR. et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16:219-223
138. Cuniasse P, Devel L, Makaritis A, Beau F, Georgiadis D, Matziari M, Yiotakis A, Dive V. Future challenges facing the development of specific active-site-directed synthetic inhibitors of MMPs. Biochimie. 2005;87:393-402
139. Fridman JS, Caulder E, Hansbury M, Liu X, Yang G, Wang Q, Lo Y, Zhou BB, Pan M, Thomas SM. et al. Selective inhibition of ADAM metalloproteases as a novel approach for modulating ErbB pathways in cancer. Clin Cancer Res. 2007;13:1892-1902
140. Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC. et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304-317
Author contact
![]() Corresponding author: Klaus Felix, Ph.D. Department of General Surgery, University of Heidelberg, Im Neuenheimer Feld 110, D - 69120 Heidelberg, Germany. Tel: +49-6221 566439, Fax: +49-6221 564830, E-mail: klaus.felixuni-heidelberg.de
Corresponding author: Klaus Felix, Ph.D. Department of General Surgery, University of Heidelberg, Im Neuenheimer Feld 110, D - 69120 Heidelberg, Germany. Tel: +49-6221 566439, Fax: +49-6221 564830, E-mail: klaus.felixuni-heidelberg.de