Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(6):688-700. doi:10.7150/ijbs.13589 This issue Cite
Research Paper
Ubisol-Q10 Prevents Glutamate-Induced Cell Death by Blocking Mitochondrial Fragmentation and Permeability Transition Pore Opening
Santosh Kumari1,4 *, Suresh L Mehta1,3*, Gaolin Z. Milledge2, Xinyu Huang2, Haining Li5, P. Andy Li1 ![]()
1. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technology Enterprise (BRITE) College of Art and Science, North Carolina Central University, Durham, NC 27707, USA;
2. Department of Mathematics and Computer Science, North Carolina Central University, Durham, NC 27707, USA;
3. Department of Neurological Surgery, University of Wisconsin, School of Medicine and Public Health, Madison, WI 53792, USA;
4. Department of Medicine, University of Wisconsin, School of Medicine and Public Health, Madison, WI 53792, USA.
5. Neurological Center of the General Hospital of Ningxia Medical University, Ningxia Key Laboratory for Cerebrocranial Diseases, Ningxia Medical University, Yinchuan, 750004, P.R. China.
*Equal contribution
Received 2015-8-17; Accepted 2015-10-1; Published 2016-4-27
Abstract

Mitochondrial dysfunction and oxidative stress are the major events that lead to the formation of mitochondrial permeability transition pore (mPTP) during glutamate-induced cytotoxicity and cell death. Coenzyme Q10 (CoQ10) has widely been used for the treatment of mitochondrial disorders and neurodegenerative diseases. Comparing to traditional lipid-soluble CoQ10, water soluble CoQ10 (Ubisol-Q10) has high intracellular and intra-mitochondrial distribution. The aims of the present study are to determine the neuroprotective effects of Ubisol-Q10 on glutamate-induced cell death and to explore its functional mechanisms. HT22 neuronal cells were exposed to glutamate. Cell viability was measured and mitochondrial fragmentation was assessed by mitochondrial imaging. The mPTP opening was determined by mitochondrial membrane potential and calcium retention capacity. The results revealed that the anti-glutamate toxicity effects of Ubisol-Q10 was associated with its ability to block mitochondrial fragmentation, to maintain calcium retention capacity and mitochondrial membrane potential, and to prevent mPTP formation, AIF release, and DNA fragmentation. We concluded that Ubisol-Q10 protects cells from glutamate toxicity by preserving the integrity of mitochondrial structure and function. Therefore, adequate CoQ10 supplementation may be beneficial in preventing cerebral stroke and other disorders that involve mitochondrial dysfunction.
Keywords: Coenzyme Q10, glutamate toxicity, neuronal cell death, AIF, mitochondrial fragmentation, calcium retention.
Introduction
Mitochondrial dysfunction due to glutamate toxicity is one of the major events that accelerates neuronal cell death during acute brain injury such as ischemia/stroke, traumatic brain injury, and chronic neurodegenerative diseases including Alzheimer disease, Parkinson's disease, and amyotrophic lateral sclerosis [1]. Mitochondrial dysfunction due to glutamate toxicity is mediated by receptor-initiated excitotoxicity [2, 3] and non-receptor mediated oxidative toxicity as a result of depletion of intracellular cysteine and glutathione [4]. This leads to excessive accumulation of reactive oxygen species, resulting in oxidative stress [4, 5]. Emerging evidence indicates that oxidative stress due to excessive generation of reactive oxygen species can affects mitochondria adversely [6]. Mitochondrial dysfunction releases various factors including cytochrome c, apoptosis-inducing factor (AIF), Smac/Diablo (second mitochondria-derived activator of caspase/direct IAP binding protein with low PI), or endonuclease G which are responsible for activation of caspase-dependent or caspase-independent mechanisms of DNA degradation and cell death [7]. One of the major sites of reactive oxygen species (ROS) production in the cell is the mitochondria. Mitochondrial ROS is produced upon leakage of electrons through complex I (NADH-ubiquinone oxidoreductase) or III (ubiquinone-cytochrome c oxidoreductase) of the mitochondrial respiratory chain [8]. The products of this leakage are superoxide anions and hydrogen peroxide. Superoxide is converted by the antioxidant enzyme superoxide dismutase (SOD) into hydrogen peroxide in the mitochondria (SOD2 or MnSOD) or in the cytosol (SOD1 or CuZnSOD). Hydrogen peroxide is further simplified to water by glutathione peroxidase. Oxidative stress due to depletion of antioxidant or excessive accumulation of ROS has detrimental effect on mitochondrial structure and function. Moreover, oxidative stress and mitochondrial dysfunction are the primary events in glutamate-induced toxicity [9].
Mitochondria in healthy neurons resemble long, yet remarkably dynamic filaments undergoing cycles of mitochondrial fission and fusion events. Fission processes are regulated by dynamin-related protein 1 (Drp1), Mitochondrial fission 1 protein (Fis1), and endophilin B1 (Endo B1); while fusion process is mediated by mitofusin-1 and -2 (Mfn1, -2) and optic atrophy 1 (OPA1) [10]. Disturbance in mitochondrial dynamics toward continuous fission leads to mitochondrial fragmentation, formation of mPTP, and activation of apoptosis and neurodegeneration [11-15]. Drp1, which induces mitochondrial fission, has been reported to stimulate Bid-induced Bax oligomerization and cytochrome c release by promoting tethering and hemifusion of membranes [16]. Bax knockdown drastically inhibited the mitochondrial accumulation of Drp 1 and Drp1 knockdown attenuated cell apoptosis [17]. Drp1/Bax activation promotes mitochondrial fragmentation, reduces mitochondrial number, and activates apoptotic cell death [18]. We have recently demonstrated that glutamate exposure induces mitochondrial fragmentation [5]. It is not clear whether coenzyme Q10 (CoQ10) is capable of preventing mitochondrial fragmentation caused by glutamate exposure.
