Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results and Discussion
Conclusion
Methods and Materials
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(7):768-775. doi:10.7150/ijbs.14718 This issue Cite
Research Paper
Inhibition of wnt/β-catenin Signaling in Hepatocellular Carcinoma by an Antipsychotic Drug Pimozide
Valerie Fako1, Zhipeng Yu1, Curtis J. Henrich2, 3, Tanya Ransom2, Anuradha S. Budhu1 ![]() , Xin W. Wang1
, Xin W. Wang1 ![]()
1. Laboratory of Human Carcinogenesis, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland 20892, USA;
2. Molecular Targets Laboratory, National Cancer Institute, Frederick, Maryland 21702, USA;
3. Basic Research Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21701, USA.
Received 2015-12-16; Accepted 2016-3-11; Published 2016-4-28
Abstract

Hepatocellular carcinoma (HCC) is one of the most common forms of malignant cancers in the world, yet very few effective systemic treatments for HCC patients exist. Thus, the development of new treatment modalities presents a great need. The wnt/β-catenin signaling pathway is highly activated in stem cell-like aggressive HCC, which is associated with chemoresistance and poor survival in HCC patients. In a previous study, we found that an FDA-approved psychiatric drug, pimozide (PMZ), has anti-cancer properties in HCC cell lines that express epithelial cell adhesion molecule (EpCAM), a hepatic stem cell marker that is a functional down-stream target of the wnt/β-catenin pathway. In this study, we demonstrate that PMZ effectively inhibits cell growth of HCC cells by disrupting the wnt/β-catenin signaling pathway and reducing EpCAM expression. Thus, PMZ may be a useful molecular entity that could be repurposed as an anti-cancer therapy for treatment of HCC.
Keywords: Pimozide, wnt/β-catenin, EpCAM, HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death worldwide, and the 5-year overall survival rate in the United States is less than 20%, due in part to the lack of systemic treatment options [1, 2]. Although several therapeutic approaches exist for patients with early or intermediate stage HCC (such as surgical resection, ablation or liver transplantation) [3], most patients are diagnosed at an advanced stage when the tumor is unresectable, due to poor screening tools and lack of symptoms during early stage disease, especially in developing countries [4]. HCC is intrinsically resistant to a number of cytotoxic agents [5], and currently the multi-kinase inhibitor sorafenib is the only approved systemic chemotherapeutic agent for the treatment of HCC. However, sorafenib treatment has been shown to give only a small survival advantage (~2.8 months) compared to supportive care for patients with advanced stage HCC [6]. Additionally, acquired resistance to sorafenib over the course of therapy is likely [7], thus, it is clear that the development of new systemic treatment options for patients with HCC is a growing priority.
One hallmark of HCC is a high tumor heterogeneity, including the presence of HCC tumor-initiating stem cells within tumors, which makes the design of new treatment modalities especially difficult [8]. A number of pathways have been implicated in the maintenance of HCC stem cells including RAF/MEK/ERK; PI3K/AKT/mTOR; and wnt/β-catenin signaling [9]. Epithelial cell adhesion molecule (EpCAM) is a marker of hepatic progenitor cells, as well as HCC tumor-initiating cells and aggressive disease [10, 11]. EpCAM is also a direct down-stream transcriptional target of wnt/β-catenin signaling [12], demonstrating that the wnt/β-catenin signaling pathway is an especially attractive target for the development of new chemotherapeutics.
The repositioning of non-cancer drugs by screening for novel anti-cancer activity provides a powerful strategy to rapidly identify new cancer therapeutics from the existing pool of small molecules that have already undergone rigorous testing via clinical trials for human safety, thus potentially fast-tracking the development and approval process, while reducing risk [13]. Recently, in a high-throughput drug screen designed to identify inhibitors of EpCAM dependent growth, our laboratory discovered that an FDA-approved drug pimozide (PMZ), a psychotropic dopamine receptor antagonist [14], preferentially inhibited proliferation in several EpCAM expressing HCC cell lines, compared to EpCAM negative HCC cell lines [15].
