Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
I. Rare highly penetrant...
II. Common low risk pancreatic...
Common pancreatic cancer risk...
Pathway analyses of pancreatic...
Functional understanding of...
Summary and future perspectives
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):314-325. doi:10.7150/ijbs.15001 This issue Cite
Review
Pancreatic Cancer Genetics
Laufey T. Amundadottir ![]()
Laboratory of Translational Genomics, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Published 2016-1-28
Abstract
Although relatively rare, pancreatic tumors are highly lethal [1]. In the United States, an estimated 48,960 individuals will be diagnosed with pancreatic cancer and 40,560 will die from this disease in 2015 [1]. Globally, 337,872 new pancreatic cancer cases and 330,391 deaths were estimated in 2012 [2]. In contrast to most other cancers, mortality rates for pancreatic cancer are not improving; in the US, it is predicted to become the second leading cause of cancer related deaths by 2030 [3, 4]. The vast majority of tumors arise in the exocrine pancreas, with pancreatic ductal adenocarcinoma (PDAC) accounting for approximately 95% of tumors. Tumors arising in the endocrine pancreas (pancreatic neuroendocrine tumors) represent less than 5% of all pancreatic tumors [5]. Smoking, type 2 diabetes mellitus (T2D), obesity and pancreatitis are the most consistent epidemiological risk factors for pancreatic cancer [5]. Family history is also a risk factor for developing pancreatic cancer with odds ratios (OR) ranging from 1.7-2.3 for first-degree relatives in most studies, indicating that shared genetic factors may play a role in the etiology of this disease [6-9]. This review summarizes the current knowledge of germline pancreatic cancer risk variants with a special emphasis on common susceptibility alleles identified through Genome Wide Association Studies (GWAS).
Keywords: pancreatic tumors
I. Rare highly penetrant mutations and familial cancer syndromes
A small proportion, approximately 5-10% of the familial aggregation of pancreatic cancer is thought to be explained by hereditary cancer syndromes and inherited forms of pancreatitis, caused by rare high-risk inherited mutations [10-18]. Multiple genes have been identified as carrying mutations that increase risk of pancreatic cancer, most often within multi-cancer familial cancer syndromes, but also in patients with inherited mutations that cause hereditary pancreatitis. While readers are referred to excellent reviews of high risk genes that predispose to pancreatic cancer [19-21], this review summarizes the most consistent findings:
Peutz-Jeghers syndrome is a rare autosomal dominant disease characterized by melanocytic macules of lips, buccal mucosa and digits, and benign intestinal polyps with a greatly increased risk of multiple malignancies that include gastrointestinal, breast and gynecological cancers [22]. Mutations in the STK11 gene (chr19p13.3) which encodes the tumor suppressor serine/threonine protein kinase STK11 cause Peutz-Jeghers syndrome. They are associated with a high risk of pancreatic cancer (relative risk estimates range from 76-132) in studies conducted in the U.S. and Europe [23, 24]. STK11 regulates diverse processes, such as cell growth, cell polarity, energy metabolism and apoptosis, mainly via regulation of AMPK/mTOR signaling [22].
Familial atypical multiple mole and melanoma syndrome (FAMMM). Inactivating mutations in the CDKN2A tumor suppressor gene (chr9p21.3) are associated with familial melanoma, an autosomal dominantly inherited cancer syndrome termed FAMMM [25]. Other cancers, including pancreatic cancer, are seen at a higher than expected frequency in a subset of FAMMM families [26, 27]. The CDKN2A gene encodes cyclin-dependent kinase inhibitor 2A (also called p16), an important cell cycle regulator that negatively regulates cell proliferation [25]. Among pancreatic cancer cases, either unselected or selected for positive pancreatic cancer family history, 0.6-3.3% have been described to carry deleterious germline mutations in CDKN2A, respectively [28, 29]. Specific CDKN2A mutations appear to be associated with a high frequency of pancreatic cancer. An example is the Dutch Leiden founder mutation (19 bp deletion in CDKN2A) which is associated with an estimated 48 fold increased risk of pancreatic cancer [30, 31].
Hereditary nonpolyposis colorectal cancer (HNPCC), or Lynch syndrome, is characterized by germline mutations in DNA mismatch repair (MMR) genes that include MLH1 (chr3p22.2), MSH2 (chr2p21), MSH6 (chr2p16.3) and PMS2 (chr7p22.1). Bi-allelic loss of an MMR gene leads to genomic instability which is often manifested by microsatellite instability in colorectal tumors. Mutation carriers have an increased risk of multiple cancer types, most notably colorectal and endometrial cancer, but also pancreatic cancer [32-34]. A large study that included 147 families (over 6,000 individuals) with mutations in MMR genes, noted at least one case of pancreatic cancer in ~21% families. The cumulative risk of pancreatic cancer up to age 70 was 3.68% in this study, or close a 9 fold increased risk as compared to the general population [14]. Familial adenomatous polyposis (FAP) syndrome is characterized by numerous (often thousands) colorectal polyps and a greatly increased risk of colon cancer. This syndrome is caused by mutations in the APC tumor suppressor gene on chr5q22.2, which encodes a negative regulator of the WNT signaling pathway [35]. Pancreatic cancer has been observed at a higher than expected frequency in affected individuals with an estimated relative risk (RR) of 4.46 [36, 37].
Hereditary breast and ovarian cancer syndrome (HBOC) is characterized by germ-line mutations in the BRCA1 and BRCA2 genes and an increased risk of breast, ovarian, prostate and pancreatic cancer [38, 39]. These two genes encode tumor suppressor proteins that play important roles in DNA damage response and DNA repair to protect genome integrity [40]. Mutations in BRCA2 (chr13q13.1) have been associated with an increased risk (~4-6 fold) of pancreatic cancer in multiple studies [41-43]. Results for BRCA1 (chr17q21.31) have been less consistent across studies and range from no effect to ~4 fold increased risk [42, 44]. Two additional genes that increase risk of breast cancer, namely PALB2 (chr16p12.2) and ATM (11q22.3) have also been found mutated in pancreatic cancer families [13, 29, 45-48]. PALB2 encodes partner and localizer of BRCA2, a protein that interacts with BRCA1 and BRCA2 and is required for localization of BRCA2 to nuclear double strand DNA break repair foci [49]. ATM encodes a serine/threonine kinase member of the phosphatidylinositol 3-kinase family which plays a central role in the repair of double-strand DNA breaks and cell cycle checkpoint control [50].
Hereditary pancreatitis (HP). Pancreatitis is an inflammatory condition of the pancreas resulting from premature activation, or lack of inhibition, of digestive enzymes in the pancreas that can lead to exocrine damage and pancreatic insufficiency [51]. Inherited forms of pancreatitis (hereditary pancreatitis, HP) are characterized by recurrent episodes of acute pancreatitis that often start in childhood or early adolescence and eventually develop into chronic pancreatitis in adulthood [19, 21]. The most common causes of HP are germline gain of function mutations in the serine-1 protease gene, PRSS1 (chr7q34). This gene encodes trypsin-1 (cationic trypsinogen), a digestive enzyme normally synthesized in large amounts by the acinar cells of the exocrine pancreas as an inactive pro-enzyme that is cleaved and activated in the gastrointestinal tract. In HP patients, the gain of function mutations lead to enhanced trypsinogen auto-activation and, in some cases, reduce its inactivation [52]. Other genes have been reported to influence susceptibility to pancreatitis, including SPINK1 (encoding pancreatic secretory trypsin inhibitor), CFTR (cystic fibrosis transmembrane conductance regulator), CTRC (chymotrypsin-C) and CASR (extracellular calcium-sensing receptor) [18, 53]. A recent meta-analysis estimated that chronic pancreatitis and hereditary pancreatitis were associated with 13.3 and 69.0 fold increased risk of pancreatic cancer, respectively [54].