Ubiquinone CoQ10 (coenzyme Q10, CoQ10) is a well-known electron transporter of the mitochondrial respiratory chain that shuttles electrons from complex I and II (succinate-ubiquinone oxidoreductase) to complex III during oxidative phosphorylation and energy production. It is normally synthesized in the body. However, the levels of CoQ10 decrease with genetic mutation, cancer, ageing in human and animal tissues. Whole body concentration of CoQ10 is also affected by certain drugs such as statins that inhibit the synthesis of CoQ10 in patients with a risk of cardiovascular diseases and stroke [19-21]. Deficiency of CoQ10 can cause varying clinical syndromes, including encephalomyopathy, mental retardation, recurrent myoglobinuria, isolated myopathy, etc. [22-24]. Supplementation of CoQ10 can restore normal mitochondrial concentration levels in aging [25-27]. Available evidence suggests that besides the above essential role, CoQ10 also acts as a ubiquitous free radical scavenger and has been shown to ameliorate cell death and protects cells under various stress conditions including neurodegeneration diseases [25, 28, 29]. Although, it is not clear whether the beneficial effects of CoQ10 is only limited to its antioxidant property, we and others have reported that CoQ10 prevents apoptosis activated upon mitochondrial dysfunction [29-35]. Similarly, evidence of CoQ10 against glutamate-induced cell death is lacking. Therefore, in the present study we investigated whether CoQ10 curtails glutamate toxicity, prevents cell death, and offers neuroprotective role. Further, we also explored whether the beneficial effects of CoQ10 against glutamate toxicity are mediated through regulating calcium fluctuations, mPTP formation, mitochondrial membrane potential, and mitochondrial dynamic balance. Water-soluble CoQ10 (aka Ubisol-Q10) was developed to overcome low bioavailability of traditional oil-soluble CoQ10 when taken orally [36]. Because traditional oil-soluble CoQ10 has a low bioavailability when taken orally, we decided to use the water soluble Q10 (aka Ubisol-Q10) for the present study. Ubisol-Q10 has better bioavailability and increases cellular and mitochondrial uptake by 20 to 30 fold over the oil-soluble formulation. In addition, it is able to pass through the Blood-Brain Barrier (BBB) [37, 38]. Our data indicates that glutamate challenge induces mitochondrial dysfunction through ROS generation and mitochondrial membrane potential alteration. This further activates mitochondrial fission that results in mitochondrial fragmentation and mPTP formation. These events lead to AIF nuclear translocation, DNA fragmentation, and cell death. Ubisol-Q10 pre-treatment prevents mitochondrial dysfunction, stabilizes the mitochondrial membrane potential, blocks mPTP formation, prevents AIF nuclear translocation and nuclear DNA fragmentation, and subsequent cell death. These results clearly indicate that Ubisol-Q10 offers neuroprotective benefits partly through preserving mitochondrial dynamic balance and function and preventing apoptotic cell damage.
Materials and Methods
Cell culture, experimental treatments and viability assays
Murine hippocampal HT22 neuronal cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, and 200 mM streptomycin/penicillin (Invitrogen) and maintained at 90%-95% relative humidity in 5% CO2 at 37 °C. Glutamate (Sigma) and Ubisol-Q10 (Zymes LLC) were dissolved in water. The stock solutions were diluted with cell culture medium for each experiment. Cells were subjected to glutamate stress 24 h after Ubisol-Q10 treatment. Varying concentrations (1-5 mM) of glutamate [5] and Ubisol-Q10 (10-25 µg) were tested and evaluated for cell viability after 24 h of treatment. Quantification of cell viability in HT22 cells was performed in 96-well plates with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HT22 cells were seeded at a density of 10,000 cells per well. After 24 h of treatment, cells in each well were incubated with 10 μl MTT (5 mg/ml) at a final concentration of 500 μg/ml. After 2 h the medium was removed from the wells and formazan formed was dissolved in dimethyl sulfoxide (DMSO, 80 μl). The absorbance was read with a dual-monochromator, multi-detection microplate reader (SpectraMax M5 Molecular Device, Sunnyvale, CA) at 540 nm. Cell viability was expressed as a percent of the control.
For drug treatment, antioxidant Trolox, hydrogen peroxide (H2O2), staurosporine, cyclosporine, and sodium azide were used to delineate the effects of Ubisol-Q10.
Detections of ROS levels and calcium retention capacity
Intracellular superoxide anion production was determined using dihydroethidium (DHE) as a fluorescent probe. Briefly, cells (2x106/ml) were incubated with the DHE (2.5 μM) for 30 min at 37ºC, after which they were washed, resuspended in phosphate buffered saline (PBS), and analyzed for fluorescent signal intensity using Fluoromax-4 spectrofluorometer at the excitation and emission wavelengths of 480 nm and 590 nm respectively. The florescence was recorded and represented as relative fluorescence intensity (RFI).
For measuring calcium retention capacity of the mitochondria, HT22 cells (5×106) were harvested, washed with PBS and permeabilized immediately with digitonin (50 µg/ml) for 2 min at 25°C in a Ca2+ free KCl medium (125 mM KCl, 2 mM K2HPO4, 1 mM MgCl2, 20 mM HEPES, pH 7.0) [37] Measurements of Ca2+ were performed fluorimetrically at 37°C with a FluoroMax-4 spectrofluorometer equipped with magnetic stirring and thermostatic controls. Extra-mitochondrial Ca2+ was measured in the presence of 1 µM Calcium Green-5N, 10 mM glutamate, 0.5 mM malate or 10mM succinate with excitation and emission wavelengths set at 506 and 530 nm respectively. The free calcium (Ca2+) uptake and Ca2+ release of digitonin-permeabilized cells were measured by loading cells with Ca2+ pulses at constant time intervals.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was measured using JC-1 dye. JC-1 exhibits potential-dependent accumulation in mitochondria with a fluorescence emission shift from green (~529 nm) to red (~590 nm). Briefly, HT22 cells were grown on Lab-Tek II Chamber Slide (Thermo Scientific) and incubated with JC-1 dye (5 µg/ml). JC-1 was excited at 488 nm and emission of JC-1 monomer (depolarized) and J-aggregate (polarized mitochondria) forms were captured at 525/50 and 605/75 nm, respectively, with Nikon laser-scanning confocal microscope (Nikon Eclipse C1). To avoid the photobleaching, laser power was attenuated to the minimal level. Minimum three fields per group were captured to determine the changes in mitochondrial membrane potential. Carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP 5 µM) was used as a control to dissipate mitochondrial potential.