Given that EpCAM is a functional downstream target of wnt/β-catenin signaling and that PMZ can inhibit EpCAM dependent growth, we hypothesized that PMZ may act as a novel inhibitor of the wnt/β-catenin signaling pathway. Accumulation of β-catenin protein, required for active β-catenin signaling, is regulated by phosphorylation of Ser45 by casein kinase 1 (CK1), followed by phosphorylation of residues Ser33, Ser37 and Thr41 by glycogen synthase kinase 3 (GSK3), which targets β-catenin for ubiquitination and degradation. Stabilization of β-catenin protein in the cytoplasm, followed by translocation into the nucleus and transcriptional activation of down-stream target genes, is influenced by additional phosphorylation events. Several phosphorylated residues, including Ser191/Ser605 by JNK2, Ser552 by AKT, and Ser675 by protein kinase A (PKA), are important for β-catenin stabilization and nuclear localization (reviewed in [16]). In this study, we examined the ability of PMZ to inhibit wnt/β-catenin signaling using Hep3B and HepG2 HCC cell lines, both of which contain a homogenous population of cells that are positive for the HCC stem-cell marker EpCAM [12]. Hep3B cells contain non-mutated β-catenin protein with active wnt/β-catenin signaling [12], but HepG2 cells contain an exon 3-4 truncation mutation in the CTNNB1 gene. This truncation corresponds to amino acids 25-140 in the β-catenin protein, and thus removal of the key regulatory sites discussed above, which allows for the aberrant accumulation of β-catenin for constitutive activation of the wnt/β-catenin pathway [16, 17]. We found that treatment with PMZ inhibited cellular proliferation, viability, colony forming ability, and spheroid formation, while increasing apoptosis, in both Hep3B and HepG2 cells. PMZ also down-regulated the transcriptional activity, gene expression and protein expression of β-catenin and EpCAM. Therefore, PMZ likely acts as a novel inhibitor of the wnt/β-catenin signaling pathway, and may provide a viable strategy for the treatment of aggressive HCC.
Results and Discussion
Pimozide reduces cellular proliferation and viability and increases apoptosis induction in HCC cells
To characterize the effect of PMZ on wnt/β-catenin signaling, we first examined HCC cell growth in the presence of PMZ and determined an optimal PMZ treatment concentration for our studies using Hep3B and HepG2 HCC cell lines with activated wnt/β-catenin signaling. As a control, we also examined the effect of PMZ on the growth of EpCAM(-) Sk-Hep-1 hepatoma cell line. PMZ treatment inhibited cellular proliferation in all cell lines in a dose-dependent manner following 48-hour treatment, but with greater potency in the EpCAM(+) cell lines (Fig 1A). We chose to use 10 µM PMZ for subsequent studies, as 10 µM treatment occurred at ~IC60-IC70 concentration in both EpCAM(+) cell lines, but only the ~IC25 in Sk-Hep-1 cells.
Pimozide treatment decreases cellular proliferation and cell viability and increases apoptosis induction in EpCAM (+) cells. (A) Representative dose-response curves show inhibition of cellular proliferation following PMZ treatment in Hep3B (dashed line, open square points), HepG2 cells (solid line, open diamond points), and Sk-Hep-1 cells (dotted line, open circle points). (B) Representative graphs showing cell index with DMSO (black bars) or PMZ (gray bars) treatment in Hep3B, HepG2, Sk-Hep-1 and HHT4 cells at 6 (left panel), 12 (middle panel) and 24 hours (left panel). (C) Representative bar graphs showing the relative induction of active caspase 3/7 in Hep3B (left panel) and HepG2 cells (right panel) following DMSO (black bars) or PMZ (gray bars) treatment. Error bars represent ± SD, with at least triplicate determinations per point. * p<0.05, ** p<0.01, # <0.0001.