II. Common low risk pancreatic cancer susceptibility loci
While linkage analysis and sequencing approaches (candidate gene, exome or whole genome) have been successful in identifying germline mutations in high risk cancer susceptibility genes that influence the risk of pancreatic cancer, these methods are less suitable for the identification of common germline variation that infers low risk to carriers. The approach used successfully in the last decade to uncover this class of susceptibility alleles for many diseases and traits has been to scan hundreds of thousands - to millions - common germline variants (mostly single nucleotide polymorphisms, SNPs) across the genome using microarrays. These scans, termed Genome Wide Association Studies (GWAS), are agnostic gene mapping approaches conducted in large numbers of case and control subjects, and, due to the multiple testing aspect, require stringent thresholds of significance (generally P<1x10-8 or 5x10-8) and validation in independent sample sets [55, 56]. Notably, due to the design of these studies, the GWAS approach identifies germline variation that, in most cases, is non-coding and therefore does not alter the amino acid sequences of proteins but rather, may influence expression of genes located close by or even at a considerable distance.
The first pancreatic cancer GWAS were performed by the Pancreatic Cancer Cohort Consortium (PanScan) and the Pancreatic Cancer Case Control Consortium (PanC4) with the aim of identifying common susceptibility markers for this deadly disease. Four GWAS phases have now been reported: PanScan I in 2009, PanScan II in 2010, PanScan III in 2014 and PanC4 in 2015 [57-60]. The first two phases of PanScan consisted of twelve case-control studies nested within prospective cohort studies and eight case-control studies, including 3,851 patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) and 3,934 control subjects of European ancestry [57, 58]. Four genome wide significant pancreatic cancer risk loci were identified: chromosome 9q34.2 (in the ABO blood group gene), 1q32.1 (in NR5A2), 5p15.33 (in the CLPTM1L-TERT gene region) and 13q22.1 (in a large non-genic region flanked by KLF5 and KLF12). The third phase of PanScan (PanScan III) added 1,582 newly genotyped pancreatic cancer cases and 5,203 control subjects with replication in samples from the European PANDoRA case-control consortium [61] resulting in a total of 7,683 cases and 14,397 control subjects, adding 5 GWAS significant (and one suggestive) risk loci on chromosomes 5p15.33 (second independent risk locus in the CLPTM1L-TERT gene region), 7q23.2 (LINC-PINT), 16q23.1 (BCAR1), 13q12.2 (PDX1), 22q12.1 (ZNRF3) and 8q24.1 (nongenic) [59]. The most recent GWAS, led by PanC4, added 4,164 newly scanned pancreatic cancer cases and 3,792 control subjects. With replication phases, it included a total of 9,925 cases and 11,569 control subjects of European ancestry (including PanScan I and II). After replication of the top hits from stage I in PANDoRA, four new GWAS significant loci were uncovered on chromosomes 17q24.3 (LINC00673), 2p14 (ETAA1), 7p14.1 (SUGCT) and 3q28 (TP63) [60]. Thus, a total of 13 common germline risk loci that have reached a GWAS significance threshold (P<5x10-8) are currently known for pancreatic cancer risk (Table 1, Figure 1) in populations of European ancestry.
GWAS significant pancreatic cancer susceptibility loci discovered in PanScan I, II and III and PanC4
| Chr | Gene(s) | SNP | Location | Minor/major allele | OR (95% CI) | MAF | P |
|---|---|---|---|---|---|---|---|
| 9q34.1 | ABO | rs687289 | 136,137,106 | T/C | 1.27 (1.20-1.35) | 0.37 | 1.6 x 10-16 |
| 13q22.1 | KLF5, KLF12 | rs9543325 | 73,916,628 | C/T | 1.23 (1.18-1.30) | 0.39 | 4.3 x 10-14 |
| 17q24.3 | LINC00673 | rs11655237 | 70,400,166 | T/C | 1.26 (1.19 1.34) | 0.11 | 1.4 x 10-14 |
| 5p15.33 | TERT, CLPTM1L | rs2736098 | 1,294,086 | T/C | 0.80 (0.76-0.85) | 0.23 | 9.8 x 10-14 |
| 7q32.3 | LINC-PINT | rs6971499 | 130,680,521 | C/T | 0.79 (0.74-0.84) | 0.13 | 3.0 x 10-12 |
| 5p15.33 | CLPTM1L, TERT | rs31490# | 1,344,458 | A/G | 1.20 (1.14-1.27) | 0.44 | 2.0 x 10-11 |
| 1q32.1 | NR5A2 | rs10919791 | 199,965,168 | A/G | 0.79 (0.75-0.85) | 0.22 | 1.4 x 10-11 |
| 16q23.1 | BCAR1, CTRB1, CTRB2 | rs7190458 | 75,263,661 | A/G | 1.46 (1.30-1.65) | 0.05 | 1.1 x 10-10 |
| 13q12.2 | PDX1 | rs9581943 | 28,493,997 | A/G | 1.15 (1.10-1.20) | 0.43 | 2.4 x 10-9 |
| 2p14 | ETAA1 | rs1486134 | 67,639,769 | G/T | 1.14 (1.09-1.19) | 0.27 | 3.4 x 10-9 |
| 22q12.1 | ZNRF3 | rs16986825 | 29,300,306 | T/C | 1.18 (1.12-1.25) | 0.16 | 1.2 x 10-8 |
| 7p14.1 | SUGCT | rs17688601 | 40,866,663 | A/C | 0.88 (0.84-0.92) | 0.25 | 1.4 x 10-8 |
| 3q28 | TP63 | rs9854771 | 189,508,471 | A/G | 0.89 (0.85-0.93) | 0.39 | 2.4 x 10-8 |
| 8q24.21 | LINC00824, MIR1208, PVT1 | rs1561927* | 129,568,078 | C/T | 0.87 (0.83-0.92) | 0.25 | 1.3 x 10-7 |
| 12q24.31 | HNF1A | rs7310409* | 121,424,861 | A/G | 1.11 (1.06-1.15) | 0.42 | 6.3 x 10-7 |
Association results from GWAS studies conducted in pancreatic cancer cases and controls of European ancestry. PanScan I, II and III: 7,683 case and 14,397 control subjects. PanC4: 9,925 case and 11,569 control subjects. Note that PanScan I and II are common to both PanScan and PanC4 GWAS. Gene: closest RefSeq gene(s). SNP: rs number of SNP reported as most significantly associated with pancreatic cancer risk for each risk locus. Location: position of SNP in NCBI genome build 37 (Hg19). Minor and major alleles are listed. MAF: minor allele frequency in 1000G European (EUR) population as per 1000G Phase 3 version 1. # This locus was originally tagged by rs401681 and has now been fine-mapped to rs451360 and correlated variants. * Suggestive risk loci in both PanScan III and PanC4.
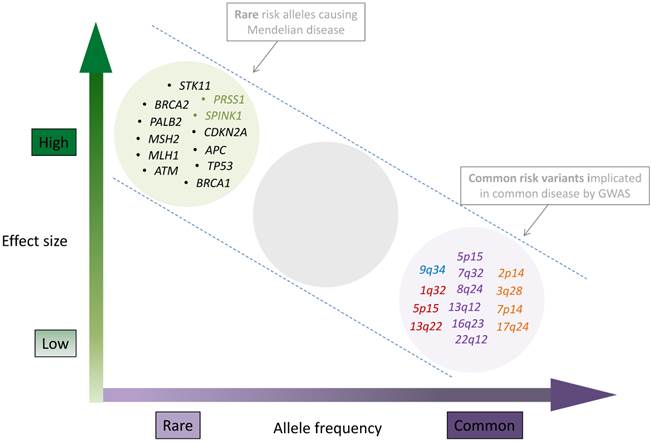
The current landscape of inherited pancreatic cancer risk variants. Allele frequencies ranging from rare to common are shown on x-axis, and effect sizes ranging from low to high on the y-axis. The clusters represent genes with germline mutations identified through linkage, candidate gene and sequencing approaches (top left) and chromosomal band locations for common susceptibility loci identified through GWAS (lower right). Genes that are mutated as part of multi cancer syndromes are listed in black, genes that influence hereditary pancreatitis are listed in green. Common, low risk pancreatic cancer risk loci are shown in the lower right hand part of the figure and colored according to the GWAS phase they were identified: PanScan I (blue), PanScan II (red), PanScan III (purple) and PanC4 (orange). GWAS loci from non-European populations were not included in the figure but are listed in tables 2 and 3. Figure layout was adapted from Manolio T. et.al, Nature 2009 [56] with permission.