Subcellular fractions and Immunoblotting
Mitochondrial and cytosolic fractions were used in the study and the fractions were isolated as described previously [5]. Briefly, cells were washed with PBS and resuspended in cytosolic lysis buffer (250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4 pH 7.2, 200 μg/ml digitonin, 100 mM PMSF, protease inhibitor cocktail (Halt, Thermo Scientific) for 5 min on ice. These cells were centrifuged at 1,000 × g for 5 min to separate the supernatant as a cytosolic fraction and the pellet was resuspended in two volumes of mitochondrial lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% (v/v) Triton X-100, 0.3% NP-40, PMSF, protease inhibitor cocktail) for 5 min on ice. The resulting suspension was centrifuged at 10,000 × g for 10 min and the supernatant was collected as the mitochondrial fraction. The purity of the fractions has been previously verified [39]. Protein concentration was determined using the Bradford assay and same amount of protein (20 μg) was loaded into each lane of a 4-12% NuPAGE gel (Invitrogen). Following electrophoresis, proteins were transferred to a nitrocellulose membrane (Invitrogen) and blocked for nonspecific binding with 5% skim milk. The membrane was incubated for target proteins using primary antibodies specific for Drp1 (1:1000, 8570, Cell Signaling USA), phospho-Drp1 (Ser616 1:800, 3455, Cell Signaling USA), Fis1 (1:500, sc-98900, MBL,MA USA), AIF (1:500, sc-9416, Santa Cruz Biotechnology, CA, USA), cytochrome C (1:500, sc-13560, Santa Cruz Biotechnology, CA, USA), VDAC (1 µg/ml ab14744, Abcam Inc, MA, USA) or Actin (1:1000,sc-1616, Santa Cruz Biotechnology, CA, USA) overnight at 4ºC. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The blots were then developed using the Pierce ECL Western Blotting Substrate (Thermo Scientific). The β-actin or VDAC bands were used as internal loading controls and the ratios of the targeted proteins and loading control were calculated and presented as final results.
Mitochondrial morphometrics
HT22 cells were grown on Lab-Tek II Chamber Slide (Thermoscientific). Mitochondrial staining was carried out with MitoTracker®Red CM-XRos (Invitrogen) as described previously [5]. DAPI was used to label nuclei. Minimum 3 images were captured in each well for analyses using the Nikon laser-scanning confocal microscope at the magnification of 63X. An image process program was designed to identify and quantify mitochondria structures from the RGB images according to the published method [40, 41]. In brief, we first created a mask to filter out the nucleus regions that are mostly blue. Next, we extracted only the red channel and converted it to a gray-scale image. The resulting image was adjusted to increase contrast. A "top-hat" spatial filter was applied in order to isolate bright features from a dark background using a flat, disk-shaped structuring element with a radius of 2 pixels. In order to reduce noise while preserving edges, a 2-D median filtering with a dimension of 3 x 3 pixels was applied twice. An additional threshold was applied to remove darker pixels.
For morphological analysis, three parameters were calculated for each mitochondrial object: the form factor F (perimeters2/(4π• area)), mitochondrial perimeter and mitochondrial area. We also computed the total number of mitochondria per cell. Form factor is independent of image magnification and has a minimum value of 1 (corresponding to a perfect circle). For statistical analysis, we calculated the average values of F for each image.
AIF immunostaining
HT22 cells were grown on Lab-Tek II Chamber Slides (Thermo Scientific). Cells were fixed 18 h after glutamate exposure with 4% paraformaldehyde for 20 min, washed with PBS, and permeabilized in Triton X-100 (0.3%) for 5 min. The cells were blocked with 10% donkey serum for 1 h at room temperature. After blocking, cells were incubated overnight at 4°C with primary antibodies against AIF (1:200, Santa Cruz Biotechnology CA). Cells were washed and incubated for 2 h at room temperature with secondary antibodies conjugated with Alexa Fluor 488 (1:500 dilution; Invitrogen). Finally, slides were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories). Slides were scanned with a Nikon laser-scanning confocal microscope. The experiment was repeated thrice and, at minimum, three fields per group were captured and processed for analysis.
DNA fragmentation assay
DNA fragmentation due to apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end-labeling (TUNEL) assay using a DeadEnd Fluorometric TUNEL system kit (Promega, USA) per the kit instructions. A minimum of three random fields per group were captured. TUNEL positive cells were counted and presented as percent of total cells.
Statistical analysis
Data are presented as mean±SD and analyzed with GraphPad Prism 5 software. One-way ANOVA followed by Tukey's multiple comparison test and two-way ANOVA followed by Bonferoni test were used when appropriate. A p<0.05 is considered statistically significant.
Results
Pretreatment of HT22 cells with Ubisol-Q10 blocked glutamate-induced cell death
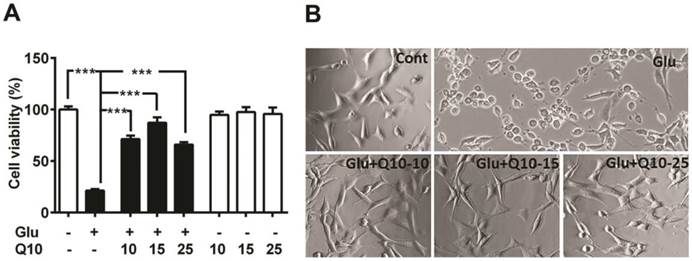
To determine the neuroprotective role of Q10, we exposed Ubisol-Q10 pretreated and untreated HT22 neuronal cells to glutamate and cell survival was determined by MTT assay and cellular morphology (Fig. 1). Glutamate induced cell death in HT22 cells in a dose (data not shown) and time dependent manner [5]. Therefore, the cell survival was limited to around 20 % of control (Fig. 1A and B) with 4 mM of glutamate after 24 h of exposure [5]. In contrast, Ubisol-Q10 alone up to the 25 µg/ml range was well tolerated by HT22 neurons without any influence on cell viability (Fig. 1A). Interestingly, Ubisol-Q10 (10-25 µg/ml) blocked glutamate-induced death in HT22 cells; thereby significantly (p<0.001) improved the cell survival (from 21 to 87 %) of control HT22 cells (Fig. 1A and B). The Ubisol-Q10-induced protection was not transiently limited to 24 h as the rescue effects were also noticed up to 48 h of glutamate exposure (data not shown), suggesting the long-term protective effect of Ubisol-Q10.
Ubisol-Q10 blocks glutamate toxicity-induced cell death. (A) Concentration-dependent effect of water soluble Ubisol-Q10 on HT22 cell viability alone and with 4 mM glutamate. Ubisol-Q10 was added to the cell media prior to the glutamate exposure and cell viability was estimated 24 h after addition. Ubisol-Q10 alone upto 25 μg had no effect on cell viability whereas glutamate exposure significantly reduced the survival of cells to nearly 20 % of control. Pretreatment of cells with Ubisol-Q10 resisted the effect of glutamate toxicity thereby showed significant reduction in cell mortality. Ubisol-Q10 concentration of 15 μg showed maximum protection to glutamate induced cell death in HT222 cells. (B) Representative photomicrograph of HT22 cells treated with Ubisol-Q10, glutamate and both. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significant levels is shown by ***=p<0.001 vs. respective group. Glu=glutamate, Q10=Ubisol-Q10, Glu+Q10=glutamate +Ubisol-Q10 and Glu+Q10-15 to 25 = glutamate (4 mM) +Ubisol-Q10 (15-25 µg).