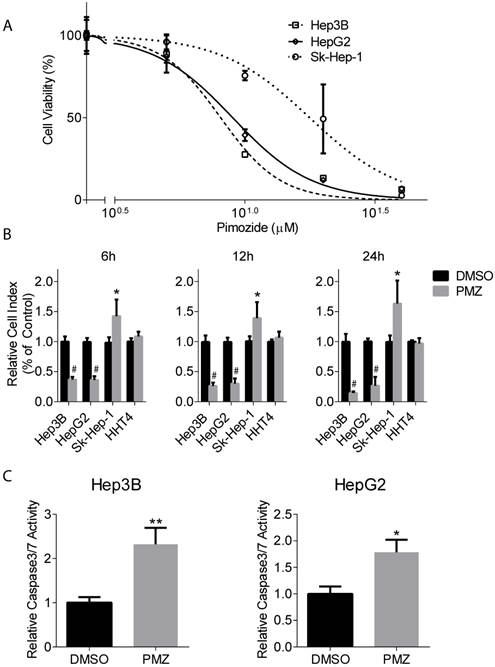
Next, we examined the effect of PMZ on HCC cell growth in real-time. We used the xCELLigence system to measure cellular viability in the presence or absence of PMZ at time points prior to 48 hours, which examines cellular viability by calculating cell index, a dimensionless parameter that measures the relative change in electrical impedance when cells are seeded in xCELLigence plates, consisting of culture wells containing electrodes. Changes in cell morphology, cell adhesion and cell viability lead to changes in cell index. We found that PMZ was effective in reducing cellular viability in both Hep3B and HepG2 cells, with a significant decrease in cell index occurring as soon as 6 hours following treatment (Fig 1B). As a control, we also examined the effect of PMZ on the growth of Sk-Hep-1 cells and immortalized normal human hepatocyte cells HHT4, and found that a similar decrease in cell index was not demonstrated (Fig 1B). Finally, we examined apoptosis induction in each cell line following PMZ treatment. We found an increased formation of active caspase 3/7, in both Hep3B and HepG2 cells, compared to vehicle control (Fig 1C), indicating that PMZ treatment causes a reduction in cellular proliferation and viability, possibly by apoptosis. In sum, these results show that PMZ exhibits anti-cancer activity, by inhibiting cancer cell growth and inducing apoptotic cell death in HCC cells, and may thus prove to be a useful strategy for targeting aggressive stem cell-like HCC.
Pimozide reduces colony formation and spheroid formation in HCC cells
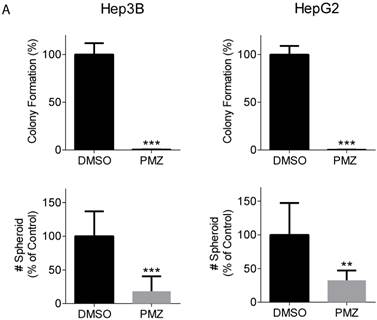
Clonogenic potential is an important feature that defines the cancer stem cell population, and the wnt/β-catenin signaling pathway has been shown to be important for maintaining stemness in HCC cancer stem cells [18, 19]. Thus, we examined the ability of PMZ to inhibit clonogenicity and spheroid formation of HCC cells. Treatment with PMZ nearly abrogated colony formation and clonogenic potential in both cell lines (Fig 2A). Additionally, we used the three-dimensional organotypic culture model for supporting spheroid growth developed in our laboratory, which is based on the AlgiMatrix culture system. This model recapitulates the biological features exhibited by cancer cells in vivo. We previously demonstrated that activated wnt/β-catenin signaling is required for formation of EpCAM(+) HCC spheroids [20], thus we used this system to examine the effect of PMZ on spheroid growth. We found that PMZ significantly reduced the number of spheroids formed in both cell lines (Fig 2B). These results provide further evidence that PMZ disrupts HCC stemness, likely by inhibiting wnt/β-catenin signaling.