Chromosome 1q32.1/NR5A2. The most significant GWAS SNP on chr1q32.1 maps to the first intron of the NR5A2 gene (rs3790844, OR=0.77, P=2.5x10-10) (Table 1) [58, 59]. This gene encodes nuclear receptor subfamily 5 group A member 2 (NR5A2), sometimes referred to as liver receptor homolog-1 (LRH-1). NR5A2 is a transcription factor that plays important roles in multiple aspects of pancreatic development and function, including cholesterol synthesis, bile acid homeostasis, steroidogenesis and in regulating stemness [62-64]. Likewise, NR5A2 is an important regulator of exocrine function in the adult pancreas where it regulates the expression of a number of acinar specific genes [65]. Nr5a2 is required for early embryonic development and mice that lack both copies of Nr5a2 die at embryonic day 7 [66]. However, heterozygous Nr5a2 mice are viable and exhibit increased rates of pancreatic acinar to ductal metaplasia (ADM) and impaired recovery after chemically induced acute pancreatitis [67, 68]. Furthermore, Nr5a2 haploinsufficiency cooperates with pancreatitis in a mouse model driven by oncogenic KRAS, increasing the number of preneoplastic PanIN (Pancreatic Intraepithelial Neoplasia) lesions and driving their progression toward PDAC [67, 68]. Thus, NR5A2 appears to be important for maintaining homeostasis in the exocrine pancreas and promote the regeneration of functional acinar cells from metaplastic duct-like cells after inflammation caused by pancreatitis, and protect the pancreas from KRAS driven pre-neoplastic changes. Although the mechanism by which variants on chr1q32.1 mediate risk of pancreatic cancer is not clear, the mouse studies described above indicate that the underlying mechanism may involve negative regulation of NR5A2 gene expression or function, perhaps in combination with inflammation in the pancreas.
Chromosome 2p14/ETAA1. A suggestive risk locus was first identified at 2p14 in a pancreatic cancer GWAS conducted in Chinese individuals (rs2035565, OR=1.33, P=5.5x10-7) [69]. A highly correlated SNP (r2=0.97 in 1000 Genomes (1000G) EUR and EAS populations [70]) was identified in the most recent GWAS conducted by the PanC4 consortium (rs1486134, OR=1.14, P=3.4x10-9) [71]. These two SNPs are located 20 kb apart, approximately 5 kb upstream and 2 kb downstream of the ETAA1 gene that encodes Ewing's tumor-associated antigen 1 (also named Ewing's tumor-associated antigen 16), a 926 amino acid cell surface antigen of relatively unknown function [72].
Chromosome 3q28/TP63. The most significant association at 3q28 was seen for rs9854771 (OR=0.89, P=2.4x10-8), in TP63 [71]. This gene encodes tumor protein p63, a member of the p53 family of transcription factors (that includes p53, p63 and p73). Although the three proteins are highly homologous and share multiple functions, they each have unique functions as well [73]. Similarly to p53, p63 (and p73) can induce cell cycle arrest and apoptosis. Alternative splicing of TP63 generates the so called ΔN p63 isoforms, that lack the acidic transactivation domain (encoded by exons 1-3) and act in a dominant-negative fashion upon p63, p53 and p73 [74]. The TP63 gene is amplified in many tumors including squamous cell lung cancers, neck and esophageal cancers that results in overexpression of the dominant negative form of the protein [73, 75]. The most significant variant at 3q28 (rs9854771) lies in the fourth intron of the full length TP63 isoforms, but in the first intron of the ΔN TP63 isoforms.
Chromosome 5p15.33/CLPTM1L/TERT. Three independent pancreatic cancer risk loci have now been identified in the multi-cancer TERT-CLPTM1L risk region on chr5p15.33. In addition to pancreatic cancer, this locus has been associated with risk of basal cell skin, bladder, breast, chronic lymphocytic leukemia, endometrial, glioma, lung, melanoma, ovarian, prostate and testicular cancer, intriguingly, with the same alleles showing increased risk for some cancers but decreased risk of other cancer types [76-89]. The TERT gene encodes the protein coding and catalytic subunit of telomerase, known for its essential role in maintaining telomere ends and the increased telomerase activity often seen in human cancers [90-92]. Telomerase also exhibits telomere independent functions, including regulation of gene expression, cell survival, epithelial to mesenchymal transition (EMT) and mitochondrial function [93]. The cleft lip and palate associated transmembrane 1 like (CLPTM1L) protein is a survival factor in lung cancer where it protects cells from apoptosis after treatment with DNA damaging agents [94, 95]. It is overexpressed in a subset of pancreatic tumors and has been shown to enhance growth of pancreatic cell lines in vitro and in vivo, as well as increase their rates of aneuploidy [96]. The first pancreatic cancer risk locus on 5p15.33 (identified in PanScan II) was marked by an intronic SNP (rs401681, OR=1.19, P=3.7x10-7) in CLPTM1L [58]. A second independent signal was identified in PanScan III, tagged by a synonymous SNP (rs2736098, OR=0.80, P=9.8x10-14) in the second exon of TERT [59]. Both SNPs were also identified in a subset based meta-analysis of multiple cancer types that identified six independent risk loci in the TERT-CLPTM1L region and fine-mapped the original signal for pancreatic cancer (marked by rs401681) to rs451360 (OR=1.29, P=2.0x10-10) and a set of highly correlated SNPs [97]. Pancreatic cancer contributed to risk in all six TERT-CLPTM1L risk loci, indicating that similar approaches across cancer types in other multi-cancer risk loci may prove useful to identify additional pancreatic cancer susceptibility variants that may not reach genome wide significance in single cancer GWAS due to lack of statistical power. The synonymous SNP in TERT, and several correlated variants in this genomic region, have been associated with telomere length in white blood cells and with TERT promoter activity in vitro [82, 98, 99]. Recently, a third independent risk locus was identified in this genomic region through a candidate gene analysis of the TERT and TERC genes in 5,550 pancreatic cancer cases and 7,585 control subjects from PANDoRA and PanScan. The most significant SNP (rs2853677, OR=0.85, P=8.3x10-8) was also described in the multi cancer fine-mapping study described above (as Region 3) [97, 100].
Chromosome 7p14.1/SUGCT. This risk locus is tagged by rs17688601 (OR=0.88, P=1.4x10-8), a SNP in the succinyl-CoA:glutarate-CoA transferase (SUGCT) gene, previously named C7orf10 (chromosome 7 open reading frame 10) [71]. This gene encodes succinate-hydroxymethylglutarate CoA-transferase which catalyzes the succinyl-CoA-dependent conversion of glutarate to glutaryl-CoA [101].