Ubisol-Q10 decreased ROS levels and stabilized mitochondrial membrane potential
We have previously shown that glutamate exposure alters ROS levels and mitochondrial membrane potential [5]. In the present study, we set the baseline level of ROS production in control cells as 100%. Exposure of HT22 neurons to glutamate resulted in enhanced ROS formation when measured after 18 h (p<0.05). Presently, we determined whether Ubisol-Q10 treatment could prevent the increase in ROS production and whether this effect is associated with stabilizing mitochondrial potential (Fig. 2). Results revealed that glutamate induced the generation of ROS whereas Ubisol-Q10 treatment curtailed it (Fig. 2A).
Ubisol-Q10 prevents glutamate induced ROS production and mitochondrial potential loss. (A) ROS production were measured using DHE in Ubisol-Q10 pretreated HT22 cells exposed to glutamate. Ubisol-Q10 normalized the glutamate exposure increased levels of ROS. (B) Micrograph of HT22 cells shows potential loss after glutamate exposure (arrow). Ubisol-Q10 pretreatment prevented glutamate induced depolarization of mitochondrial membrane potential. Mitochondrial potential measured with JC-1, shows red staining as normal potential whereas green staining represents depolarization and potential loss. (C) Summarized graph shows the effect of Antioxidant Trolox and Ubisol-Q10 on glutamate-induced cell death. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significant levels is shown by *=p<0.05 and ***=p<0.001 vs. respective group. Glu=glutamate, Q10=Ubisol-Q10 and Glu+Q10 = glutamate+Ubisol-Q10.
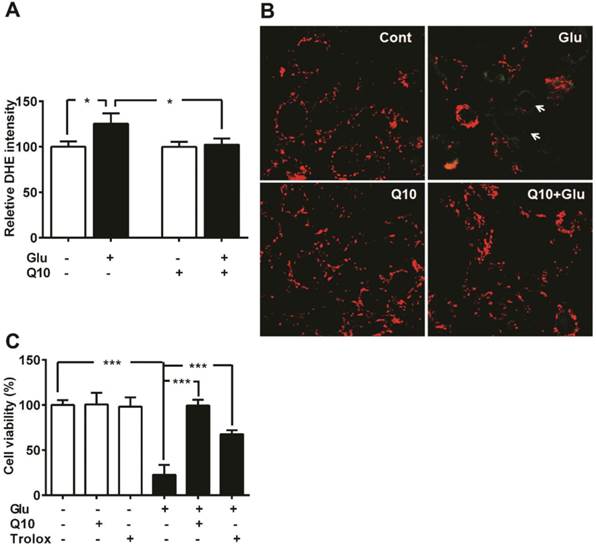
Likewise, glutamate also affected mitochondrial membrane potential. Glutamate exposure resulted in membrane depolarization as reflected by the increase in green fluorescence of JC-1 monomer. Pretreatment with Ubisol-Q10 prevented glutamate-induced mitochondrial membrane depolarization as reflected by the predominant red fluorescence of JC-1 aggregates that were taken by normal polarized mitochondria. Non-treated control cells and cells treated with Ubisol-Q10 alone maintained normal mitochondrial potential (Fig. 2B).
To determine whether glutamate-induced cell death is mediated by increased free ROS production and loss of mitochondrial potential through formation of mPTP, we used free radical scavenger Trolox and Cyp-D /ANT channel inhibitor cyclosporine. Cell viability study results revealed that Trolox significantly ameliorated glutamate toxicity (Fig. 2C), while cyclosporine A failed to prevent glutamate induced cell death in HT22 cells (Data not shown). These results suggest that glutamate-induced cell death is mediated through formation of ROS and mPTP independent of Cyp-D /ANT.
Ubisol-Q10 prevented glutamate-reduced calcium retention capacity
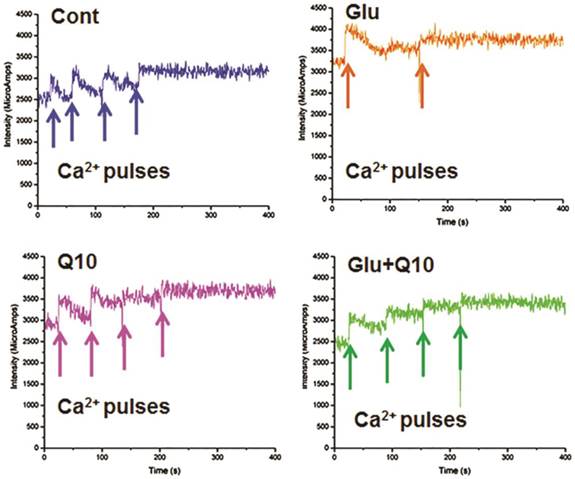
Mitochondria have been shown to transiently store calcium and thereby regulating cellular Ca2+ homeostasis. Reduction or loss of calcium retention capacity of the mitochondria leads to formation of mPTP and opening of mPTP has been shown to be a primary mediator of cell death. We investigated whether glutamate lowers calcium retention capacity and whether Ubisol-Q10 could improve it. Mitochondrial calcium uptake capacity was detected using Calcium Green-5N. Addition of calcium causes increase in extra-mitochondrial calcium and then the concentration decreased as the calcium is taken up by the mitochondria. Under normal condition, cells with Ubisol-Q10 addition did not differ in mitochondrial calcium uptake capacity as compared to control cells. After glutamate exposure, calcium retention capacity was decreased compared to control cells. Ubisol-Q10 pretreatment in glutamate-exposed cells restored the calcium retention capacity to control level (Fig. 3).