Pimozide reduces clonogenicity and stemness of EpCAM(+) cells. (A) Representative bar-graphs demonstrating abrogated clonogenic potential, as measured by colony formation assay, in Hep3B (left panel) and HepG2 cells (right panel), following PMZ treatment. (B) Representative bar-graphs showing reduction of EpCAM-dependent spheroid formation in Hep3B (left panel) and HepG2 cells (right panel), following DMSO (black bars) or PMZ (gray bars) treatment. Error bars represent ± SD, with at least triplicate determinations per point. ** p<0.01, *** p<0.001.
Pimozide reduces β-catenin and EpCAM transcriptional activity and gene expression
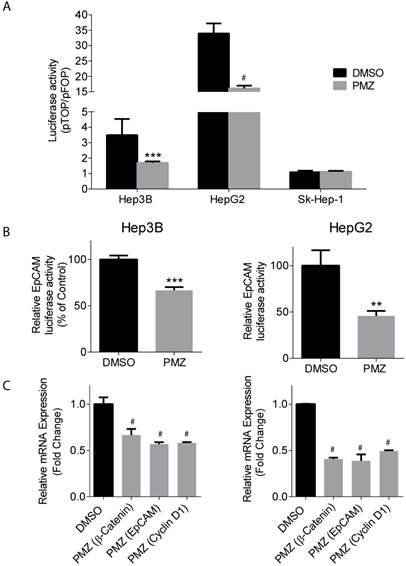
Upon activation of the wnt pathway, β-catenin is stabilized in the cytoplasm, translocates to the nucleus, and then interacts with the T-cell factor/lymphoid enhancement factor (TCF/LEF) family of transcription factors to activate the transcription of down-stream target genes, including EpCAM [21]. Thus, we sought to determine if treatment of Hep3B and HepG2 cells with PMZ could down-regulate β-catenin and EpCAM transcriptional activity and gene expression, indicating pathway inhibition by PMZ treatment. Using a pTOP reporter, which contains TCF/LEF sites upstream of the luciferase reporter, in combination with a control pFOP control reporter, containing mutated TCF/LEF sites upstream of the luciferase reporter, we found that PMZ significantly inhibited luciferase signal, and thus β-catenin transcriptional activity, in both Hep3B and HepG2 cell lines compared to control. We also found that Sk-Hep-1 did not exhibit β-catenin transcriptional activity, as noted by a pTOP/pFOP ratio of ~1, which was not affected by PMZ treatment. (Fig 3A). EpCAM promoter activity was significantly down-regulated, as measured by an EpCAM luciferase reporter, upon treatment with PMZ (Fig 3B). Using qRT-PCR analysis, we determined that PMZ effectively down-regulated the mRNA expression of β-catenin and EpCAM following treatment in both cell lines (Fig 3C). In addition, we examined the mRNA expression level of Cyclin D1, a direct target of the wnt/β-catenin pathway, which promotes cell proliferation via uncontrolled cell cycle progression [22]. PMZ treatment reduced mRNA expression level of Cyclin D1 in both Hep3B and HepG2 cells (Fig 3C). These results indicate that PMZ can effectively down-regulate the transcription of both β-catenin and its down-stream targets EpCAM and Cyclin D1. Thus PMZ is likely an effective inhibitor of the wnt/β-catenin signaling axis.
Pimozide decreases transcriptional activity and gene expression of β-catenin and wnt/ β-catenin signaling targets. (A) Representative bar-graphs showing reduction of β-catenin transcriptional activity, as measured by pTOP/pFOP luciferase assay, in Hep3B, HepG2 and Sk-Hep-1 cells following DMSO (black bars) or PMZ treatment (gray bars). (B) Representative bar-graphs showing reduction of EpCAM transcriptional activity, as measured by luciferase assay, in Hep3B (left panel) and HepG2 cells (right panel), following DMSO (black bars) or PMZ (gray bars) treatment. (C) Representative bar-graphs showing reduction of mRNA expression in Hep3B (left panel) and HepG2 cells (right panel), as measured by qRT-PCR, in β-catenin, EpCAM and Cyclin D1 following DMSO (black bars) or PMZ (gray bars) treatment. Error bars represent ± SD, with at least triplicate determinations per point. ** p<0.01, *** p<0.001, # p<0.0001.