Chromosome 7q32.3/LINC-PINT. The signal on 7q32.3 is marked by an intronic SNP (rs6971499, OR=0.79, P=3.0x10-12) in LINC-PINT, a long intergenic p53-induced non-protein coding gene involved in epigenetic silencing [59, 102]. Two additional candidate genes are located nearby: MKLN1 and KLF14. The former encodes Muskelin 1, an intracellular mediator of cellular spreading, and the latter, KLF14, encodes a member of the Kruppel-like family of transcription factors, which may suppress KRAS mediated cell growth [103-105], and acts as a master regulator of gene expression in adipose tissue [106-108].
Chromosome 8q24.21/LINC00824. A promising, near-GWAS significant risk locus was identified in PanScan III in the well-known multi-cancer risk locus on 8q24.21 (rs1561927, OR=0.87, P=1.3x10-7) [59]. The signal lies within a non-protein coding transcript, LINC00824 of unknown function. Additional candidate genes are located 400-800 kb centromeric of this SNP: MIR1205-MIR1208, TMEM75, PVT1, and MYC. Chromosome 8q24.21 is known to contain multiple cancer susceptibility loci that span over 2Mb [109, 110]. As most are located far away from this SNP, with low levels of linkage disequilibrium (LD), they are not likely to represent the same signal. Moderate LD was noted with a SNP that is 24 kb upstream of rs1561927 and reported to be associated with ovarian cancer risk (rs10088218, r2=0.33 in 1000G EUR data) indicating that it may mark the same risk locus on 8q24.21 [111].
Chromosome 9q34.2/ABO. The ABO locus was the first common pancreatic cancer risk locus identified by GWAS in 2009 [57]. The protective allele of the most significant SNP on 9q34.2 in the ABO gene (rs505922, OR=1.20, P=5.4x10-8) is in complete linkage disequilibrium (LD) with the O allele of the ABO locus (r2=1 in 1000G EUR). The ABO gene encodes a glycosyltransferase (Histo-blood group ABO system transferase) that catalyzes the transfer of specific carbohydrates to the H antigen, thereby forming the antigenic structures of the ABO blood groups as initially described by Karl Landsteiner in 1900. The A and B alleles of the ABO gene encode proteins that differ only slightly in amino acid sequence but catalyze the transfer of different carbohydrates (N-acetylgalactosamine or galactose) onto the H antigen. In contrast, the O allele contains a single base deletion that shifts the reading frame of ABO and results in a nonfunctional protein and a lack of A or B antigens [112]. A set of small studies published in the 1950s and 1960s reported an association between ABO blood type and gastrointestinal cancers [113, 114] and the discovery of the 9q32.2/ABO risk locus rekindled interest in the connection between blood groups and pancreatic cancer. Using PanScan GWAS data, individual ABO alleles were inferred and their association with pancreatic cancer risk determined showing that individuals with inferred A (OR=1.38), AB (OR=1.47) and B (OR=1.53) blood groups had an increased risk of pancreatic cancer as compared to the O group [115]. The fact that the A1 alleles conferred an increased risk of pancreatic cancer (OR=1.38), whereas A2 alleles did not, furthermore suggested that increased glycosyltransferase activity of the former, either towards the H antigen or other proteins, might explain the underlying biology of this risk locus [116, 117]. Since the PanScan GWAS report linking ABO blood groups and risk of pancreatic cancer was published in 2009, numerous studies have investigated the association of blood groups with cancer risk; the most consistently significant findings have been noted for gastric and pancreatic cancer [118-121].
Chromosome 12q24.31/HNF1A. A suggestive risk locus near the HNF1A gene was noted in PanScan III at a level approaching GWAS significance (rs1182933, OR=1.12, P=1.1x10-6) [59]. A correlated variant was observed at a similar significance in the PanC4 GWAS (rs7310409, OR=1.11, P=6.3x10-7) [71]. These two SNPs are correlated (r2=0.43 in the 1000G EUR populations) and may represent the same signal. HNF1A encodes hepatocyte nuclear factor 1α, a transcription factor known to play important developmental and homeostatic roles in the pancreas and other gastrointestinal organs [122-125]. Germ-line mutations in HNF1A cause maturity onset diabetes of the young (MODY) type 3, and account for a sizeable fraction of all MODY cases [126, 127]. Genome-wide association studies have identified common susceptibility variants in the vicinity of the HNF1A gene for T2D, cholesterol and blood levels of C-reactive protein [128-130]. Interestingly, pathway-based analyses of pancreatic cancer GWAS data have also identified potential pancreatic cancer risk SNPs in the HNF1A gene [131, 132]. A transcriptome and sub-network analysis identified HNF1A as the upstream regulator of a highly significantly deregulated expression sub-network in pancreatic tumors [133]. Overexpression of HNF1A in pancreatic cell lines resulted in cell cycle arrest, apoptosis and growth inhibition concomitant with deregulation of cell cycle and apoptosis genes indicating that HNF1A may function as a tumor suppressor gene in the pancreas [133].
Chromosome 13q22.1. The top ranked GWAS SNP (rs9543325, OR=1.26, P=3.27x10-11), on chr13q22.1 lies in a large (600 kb) non-genic region [58] suggesting that the functional variant could impact gene expression via long range intra- or inter-chromosomal interactions. This non-genic region on chr13q22.1 is flanked by two genes encoding transcription factors of the Kruppel-like family: KLF5 and KLF12. Both genes encode proteins that regulate cell growth and transformation [134-136] and KLF5 has been reported to be upregulated in pancreatic cancer [137]. Additional genes, either nearby or at a greater distance could be functionally relevant to the mechanism by which the chr13q22.1 GWAS locus confers risk of PDAC.
Chromosome 13q12.2/PDX1. A second independent signal was identified on chromosome 13 in PanScan III, on 13q12.2 (rs9581943, OR=1.15 P=2.4x10-9) [59]. The top ranked SNP is located in the promoter region of the PDX1 (pancreatic and duodenal homeobox1 protein 1) gene and is intronic to PDX1-AS1 (PDX1 antisense RNA 1), a noncoding RNA of unknown function. PDX1 plays a critical role in early pancreatic development, as well as in the differentiation of exocrine pancreas, and regulates beta-cell function in the mature pancreas [138, 139]. Mutations in this gene have been linked to agenesis of the pancreas [138] and maturity onset diabetes of the young (MODY type 4) [140]. Furthermore, PDX1 has been implicated in glucose-dependent regulation of insulin gene transcription [141], and seems a good candidate gene for this locus, although functional work is required to uncover the mechanistic basis of this association signal.
Chromosome 16q23.1/BCAR1. A synonymous SNP residing in the last exon of BCAR1 (also known as p130Cas) was noted on 16q23.1 (rs7190458, OR=1.46, P=1.1x10-10) [59]. BCAR1 functions as an adaptor protein that coordinates cell cycle control, cytoskeleton organization, and cell migration. Aberrant expression of this gene has been linked to transformation and progression of multiple cancer types [142, 143]. Two chymotrypsinogen genes, CTRB1 and CTRB2 are also located close to this SNP (5kb and 23kb, respectively) and as important members of a family of serine proteases secreted by the pancreas into the gastrointestinal tract, are plausible target genes for susceptibility variants at this locus [144]. Because of the increased risk of pancreatic cancer associated with pancreatitis and digestive enzyme dysregulation, it is tempting to speculate that the underlying molecular mechanism of this risk locus may involve the chymotrypsinogen genes. However, as in all risk loci, fine-mapping and wet-lab work is required to understand the functional consequences of carrying risk variants at 16q23.1.