Ubisol-Q10 restores glutamate reduced mitochondrial calcium retention capacity. Figures show cytosolic calcium uptake by mitochondrial in permeabilized HT22 cells. The addition of calcium (cytosolic) indicated by an arrow (25 µM of calcium pulse each) results in a rapid increase in signal that subsequently decline due to calcium uptake by mitochondria. Result shows that glutamate exposure reduced the capacity of mitochondrial to uptake cytosolic excessive calcium. Ubisol-Q10 pretreatment in contrast improved the mitochondrial calcium uptake to normal levels, thereby maintained the mitochondrial calcium uptake capacity. Cont=Control,
Ubisol-Q10 inhibits glutamate-induced mitochondrial fragmentation
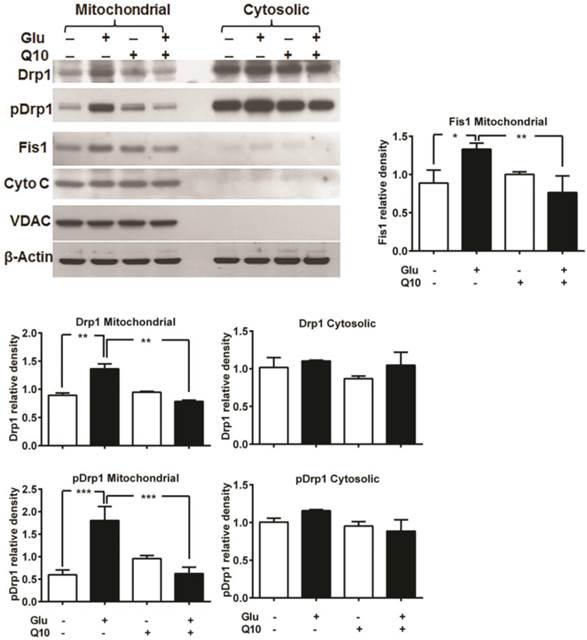
Previously, we have shown that glutamate affects mitochondrial morphology in HT22 cell by increasing mitochondrial fission, which may leads to increased ROS formation, loss of mitochondrial potential and calcium retention capacity, and adversely affects mitochondrial function [5]. We investigated whether the protective effects of Ubisol-Q10 is mediated through preventing mitochondrial fragmentation. Mitochondria are organized as a tubular network under normal control conditions. Under stress conditions, mitochondrial fission occurs and the reticular mitochondria break into short, round pieces, a process defined as mitochondrial fragmentation. Mitochondrial fission markers Drp1 and Fis1 were measured using immunoblotting. Drp1 is normally present in the cytosol of the cell, but when phosphorylated at Ser616, Drp1 translocates to the mitochondria and stimulates mitochondrial fission. The results showed that phospho-Drp1 and Fis1 were significantly increased in the mitochondrial fraction, but not in the cytosolic fraction, after 18 h of glutamate exposure. Treatment with Ubisol-Q10 completely blocked the phosphorylation of Drp1 and the increase in Fis1 (Fig. 4).
Ubisol-Q10 inhibits glutamate-induced change mitochondrial fragmentation markers. Representative Western blots and quantitative analysis of the protein band of mitochondrial fission protein Drp1, pDrp1 and Fis1. Glutamate exposure significantly increased the levels of mitochondrial fragmentation markers Drp1, pDrp1 and Fis1 without affecting the levels of VDAC and cytochrome c release after 18 h. Ubisol-Q10, in contrast blunted the corresponding elevation of Drp1, pDrp1 and Fis1 in these cells, thereby Ubisol-Q10 significantly prevented the glutamate-induced increase in these markers. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significant levels is shown by *=p<0.05, **=p<0.01 and ***=p<0.001 vs. respective group. Glu=glutamate and Q10=Ubisol-Q10.
Consistent with the protein levels of Drp1 and Fis1, mitochondrial imaging study using MitoTracker Red revealed that the glutamate caused marked mitochondrial morphologic alteration. As seen in Figure 5, the reticularity of the mitochondrial network was broken into small round shaped mitochondrial pieces after 18 h of glutamate exposure. Pretreatment with Ubisol-Q10 restored the mitochondrial tubular network in glutamate-exposed cells (Fig. 5A).
Ubisol-Q10 preserved mitochondrial morphology. (A) Photomicrograph shows mitochondrial morphology in HT22 cells treated with glutamate and Ubisol-Q10. Mitochondrial morphology was studied using MitoTracker Red with confocal microscopy. Images were processed and analyzed for structural alterations. Glutamate exposure (18 h) decreased the mitochondrial reticular network resulting into small round shaped broken mitochondria. Ubisol-Q10 pretreatment restored the mitochondrial tubular network in glutamate-exposed cells. (B) Figure shows mitochondrial perimeter and form factor (branching) analyzed from the mitochondrial micrograph. Glutamate exposure not only lowered the mitochondrial perimeter but also reduced the mitochondrial branching. Ubisol-Q10 pretreatment blocked these effects of glutamate and thereby restored normal mitochondrial morphology. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significant levels is shown by *=p<0.05 and **=p<0.01 vs. respective group. Glu=glutamate and Q10=Ubisol-Q10.
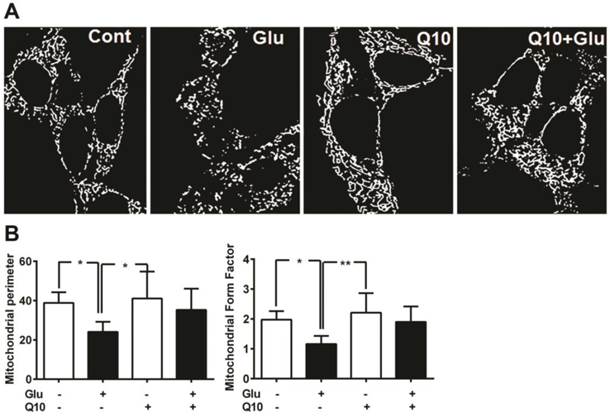
To determine the effects of glutamate exposure on mitochondrial morphology and the degree of mitochondrial network branching in cells treated with/without Ubisol-Q10, we determined the mitochondrial perimeter and form factor (branching). The result revealed that glutamate significantly (p<0.05) lowered the mitochondrial perimeter in HT22 cells (Fig. 5B). Pretreatment with Ubisol-Q10 restored the mitochondrial perimeter in glutamate-exposed cells. Similarly, glutamate exposure markedly affected the network of mitochondria. Mitochondrial was significantly (p<0.05) less branched in cells exposed to glutamate. When pretreated with Ubisol-Q10, cells recovered from glutamate toxicity and restored the form factor, which clearly suggests that Ubisol-Q10 improve cellular mitochondrial function by protecting mitochondrial morphology (Fig. 5B).