Chromosome 17q24.3/LINC00673. The signal at 17q24.3 maps to LINC00673 (long inter-genic non-protein coding RNA 673) (rs11655237, OR=1.26, P=1.42x10-14). LINC00675 is also termed HI-LNC75, and is a long noncoding RNA of unknown function that is expressed in pancreatic beta-cells [145]. An interesting candidate gene located ~ 280 kb upstream, SOX9, encodes a transcription factor that marks multipotent pancreatic progenitor cells and is important for the development of both the exocrine and endocrine pancreas. In the adult pancreas, it is expressed in ductal and centroacinar cells where it maintains homeostasis and is important for regeneration after chemically induced pancreatitis [146, 147]. Sox9 expression in pancreatic acinar cells of mouse models induces acinar to ductal metaplasia (ADM) and is required for KRAS mediated formation of preneoplastic (PanIN) lesions [148].
Chromosome 22q12.1/ZNRF3. The most significant SNP at 22q12.1, (rs16986825, OR=1.18 P=1.2x10-8) [59], is located in the first intron of the zinc and ring finger 3 (ZNRF3) gene, which encodes a transmembrane E3 ubiquitin protein ligase that negatively regulates the Wnt signaling pathway [149]. At some distance from the most notable SNP (162 kb centromeric) in this locus is CHEK2, a gene that encodes a well-known cell-cycle checkpoint kinase that cooperates with p53, BRCA1 and ATM in response to DNA damage and may therefore represent a candidate gene worthy of further follow-up [150, 151].
Common pancreatic cancer risk loci in non-European populations
In addition to GWAS performed by the Pancreatic Cohort Consortium and PanC4 in which the majority of subjects were of European ancestry, such scans have also been performed in case-control studies from China and Japan. A two phased GWAS with a combined set of 3,584 pancreatic cancer cases and 4,868 control subjects from China (ChinaPC) identified five GWAS significant risk loci on chromosomes 5p13.1/DAB2, 10q26.11/PRLHR, 21q21.3/BACH1, 21q22.3/TFF1, and 22q13.32/FAM19A5 (Table 2) [69]. A Japanese GWAS of 991 advanced pancreatic ductal adenocarcinoma cases and 5,209 control subjects reported three loci approaching GWAS significance on chromosomes 6p25.3/FOXQ1, 7q36.2/DPP6, and 12p11.21/BICD1 (Table 3) [152]. Both the Chinese and Japanese GWAS scans replicated the chr13q22.1 locus initially discovered in PanScan, and noted the risk locus on chr1q32.1/NR5A2, albeit with less significance. The risk locus on chromosome chr5p15.33/TERT/CLPTM1L (initially tagged by rs401681 and rs31490) was only noted in the Chinese GWAS scan, and 9q34.2/ABO only in the Japanese scan.
GWAS significant pancreatic cancer susceptibility loci discovered in ChinaPC GWAS
| Chr | Gene(s) | SNP | Location | Minor/major allele | OR (95% CI) | MAF | P |
|---|---|---|---|---|---|---|---|
| 21q21.3 | BACH1 | rs372883 | 30,717,737 | G/A | 0.79 (0.75-0.84) | 0.46 | 2.2 x 10-13 |
| 21q22.3 | TFF1 | rs1547374 | 43,778,895 | G/A | 0.79 (0.74-0.84) | 0.46 | 3.7 x 10-13 |
| 10q26.11 | PRLHR | rs12413624 | 120,278,944 | A/T | 1.23 (1.16-1.31) | 0.37 | 5.1 x 10-11 |
| 22q13.32 | FAM19A5 | rs5768709 | 48,929,569 | G/A | 1.25 (1.17-1.34) | 0.23 | 1.4 x 10-10 |
| 5p13.1 | DAB2 | rs2255280 | 39,394,989 | C/A | 0.81 (0.76-0.87) | 0.40 | 4.2 x 10-10 |
Association results from GWAS studies conducted in pancreatic cancer cases and controls of Chinese ancestry. ChinaPC GWAS: 3,584 case and 4,686 control subjects were included. Gene: closest RefSeq gene(s). SNP: rs number of SNP reported as most significantly associated with pancreatic cancer risk for each risk locus. Location: position of SNP in NCBI genome build 37 (Hg19). MAF: minor and major alleles are listed. Minor allele frequency (MAF) in 1000G East Asian (EAS) population as per 1000G Phase 3 version 1.
Suggestive pancreatic cancer susceptibility loci reported in a Japanese pancreatic cancer GWAS
| Chr | Gene(s) | SNP | Location | Minor/major allele | OR (95% CI) | MAF | P |
|---|---|---|---|---|---|---|---|
| 6p25.3 | FOXQ1 | rs9502893 | 1,340,189 | C/T | 1.29 (1.17-1.43) | 0.37 | 3.3 x 10-7 |
| 12p11.21 | BICD1 | rs708224 | 32,436,409 | G/A | 1.32 (1.19-1.47) | 0.35 | 3.3 x 10-7 |
| 7q36.2 | DPP6 | rs6464375* | 153,625,843 | T/C | 3.73 (2.24-6.21) | 0.11 | 4.4 x 10-7 |
Association results from a GWAS study conducted in pancreatic cancer cases and controls of Japanese ancestry. A total of 991 case and 5,209 control subjects were inlcuded. Gene: closest RefSeq gene(s). SNP: rs number of SNP reported as most significantly associated with pancreatic cancer risk for each risk locus. Location: position of SNP in NCBI genome build 37 (Hg19). Minor and major alleles are listed. Minor allele frequency (MAF) in 1000G Japanese (JPT) population as per 1000G Phase 3 version 1. *Results for recessive model is shown for this SNP.
Assessment of the Chinese and Japanese pancreatic cancer risk loci in the European PANcreatic Disease ReseArch (PANDoRA) case-control consortium and in PanScan III did not replicate any of the GWAS significant pancreatic cancer susceptibility loci identified in Asians [59, 153]. However, 2p14/ETAA1 (suggestive in ChinaPC) replicated and became GWAS significant in the PanC4 GWAS [71] indicating that added case-control samples sets may reveal more overlap between GWAS findings for pancreatic cancer across populations [154].
Pathway analyses of pancreatic cancer GWAS datasets
Pathway based analyses have been used to mine PanScan I and II GWAS data to identify genes, or groups of genes, enriched with variants whose individual effects may be too small to be detected in the GWAS [57, 58]. These approaches have highlighted pancreatic development genes including HNF1A, HNF4G, HNF1B and PDX1 [131, 132] that encode important components of the transcriptional networks that govern embryonic development of the pancreas, and maintain homeostasis in the adult gland [155, 156]. In addition, mutations in HNF1A, PDX1, and HNF1B are responsible for maturity onset diabetes of the young (MODY types 3, 4 and 5) [157-159]. Likewise, common variants in HNF1A and HNF1B have been associated with risk of type II diabetes [106, 128, 160]. With the added numbers of cases and controls in the third phase of PanScan as compared to the first two phases, the significance of two of these genes/loci (PDX1 and HNF1A) improved markedly [59], indicating that pathway analysis of GWAS datasets can tease out additional genes that influence risk of pancreatic cancer. These results furthermore suggest possible functional inter-relationships between inherited variation in genes important for pancreatic development, diabetes and pancreatic cancer risk.
Functional understanding of common pancreatic cancer risk loci
As for most other risk variants discovered through GWAS, common pancreatic cancer risk variants discovered to date are not protein coding, but rather reside in introns or intergenic regions of the genome. Much work is required after GWAS to uncover the biological mechanism(s) that explain risk at each locus. The first steps along the way are fine-mapping efforts and genomic characterization of each locus to identify the most significant tag SNPs and catalog transcribed sequences and potential functional gene regulatory regions. This is often done in conjunction with functional analysis of multiple highly correlated tag SNPs. Functional efforts involve genomic and wet lab based approaches, such as investigating gene expression levels, splicing, promoter or enhancer strength, DNA methylation, protein to DNA binding, and chromosome conformation analysis. The aim of these studies is to correlate risk genotypes to differences in specific molecular phenotypes to establish the underlying molecular mechanism at each locus.
Summary and future perspectives
It is clear that pancreatic cancer is a polygenic disease with multiple high and low risk germline susceptibility alleles. The continued search for common and rare germline variants that influence risk of pancreatic cancer holds the promise of increasing our understanding of the genetic susceptibility of this devastating disease. Initially, gene mapping in families with a high incidence of pancreatic cancer was undertaken via linkage analyses and candidate gene approaches, and more recently by whole-exome and whole-genome sequencing studies. Genes identified through this work tend to primarily influence other cancer types (including breast, ovarian, colorectal cancer and melanoma) with either moderately increased or highly increased risk of pancreatic cancer in carriers.
The search for common germline pancreatic cancer risk variants, launched by large cohort and case-control consortia in 2009, has yielded valuable insights into the etiology of pancreatic cancer. Six published pancreatic cancer GWAS studies have yielded 18 GWAS significant (and multiple suggestive) risk loci to date in European and Asian populations. As these loci only account for ~15-20% of the total heritability tagged by common SNPs, it is likely that additional pancreatic risk loci will be uncovered in future studies with larger sample sets, and in different populations around the world. Imputation and meta-analyses of existing and new GWAS datasets are expected to add to this list. Likewise, efforts in pancreatic cancer GWAS studies outside of main effects are likely to expand by investigations of susceptibility variants for survival, pharmacologic responses as well as gene-gene and gene-environmental interactions and will hopefully uncover additional risk variants [161-163]. The complex genetic architecture of pancreatic cancer will require exome and whole genome sequencing efforts in familial and sporadic cases in order to reach into the space between rare high risk and common low risk variants (as seen by the middle shaded circle in Figure 1) and the data generated is likely to become useful for imputation efforts into existing GWAS datasets. As population based sequencing studies have already shown, the number of uncommon and rare polymorphic variants in the human genome is very high [164], and may explain a substantial portion of germline risk for disease. High throughput sequencing approaches will furthermore better enable the assessment of variants not captured on GWAS platforms, such as insertion/deletion (indel) polymorphisms and copy number variants.
Although advances have been made in our understanding of the role common genetic variation plays in pancreatic cancer etiology, a considerable amount of work lies ahead in uncovering a larger number of common, uncommon and rare pancreatic cancer risk susceptibility alleles. Likewise, the identification of causative variants at each locus and the elucidation of the functional mechanism by which they mediate risk will require a great effort. A large collection of tissue samples and a wide variety of methodologies are required, including statistical, genetic, genomic, molecular, cell biology and animal model technologies. The hope is that this work may eventually help enable the development of diagnostic, prognostic, preventive and therapeutic tools to combat this highly lethal and devastating disease.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
3. Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2014. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2014;25:1650-6
4. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
5. Anderson KE MT, Silverman D. Cancer of the pancreas. In: (ed.) Schottenfeld D FJ. Cancer Epidemiology and Prevention. New York: Oxford University Press. 2006
6. Jacobs EJ, Chanock SJ, Fuchs CS, Lacroix A, McWilliams RR, Steplowski E. et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer. 2010;127:1421-8
7. Hemminki K, Li X. Familial and second primary pancreatic cancers: a nationwide epidemiologic study from Sweden. Int J Cancer. 2003;103:525-30
8. Shirts BH, Burt RW, Mulvihill SJ, Cannon-Albright LA. A population-based description of familial clustering of pancreatic cancer. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2010;8:812-6
9. Amundadottir LT, Thorvaldsson S, Gudbjartsson DF, Sulem P, Kristjansson K, Arnason S. et al. Cancer as a complex phenotype: pattern of cancer distribution within and beyond the nuclear family. PLoS medicine. 2004;1:e65
10. Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF. et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66:9818-28
11. Lynch HT, Fusaro RM, Lynch JF, Brand R. Pancreatic cancer and the FAMMM syndrome. Familial cancer. 2008;7:103-12
12. van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:1258-64 author reply 65
13. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW. et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217
14. Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM. et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA: the journal of the American Medical Association. 2009;302:1790-5
15. Consortium. TBCL. Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst. 1999;91:1310-6
16. Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK Jr, Perrault J. et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442-6
17. Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P. et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2004;2:252-61
18. Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U. et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213-6
19. Landi S. Genetic predisposition and environmental risk factors to pancreatic cancer: A review of the literature. Mutat Res. 2009;681:299-307
20. Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer-current knowledge. Nature reviews Gastroenterology & hepatology. 2012;9:445-53
21. Klein AP. Genetic susceptibility to pancreatic cancer. Molecular carcinogenesis. 2012;51:14-24
22. Korsse SE, Peppelenbosch MP, van Veelen W. Targeting LKB1 signaling in cancer. Biochimica et biophysica acta. 2013;1835:194-210
23. Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJ, Krak N. et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. Journal of medical genetics. 2013;50:59-64
24. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV. et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447-53
25. Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (p16) gene and human cancer. Mol Med. 1997;3:5-20
26. Bergman W, Watson P, de Jong J, Lynch HT, Fusaro RM. Systemic cancer and the FAMMM syndrome. Br J Cancer. 1990;61:932-6
27. Rutter JL, Bromley CM, Goldstein AM, Elder DE, Holly EA, Guerry Dt. et al. Heterogeneity of risk for melanoma and pancreatic and digestive malignancies: a melanoma case-control study. Cancer. 2004;101:2809-16
28. McWilliams RR, Wieben ED, Rabe KG, Pedersen KS, Wu Y, Sicotte H. et al. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. European journal of human genetics: EJHG. 2011;19:472-8
29. Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG. et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genetics in medicine: official journal of the American College of Medical Genetics. 2014
30. Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87:809-11
31. de Snoo FA, Bishop DT, Bergman W, van Leeuwen I, van der Drift C, van Nieuwpoort FA. et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res. 2008;14:7151-7
32. Kastrinos F, Stoffel EM. History, genetics, and strategies for cancer prevention in Lynch syndrome. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2014;12:715-27 quiz e41-3
33. Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F. et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clinical genetics. 2009;75:141-9
34. Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne SJ. et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Familial cancer. 2008;7:163-72
35. Dalavi SB, Vedpalsingh TH, Bankar SS, Ahmed MH, Bhosale DN. Familial Adenomatous Polyposis (FAP)-A Case Study and Review of Literature. Journal of clinical and diagnostic research: JCDR. 2015;9:PD05-6
36. Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, Kleibeuker JH. et al. Extra-intestinal manifestations of familial adenomatous polyposis. Annals of surgical oncology. 2008;15:2439-50
37. Giardiello FM, Offerhaus GJ, Lee DH, Krush AJ, Tersmette AC, Booker SV. et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993;34:1394-6
38. Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nature reviews Cancer. 2004;4:665-76
39. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117-30
40. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature reviews Cancer. 2012;12:68-78
41. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C. et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269-75
42. Mocci E, Milne RL, Mendez-Villamil EY, Hopper JL, John EM, Andrulis IL. et al. Risk of pancreatic cancer in breast cancer families from the breast cancer family registry. Cancer Epidemiol Biomarkers Prev. 2013;22:803-11
43. Consortium TBCL. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310-6