Ubisol-Q10 blocks mitochondrial dysfunction-induced release of AIF
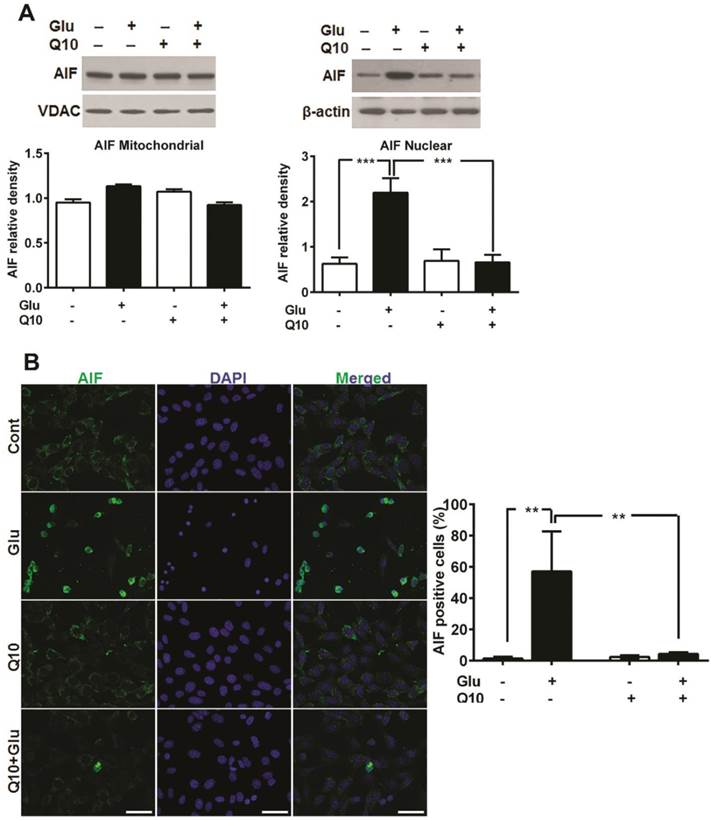
Glutamate-induced cell death in HT22 cells have been shown to be mediated by AIF dependent apoptosis [42, 43]. Formation of mPTP may lead to release of mitochondrial proapoptotic proteins such as AIF and others. To examine whether glutamate-induced mitochondrial fragmentation caused release of proapoptotic proteins and whether restoration of mitochondrial structure by Ubisol-Q10 could block the release of mitochondrial proteins, we measured the levels of cytochrome c protein levels in mitochondrial and cytosolic fractions, AIF protein in mitochondrial and nuclear fractions and AIF immunoreactivity (Fig 6). The results demonstrated that cytochrome c was not released from the mitochondria into the cytosol (Fig 4). However, AIF immunocytochemistry showed nuclear localization of AIF after glutamate treatment in HT22 cells and treatment with Ubisol-Q10 significantly blocked the AIF presence in the nuclei (Fig 6B). These results were further confirmed by immunoblotting that showed a significant increase of AIF protein in the nuclear fraction after glutamate exposure and the AIF level was markedly reduced by Ubisol-Q10 (Fig 6A).
Ubisol-Q10 prevented glutamate-induced AIF release. (A) Representative Western blots and quantitative analysis of the protein bands of AIF in mitochondrial and nuclear fractions. AIF levels were relatively similar in HT22 cells treated with glutamate and Ubisol-Q10 in mitochondrial fraction. In contrast, glutamate exposure significantly increased AIF nuclear translocation. Ubisol-Q10 pretreatment blocked the AIF nuclear translocation in cell exposed to glutamate. (B) Photomicrograph and quantitation of AIF positive cells. Cells shows peripheral punctuated staining in control untreated and Ubisol-Q10 only treated cells. AIF translocates to the nucleus following glutamate exposure as shown by diffused staining. Ubisol-Q10 blocked AIF nuclear translocation. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significant levels is shown by **=p<0.01 and ***=p<0.001 vs. respective group. Cont=Control, Glu=glutamate and Q10=Ubisol-Q10.
Ubisol-Q10 reduces glutamate-induced apoptotic injury
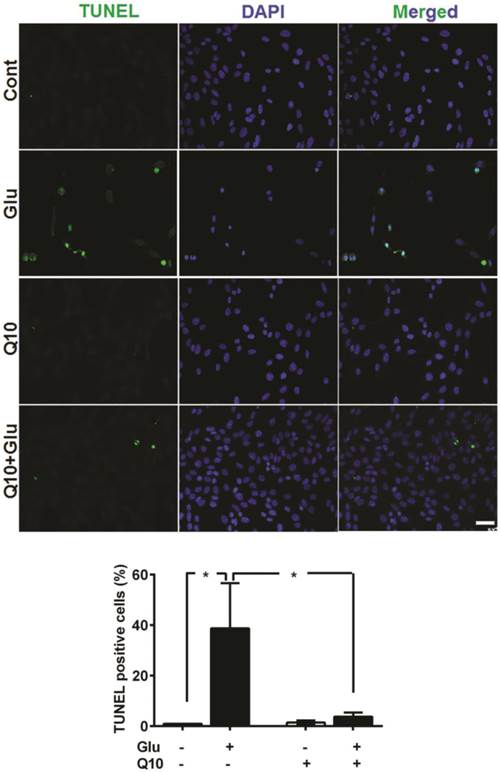
Detection of apoptotic cell death by TUNEL staining revealed that incubation of cells with 4 µM glutamate for 18 h significantly increased the number of TUNEL-positive stained cells (Fig. 7). The average percent of TUNEL positive cells reach to 38.7±17.9%. In contrast, the number of TUNEL positive cell in glutamate-exposed cells was significantly (p<0.05) reduced by Ubisol-Q10 treatment. As a result, the number of TUNEL positive cells reduced to non-glutamate control level. Few TUNEL positive cells were observed in non-treated control cells and with Q10-treatment alone. A panel of TUNEL-stained images is shown in Figure 7B and percent of TUNEL positive cells in each group is given in Figure 7B.
Ubisol-Q10 prevented DNA damage in cells treated with glutamate. Photomicrograph and quantitation of cells with DNA fragmentation determined with TUNEL staining. Glutamate increased the number of cells positive for TUNEL measure after 18 h of exposure. Ubisol-Q10 in contrast prevented the DNA fragmentation in cells exposed to glutamate. Data are the representation of 3 or more independent experiments conducted in triplicate. Values are means±SD and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. Significance is shown by *=p<0.05 vs. respective group. Cont=Control, Glu=glutamate and Q10=Ubisol-Q10.
Discussion
Mitochondrial dysfunction due to various reasons including oxidative stress plays a crucial role in activation of cell death pathways. Recently, we have shown that mitochondrial dysfunction could be the result of mitochondrial fragmentation induced by glutamate toxicity [5] and glutamate toxicity is a key step in the progression of neuronal loss in acute and chronic neurodegenerative diseases. CoQ10, in addition to carrying electrons during oxidative phosphorylation, positively influence many cellular functions presumably due to its antioxidant property [29, 32, 33, 44]. Although, it is not clear whether the beneficial effects of CoQ10 is only limited to its antioxidant property, we and others have reported that CoQ10 also prevent mitochondrial dysfunction-induced activation of apoptosis [29-32, 34, 35]. Presently we demonstrate that glutamate causes mitochondrial fragmentation, mPTP formation, calcium deregulation and mitochondrial potential loss. These events lead to AIF nuclear translocation, DNA fragmentation, and cell death. Ubisol-Q10 pretreatment prevented glutamate-induced cell death by blocking mitochondrial fragmentation, ameliorating mitochondrial dysfunction, stabilizing the mitochondrial membrane potential, blocking the mPTP formation, and preventing the AIF nuclear translocation and DNA fragmentation. These results clearly indicate that Ubisol-Q10 offers neuroprotective benefits partly through preserving mitochondrial structure and function, which thereby prevents cell damage.
In the present study, we have shown that glutamate induces cell death in HT22 cells through oxidative stress, mitochondrial dysfunction and AIF release [5, 9, 42, 43, 45, 46]. Glutamate-induced mitochondrial dysfunction could be the result of mitochondrial fragmentation due to activation of Drp1 as reported previously [5]. The fragmented mitochondria are sensitized to Bax/Bid-dependent pore formation and activation of apoptosis [16, 47, 48]. Drp1 is normally present in the cytosol, but once phosphorylated at Ser616, it translocates to the mitochondria where it localizes to potential division sites on mitochondrial outer membrane with the help of Fis1. In the present study, we show that glutamate exposure not only increased the levels of Fis1 but also increased the phosphorylation and mitochondrial translocation of Drp1 (Fig. 4), which is consistent with our previous finding [5]. Increased mitochondrial Drp1 levels were well correlated with the mitochondrial morphology (Fig. 4 and 5), suggesting that Drp1 induces mitochondrial fragmentation in cells exposed to glutamate. Likewise, mitochondrial fragmentation has been shown to be associated with increased oxidative stress and mitochondrial depolarization, whereas inhibiting Drp1 offers neuroprotection [43, 47].
Glutamate toxicity can also trigger calcium influx that leads to mitochondrial calcium overload [12] whereas, once fragmented, mitochondria reduce their calcium buffering capacity [49]. Our study showed that glutamate-exposed cells exhibited impaired mitochondrial calcium loading capacity, AIF nuclear translocation, DNA fragmentation, and cell death (Fig. 3, Fig. 6 Fig. 7 and Fig. 1). It is likely that mitochondrial calcium overload alone or in combination with increased ROS production, triggers the formation of mPTP that results in the activation of cell death pathway. Additionally, it is believed that glutamate-mediated oxidative stress due to increased ROS production has been shown to be a biphasic event in a way that glutamate induces a primary surge in ROS production after 6-8 h and followed by a secondary boost after 18 h [43]. The primary ROS could be ascribed to glutamate induced glutathione depletion as proposed previously [50], whereas the secondary boost in ROS production could be the result of mitochondrial fragmentation due to the activation of Drp1.
Furthermore, presently we showed that glutamate-induced cell death could be prevented with the supplementation of Ubisol-Q10 (Fig. 1). CoQ10, which is an endogenous electron carrier in the inner mitochondrial membrane, has been widely studied for its antioxidative properties [29, 44]. Moreover, Papucci et al. have reported that CoQ10 could also prevent apoptosis in response to apoptotic stimuli that do not generate free radicals [35]. We and others have previously shown that cells death mechanism in HT22 cells are activated due to mitochondrial dysfunction [5, 9, 42, 43] and mitochondrial dysfunction could be the result of mitochondrial fragmentation caused by Drp1. Inhibiting Drp1 improves the survival of cells in vitro and in vivo conditions of glutamate toxicity [45]. We presently reported that Ubisol-Q10 blocked Drp1 mitochondrial translocation and thereby reduced mitochondrial fragmentation upon glutamate exposure. Mitochondria appeared tubular and branched in cells treated with Ubisol-Q10 and Ubisol-Q10+glutmate. Whereas, mitochondria appeared small and rounded in glutamate only treated cells. Fragmented mitochondria are not only prone to increased oxidative stress and depolarization [51] but also are sensitized to proapoptotic Bcl-2 family protein mediated pore formation [16, 47, 48]. We found that Ubisol-Q10 supplementation not only blocked increase in ROS production but also prevented mitochondrial potential loss upon glutamate exposure (Fig 2, Fig. 4 and Fig. 5). Therefore, cells treated with Ubisol-Q10 resisted glutamate-induced decreases of mitochondrial perimeter and mitochondrial branching, and maintained normal mitochondrial morphology (Fig. 4 and 5). Although, it is not clear from the present study whether Ubisol-Q10 directly blocks mitochondrial fragmentation by inhibiting Drp1 or whether Ubisol-Q10 prevents the primary surge in ROS production which is responsible for Drp1 activation and mitochondrial fragmentation, CoQ10 has been suggested to prevent mitochondrial depolarization independent to its antioxidant property [35]. In the present study, we also showed that Ubisol-Q10 has a slightly better effect on cell survival than antioxidant Trolox (Fig. 2C). Moreover, evidence indicates that ROS production could be increased following mitochondrial fragmentation [52]. By blocking mitochondrial fragmentation, Ubisol-Q10 treatment may lower ROS production and prevent the mitochondrial depolarization. Mitochondrial depolarization is an indicator of mPTP formation during various stresses that reduce mitochondrial calcium retention capacity [50]. Glutamate exposure in the present study decreased mitochondrial calcium capacity, whereas Ubisol-Q10 pretreatment restored capacity and ultimately improved cell survival in cells exposed to glutamate (Fig. 3, Fig. 6 and Fig. 1). Likewise, mitochondrial fragmentation reduces the mitochondrial calcium retention capacity and aids in formation of mPTP though which proapoptotic protein are released. Therefore, the present study reveals that Ubisol-Q10 directly or indirectly blocks Drp1 dependent mitochondrial fragmentation, prevents mitochondrial potential loss and mPTP formation, restores mitochondrial calcium capacity, and inhibits AIF release and apoptotic DNA fragmentation, and thereby improves survival in cells exposed to glutamate. All the evidence supports that Ubisol-Q10 offers beneficial effects by preserving mitochondrial structure and function.
Conclusion
Glutamate causes cell death by inducing mitochondrial fragmentation and dysfunction, formation of mPTP, and nuclear translocation of AIF. Ubisol-Q10 prevents glutamate-induced cell death by blocking mitochondrial fragmentation, preserving mitochondrial calcium retention capacity and membrane potential, preventing mPTP formation, AIF release, and DNA fragmentation. Ubisol-Q10 supplementation may be beneficial in ameliorating stroke, brain and spinal traumatic injuries, mitochondria-associated neurodegenerative and metabolic disorders.
Acknowledgements
BRITE is partially funded by the Golden Leaf Foundation. HL is supported by the National Natural Science Foundation of China (grant # 81460179). The authors thank Mr. Brent Caligan for proofreading.
Author Contributions
Conceived and designed the experiments: SK, SLM and PAL. Performed the experiments: SK, SLM and HL. Data analysis: SK, SLM GZM, XH and PAL. Wrote the manuscript: SK, SLM and PAL.
Competing Interests
The authors declare that no competing interest exists.