44. Thompson D, Easton DF. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358-65
45. Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML. et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer discovery. 2012;2:41-6
46. Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L. et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;148:556-64
47. Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N. et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137:1183-6
48. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N. et al. PALB2 mutations in European familial pancreatic cancer families. Clinical genetics. 2010;78:490-4
49. Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO reports. 2009;10:990-6
50. Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harbor symposia on quantitative biology. 2005;70:99-109
51. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252-61
52. Sahin-Toth M, Toth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun. 2000;278:286-9
53. Whitcomb DC. Genetic risk factors for pancreatic disorders. Gastroenterology. 2013;144:1292-302
54. Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349-58
55. (WTCCC) TWTC-CC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661-78
56. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ. et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747-53
57. Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA. et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986-90
58. Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB. et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224-8
59. Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z. et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46:994-1000
60. Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M. et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911-6
61. Campa D, Rizzato C, Capurso G, Giese N, Funel N, Greenhalf W. et al. Genetic susceptibility to pancreatic cancer and its functional characterisation: the PANcreatic Disease ReseArch (PANDoRA) consortium. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2013;45:95-9
62. Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends in cell biology. 2004;14:250-60
63. Kelly VR, Xu B, Kuick R, Koenig RJ, Hammer GD. Dax1 up-regulates Oct4 expression in mouse embryonic stem cells via LRH-1 and SRA. Mol Endocrinol. 2010;24:2281-91
64. Heng JC, Feng B, Han J, Jiang J, Kraus P, Ng JH. et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell stem cell. 2010;6:167-74
65. Holmstrom SR, Deering T, Swift GH, Poelwijk FJ, Mangelsdorf DJ, Kliewer SA. et al. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes & development. 2011;25:1674-9
66. Pare JF, Malenfant D, Courtemanche C, Jacob-Wagner M, Roy S, Allard D. et al. The fetoprotein transcription factor (FTF) gene is essential to embryogenesis and cholesterol homeostasis and is regulated by a DR4 element. J Biol Chem. 2004;279:21206-16
67. Flandez M, Cendrowski J, Canamero M, Salas A, Del Pozo N, Schoonjans K. et al. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRasG12V-driven pancreatic tumourigenesis. Gut. 2013
68. von Figura G, Morris JPt, Wright CV, Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63:656-64
69. Wu C, Miao X, Huang L, Che X, Jiang G, Yu D. et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2012;44:62-6
70. Genomes. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061-73
71. Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M. et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015
72. Borowski A, Dirksen U, Lixin L, Shi RL, Gobel U, Schneider EM. Structure and function of ETAA16: a novel cell surface antigen in Ewing's tumours. Cancer immunology, immunotherapy: CII. 2006;55:363-74
73. Graziano V, De Laurenzi V. Role of p63 in cancer development. Biochimica et biophysica acta. 2011;1816:57-66
74. Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harbor perspectives in biology. 2010;2:a004887
75. Petitjean A, Hainaut P, Caron de Fromentel C. TP63 gene in stress response and carcinogenesis: a broader role than expected. Bulletin du cancer. 2006;93:E126-35
76. Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M. et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85:679-91
77. McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G. et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40:1404-6
78. Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616-22
79. Broderick P, Wang Y, Vijayakrishnan J, Matakidou A, Spitz MR, Eisen T. et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69:6633-41
80. Hsiung CA, Lan Q, Hong YC, Chen CJ, Hosgood HD, Chang IS. et al. The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet. 2010:6
81. Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M. et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. 2011;43:785-91
82. Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A. et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221-7
83. Chung CC, Ciampa J, Yeager M, Jacobs KB, Berndt SI, Hayes RB. et al. Fine mapping of a region of chromosome 11q13 reveals multiple independent loci associated with risk of prostate cancer. Hum Mol Genet. 2011;20:2869-78
84. Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B. et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899-904
85. Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D. et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet. 2010;42:604-7
86. Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G, Jakobsdottir M. et al. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet. 2009;41:909-14
87. Barrett JH, Iles MM, Harland M, Taylor JC, Aitken JF, Andresen PA. et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43:1108-13
88. Berndt SI, Skibola CF, Joseph V, Camp NJ, Nieters A, Wang Z. et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat Genet. 2013;45:868-76
89. Carvajal-Carmona LG, O'Mara TA, Painter JN, Lose FA, Dennis J, Michailidou K. et al. Candidate locus analysis of the TERT-CLPTM1L cancer risk region on chromosome 5p15 identifies multiple independent variants associated with endometrial cancer risk. Human genetics. 2015;134:231-45
90. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB. et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349-52
91. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464-8
92. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL. et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011-5
93. Ding D, Zhou J, Wang M, Cong YS. Implications of telomere-independent activities of telomerase reverse transcriptase in human cancer. The FEBS journal. 2013;280:3205-11
94. Yamamoto K, Okamoto A, Isonishi S, Ochiai K, Ohtake Y. A novel gene, CRR9, which was up-regulated in CDDP-resistant ovarian tumor cell line, was associated with apoptosis. Biochem Biophys Res Commun. 2001;280:1148-54
95. James MA, Wen W, Wang Y, Byers LA, Heymach JV, Coombes KR. et al. Functional characterization of CLPTM1L as a lung cancer risk candidate gene in the 5p15.33 locus. PloS one. 2012;7:e36116
96. Jia J, Bosley AD, Thompson A, Hoskins JW, Cheuk A, Collins I. et al. CLPTM1L promotes growth and enhances aneuploidy in pancreatic cancer cells. Cancer Res. 2014;74:2785-95
97. Wang Z, Zhu B, Zhang M, Parikh H, Jia J, Chung CC. et al. Imputation and subset-based association analysis across different cancer types identifies multiple independent risk loci in the TERT-CLPTM1L region on chromosome 5p15.33. Hum Mol Genet. 2014;23:6616-33
98. Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP. et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371-84
99. Kote-Jarai Z, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Dadaev T, Jugurnauth-Little S. et al. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum Mol Genet. 2013;22:2520-8
100. Campa D, Rizzato C, Stolzenberg-Solomon R, Pacetti P, Vodicka P, Cleary SP. et al. TERT gene harbors multiple variants associated with pancreatic cancer susceptibility. Int J Cancer. 2015
101. Marlaire S, Van Schaftingen E, Veiga-da-Cunha M. C7orf10 encodes succinate-hydroxymethylglutarate CoA-transferase, the enzyme that converts glutarate to glutaryl-CoA. Journal of inherited metabolic disease. 2014;37:13-9
102. Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V. et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome biology. 2013;14:R104
103. Adams JC, Seed B, Lawler J. Muskelin, a novel intracellular mediator of cell adhesive and cytoskeletal responses to thrombospondin-1. The EMBO journal. 1998;17:4964-74
104. Fernandez-Zapico ME, Lomberk GA, Tsuji S, DeMars CJ, Bardsley MR, Lin YH. et al. A functional family-wide screening of SP/KLF proteins identifies a subset of suppressors of KRAS-mediated cell growth. The Biochemical journal. 2011;435:529-37
105. Valiyaveettil M, Bentley AA, Gursahaney P, Hussien R, Chakravarti R, Kureishy N. et al. Novel role of the muskelin-RanBP9 complex as a nucleocytoplasmic mediator of cell morphology regulation. The Journal of cell biology. 2008;182:727-39
106. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP. et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579-89
107. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707-13
108. Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A. et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561-4
109. Grisanzio C, Freedman ML. Chromosome 8q24-Associated Cancers and MYC. Genes & cancer. 2010;1:555-9
110. Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: an oasis of non-coding transcriptional activity. Frontiers in genetics. 2012;3:69
111. Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K. et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362-70 70e1-2
112. Hakomori S. Antigen structure and genetic basis of histo-blood groups A, B and O: their changes associated with human cancer. Biochimica et biophysica acta. 1999;1473:247-66
113. Marcus DM. The ABO and Lewis blood-group system. Immunochemistry, genetics and relation to human disease. N Engl J Med. 1969;280:994-1006
114. Aird I, Bentall HH, Roberts JA. A relationship between cancer of stomach and the ABO blood groups. Br Med J. 1953;1:799-801
115. Wolpin BM, Kraft P, Gross M, Helzlsouer K, Bueno-de-Mesquita HB, Steplowski E. et al. Pancreatic cancer risk and ABO blood group alleles: results from the pancreatic cancer cohort consortium. Cancer Res. 2010;70:1015-23
116. Wolpin BM, Kraft P, Xu M, Steplowski E, Olsson ML, Arslan AA. et al. Variant ABO blood group alleles, secretor status, and risk of pancreatic cancer: results from the pancreatic cancer cohort consortium. Cancer Epidemiol Biomarkers Prev. 2010;19:3140-9
117. Yamamoto F, McNeill PD, Hakomori S. Human histo-blood group A2 transferase coded by A2 allele, one of the A subtypes, is characterized by a single base deletion in the coding sequence, which results in an additional domain at the carboxyl terminal. Biochem Biophys Res Commun. 1992;187:366-74
118. Zhang BL, He N, Huang YB, Song FJ, Chen KX. ABO blood groups and risk of cancer: a systematic review and meta-analysis. Asian Pacific journal of cancer prevention: APJCP. 2014;15:4643-50
119. Franchini M, Liumbruno GM. ABO blood group: old dogma, new perspectives. Clinical chemistry and laboratory medicine: CCLM / FESCC. 2013;51:1545-53
120. Khalili H, Wolpin BM, Huang ES, Giovannucci EL, Kraft P, Fuchs CS. et al. ABO blood group and risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:1017-20
121. Gates MA, Wolpin BM, Cramer DW, Hankinson SE, Tworoger SS. ABO blood group and incidence of epithelial ovarian cancer. Int J Cancer. 2011;128:482-6
122. Courtois G, Morgan JG, Campbell LA, Fourel G, Crabtree GR. Interaction of a liver-specific nuclear factor with the fibrinogen and alpha 1-antitrypsin promoters. Science. 1987;238:688-92
123. Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL. et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378-81
124. D'Angelo A, Bluteau O, Garcia-Gonzalez MA, Gresh L, Doyen A, Garbay S. et al. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development. 2010;137:1573-82
125. Friedlander G, Runembert I, Vrtovsnik F, Terzi F. Renal tubular cells cultured from genetically modified animals. Experimental nephrology. 1999;7:407-12
126. Ellard S, Colclough K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Human mutation. 2006;27:854-69
127. Giuffrida FM, Reis AF. Genetic and clinical characteristics of maturity-onset diabetes of the young. Diabetes, obesity & metabolism. 2005;7:318-26
128. Holmkvist J, Cervin C, Lyssenko V, Winckler W, Anevski D, Cilio C. et al. Common variants in HNF-1 alpha and risk of type 2 diabetes. Diabetologia. 2006;49:2882-91
129. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T. et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234-44
130. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S. et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274-83
131. Pierce BL, Ahsan H. Genome-wide "pleiotropy scan" identifies HNF1A region as a novel pancreatic cancer susceptibility locus. Cancer Res. 2011;71:4352-8
132. Li D, Duell EJ, Yu K, Risch HA, Olson SH, Kooperberg C. et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33:1384-90
133. Hoskins JW, Jia J, Flandez M, Parikh H, Xiao W, Collins I. et al. Transcriptome analysis of pancreatic cancer reveals a tumor suppressor function for HNF1A. Carcinogenesis. 2014;35:2670-8
134. McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiological reviews. 2010;90:1337-81
135. Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66:2691-706
136. Nakamura Y, Migita T, Hosoda F, Okada N, Gotoh M, Arai Y. et al. Kruppel-like factor 12 plays a significant role in poorly differentiated gastric cancer progression. Int J Cancer. 2009;125:1859-67
137. Mori A, Moser C, Lang SA, Hackl C, Gottfried E, Kreutz M. et al. Up-regulation of Kruppel-like factor 5 in pancreatic cancer is promoted by interleukin-1beta signaling and hypoxia-inducible factor-1alpha. Mol Cancer Res. 2009;7:1390-8
138. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106-10
139. MacDonald RJ, Swift GH, Real FX. Transcriptional control of acinar development and homeostasis. Progress in molecular biology and translational science. 2010;97:1-40
140. Vaxillaire M, Bonnefond A, Froguel P. The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best practice & research Clinical endocrinology & metabolism. 2012;26:171-87
141. Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. The EMBO journal. 1993;12:4251-9
142. Barrett A, Pellet-Many C, Zachary IC, Evans IM, Frankel P. p130Cas: a key signalling node in health and disease. Cellular signalling. 2013;25:766-77
143. Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer. 2010;10:858-70
144. Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Digestive diseases and sciences. 2007;52:1-17
145. Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T. et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell metabolism. 2012;16:435-48
146. Grimont A, Pinho AV, Cowley MJ, Augereau C, Mawson A, Giry-Laterriere M. et al. SOX9 regulates ERBB signalling in pancreatic cancer development. Gut. 2014
147. Seymour PA. Sox9: a master regulator of the pancreatic program. The review of diabetic studies: RDS. 2014;11:51-83
148. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt. et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer cell. 2012;22:737-50
149. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M. et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012;485:195-200
150. Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer. 2007;7:925-36
151. Gronwald J, Cybulski C, Piesiak W, Suchy J, Huzarski T, Byrski T. et al. Cancer risks in first-degree relatives of CHEK2 mutation carriers: effects of mutation type and cancer site in proband. Br J Cancer. 2009;100:1508-12
152. Low SK, Kuchiba A, Zembutsu H, Saito A, Takahashi A, Kubo M. et al. Genome-wide association study of pancreatic cancer in Japanese population. PloS one. 2010;5:e11824
153. Campa D, Rizzato C, Bauer AS, Werner J, Capurso G, Costello E. et al. Lack of replication of seven pancreatic cancer susceptibility loci identified in two Asian populations. Cancer Epidemiol Biomarkers Prev. 2013;22:320-3
154. Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566
155. Maestro MA, Cardalda C, Boj SF, Luco RF, Servitja JM, Ferrer J. Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocrine development. 2007;12:33-45
156. Martin M, Hauer V, Messmer M, Orvain C, Gradwohl G. Transcription factors in pancreatic development. Animal models. Endocrine development. 2007;12:24-32
157. Glucksmann MA, Lehto M, Tayber O, Scotti S, Berkemeier L, Pulido JC. et al. Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes. 1997;46:1081-6
158. Carette C, Vaury C, Barthelemy A, Clauin S, Grunfeld JP, Timsit J. et al. Exonic duplication of the hepatocyte nuclear factor-1beta gene (transcription factor 2, hepatic) as a cause of maturity onset diabetes of the young type 5. The Journal of clinical endocrinology and metabolism. 2007;92:2844-7
159. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M. et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384:455-8
160. Furuta H, Furuta M, Sanke T, Ekawa K, Hanabusa T, Nishi M. et al. Nonsense and missense mutations in the human hepatocyte nuclear factor-1 beta gene (TCF2) and their relation to type 2 diabetes in Japanese. The Journal of clinical endocrinology and metabolism. 2002;87:3859-63
161. Wu C, Kraft P, Stolzenberg-Solomon R, Steplowski E, Brotzman M, Xu M. et al. Genome-wide association study of survival in patients with pancreatic adenocarcinoma. Gut. 2012
162. Willis JA, Olson SH, Orlow I, Mukherjee S, McWilliams RR, Kurtz RC. et al. A replication study and genome-wide scan of single-nucleotide polymorphisms associated with pancreatic cancer risk and overall survival. Clin Cancer Res. 2012;18:3942-51
163. Innocenti F, Owzar K, Cox NL, Evans P, Kubo M, Zembutsu H. et al. A genome-wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clin Cancer Res. 2012;18:577-84
164. Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF. et al. The functional spectrum of low-frequency coding variation. Genome biology. 2011;12:R84
Author contact
![]() Corresponding author: Advanced Technology Center, 8717 Grovemont Circle, Bethesda, MD 20892. amundadottirlnih.gov
Corresponding author: Advanced Technology Center, 8717 Grovemont Circle, Bethesda, MD 20892. amundadottirlnih.gov