References
1. Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19:351-69
2. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta pharmacologica Sinica. 2009;30:379-87
3. Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Molecular neurobiology. 2001;24:107-29
4. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547-58
5. Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PloS one. 2012;7:e39382
6. Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of neurochemistry. 2009;109(Suppl 1):133-8
7. Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain research reviews. 2007;54:34-66
8. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox biology. 2013;1:304-12
9. Fukui M, Song JH, Choi J, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. European journal of pharmacology. 2009;617:1-11
10. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Molecular biology of the cell. 2004;15:5001-11
11. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A. et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nature genetics. 2000;26:211-5
12. Chan DC. Mitochondrial fusion and fission in mammals. Annual review of cell and developmental biology. 2006;22:79-99
13. Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Human molecular genetics. 2009;18:R169-76
14. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. The Journal of biological chemistry. 2005;280:26185-92
15. Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Molecular and cellular biology. 2003;23:5409-20
16. Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R. et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889-901
17. Wu S, Zhou F, Zhang Z, Xing D. Bax is essential for Drp1-mediated mitochondrial fission but not for mitochondrial outer membrane permeabilization caused by photodynamic therapy. Journal of cellular physiology. 2011;226:530-41
18. Leinninger GM, Backus C, Sastry AM, Yi YB, Wang CW, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiology of disease. 2006;23:11-22
19. Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Molecular aspects of medicine. 1997;18(Suppl):S137-44
20. Qu J, Kaufman Y, Washington I. Coenzyme Q10 in the human retina. Investigative ophthalmology & visual science. 2009;50:1814-8
21. Rundek T, Naini A, Sacco R, Coates K, DiMauro S. Atorvastatin decreases the coenzyme Q10 level in the blood of patients at risk for cardiovascular disease and stroke. Archives of neurology. 2004;61:889-92
22. Garcia-Corzo L, Luna-Sanchez M, Doerrier C, Garcia JA, Guaras A, Acin-Perez R. et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Human molecular genetics. 2013;22:1233-48
23. Quinzii CM, Hirano M. Primary and secondary CoQ(10) deficiencies in humans. BioFactors. 2011;37:361-5
24. Salviati L, Trevisson E, Rodriguez Hernandez MA, Casarin A, Pertegato V, Doimo M. et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. Journal of medical genetics. 2012;49:187-91
25. Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8892-7
26. Reahal S, Wrigglesworth J. Tissue concentrations of coenzyme Q10 in the rat following its oral and intraperitoneal administration. Drug metabolism and disposition: the biological fate of chemicals. 1992;20:423-7
27. Yan J, Fujii K, Yao J, Kishida H, Hosoe K, Sawashita J. et al. Reduced coenzyme Q10 supplementation decelerates senescence in SAMP1 mice. Experimental gerontology. 2006;41:130-40
28. Ostrowski RP, Piotrowski P, Pankowska T, Smialek M. Evaluation of morphological changes after treatment with coenzyme Q10 (CoQ10) in endothelin-1 induced experimental ischemia in the rat. Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences. 1998;36:185-8
29. Jing L, He M-T, Chang Y, Mehta SL, He Q-P, Zhang J-Z. et al. Coenzyme Q10 Protects Astrocytes from ROS-Induced Damage through Inhibition of Mitochondria-Mediated Cell Death Pathway. Intl J Biol Sci. 2015;11:59-66
30. Belliere J, Devun F, Cottet-Rousselle C, Batandier C, Leverve X, Fontaine E. Prerequisites for ubiquinone analogs to prevent mitochondrial permeability transition-induced cell death. Journal of bioenergetics and biomembranes. 2012;44:207-12
31. Devun F, Walter L, Belliere J, Cottet-Rousselle C, Leverve X, Fontaine E. Ubiquinone analogs: a mitochondrial permeability transition pore-dependent pathway to selective cell death. PloS one. 2010;5:e11792
32. Jing L, Kumari S, Mendelev N, Li PA. Coenzyme q10 ameliorates ultraviolet B irradiation induced cell death through inhibition of mitochondrial intrinsic cell death pathway. International journal of molecular sciences. 2011;12:8302-15
33. Li H, Chen G, Ma W, Li PA. Water-soluble coenzyme q10 inhibits nuclear translocation of apoptosis inducing factor and cell death caused by mitochondrial complex I inhibition. International journal of molecular sciences. 2014;15:13388-400
34. Orsucci D, Mancuso M, Ienco EC, LoGerfo A, Siciliano G. Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Current medicinal chemistry. 2011;18:4053-64
35. Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A. et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. The Journal of biological chemistry. 2003;278:28220-8
36. Spindler M, Beal MF, Henchcliffe C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatric disease and treatment. 2009;5:597-610
37. Constantinescu R, McDermott MP, Dicenzo R, de Blieck EA, Hyson HC, Beal MF. et al. A randomized study of the bioavailability of different formulations of coenzyme Q(10) (ubiquinone). Journal of clinical pharmacology. 2007;47:1580-6
38. Muthukumaran K, Leahy S, Harrison K, Sikorska M, Sandhu JK, Cohen J. et al. Orally delivered water soluble Coenzyme Q10 (Ubisol-Q10) blocks on-going neurodegeneration in rats exposed to paraquat: potential for therapeutic application in Parkinson's disease. BMC neuroscience. 2014;15:21
39. Mendelev N, Mehta SL, Witherspoon S, He Q, Sexton JZ, Li PA. Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion. 2011;11:76-82
40. De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Current biology: CB. 2005;15:678-83
41. Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. American journal of physiology Cell physiology. 2005;289:C881-90
42. Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S. et al. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell death and differentiation. 2008;15:1553-63
43. Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell death and differentiation. 2011;18:282-92
44. Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiology of disease. 2005;18:618-27
45. Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J. et al. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell death and differentiation. 2012;19:1446-58
46. Koh PO. 17Beta-estradiol prevents the glutamate-induced decrease of Akt and its downstream targets in HT22 cells. The Journal of veterinary medical science / the Japanese Society of Veterinary Science. 2007;69:285-8
47. Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. American journal of physiology Cell physiology. 2011;300:C447-55
48. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T. et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Developmental cell. 2008;14:193-204
49. Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Annals of the New York Academy of Sciences. 2008;1147:283-92
50. Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9893-8
51. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ. et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Molecular and cellular biology. 2012;32:309-19
52. Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. Journal of cell science. 2013;126:789-802
Author contact
![]() Corresponding author: P. Andy Li, Department of Pharmaceutical Sciences , North Carolina Central University, BRITE Building 2025, 302 East Lawson Street, Durham, NC 27707. Fax: 919-530-6600 E-mail: pliedu.
Corresponding author: P. Andy Li, Department of Pharmaceutical Sciences , North Carolina Central University, BRITE Building 2025, 302 East Lawson Street, Durham, NC 27707. Fax: 919-530-6600 E-mail: pliedu.