Pimozide decreases β-catenin and EpCAM protein expression
Finally, to confirm that PMZ effectively down-regulates the wnt/β-catenin pathway at the protein level, we first determined if EpCAM protein expression is decreased following PMZ treatment. We found that in both Hep3B and HepG2 cell lines, EpCAM protein was down-regulated following treatment with PMZ (Fig 4A), indicating down-stream inhibition of wnt/β-catenin signaling.
Pimozide reduces EpCAM and β-catenin protein levels in EpCAM(+) cells. (A) Representative Western blots showing reduction in EpCAM protein expression in Hep3B and HepG2 cells following PMZ treatment. β-catenin and phosphorylated β-catenin (S675) protein expression is reduced following PMZ treatment in HepG2 cells. (B) Representative Western blot showing reduction of nuclear β-catenin following PMZ treatment in Hep3B cells. Phosphorylated GSK3β abundance is reduced with PMZ treatment.
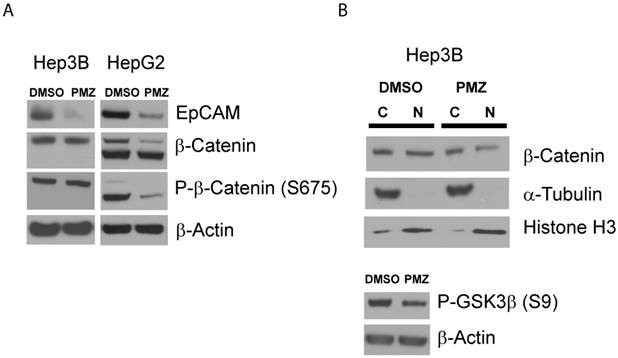
We next sought to investigate if PMZ treatment could down-regulate β-catenin at the protein level. The abundance of β-catenin is controlled by a number of post-translational modifications, with key regulatory sites removed in β-catenin protein expressed in HepG2 via truncation mutation. Therefore, due to the differences in β-catenin protein in Hep3B and HepG2 cells, it is plausible that each cell line has adapted differential regulation of relative β-catenin abundance, thus PMZ may affect these cells in different ways. Therefore, we first chose to examine if PMZ treatment affects the global abundance of β-catenin in the whole-cell lysate of Hep3B and HepG2 cells, as well as the abundance of phospho-β-catenin Ser675, which stabilizes β-catenin by inhibiting destruction by ubiquitination [23]. We elected to examine phospho-β-catenin Ser675, because phosphorylation of β-catenin by PKA during wnt signaling has been confirmed to play an important role in liver regeneration and hepatocyte proliferation [24, 25]. We found that total β-catenin protein was down-regulated following PMZ treatment in HepG2 cells, but remained unchanged in Hep3B cells (Fig 4A). PMZ treatment also decreased the total abundance of phospho-β-catenin Ser675 in HepG2 cells, but not Hep3B cells (Fig 4A). To further investigate the effect of PMZ on β-catenin expression in Hep3B cells, cytoplasmic-nuclear fractionation was used to examine β-catenin sub-cellular localization, which showed that PMZ reduced the amount of β-catenin in the nucleus (Fig 4B).
One possible explanation for these results is that PMZ inhibits the PKA signaling cascade. In Hep3B cells, although the total amount of β-catenin and phospho-β-catenin appear to be unchanged following PMZ treatment, nuclear levels are decreased. PKA has been shown to phosphorylate GSK3α and β at Ser21 and Ser9, respectively, thus activating β-catenin signaling by preventing GSK3 from phosphorylating β-catenin at Ser33, Ser37 and Thr41, which leads to β-catenin ubiquitination and destruction [26]. Therefore, treatment with PMZ may inhibit PKA signaling, thereby preventing the inhibition of GSK3 and leading to reduction of β-catenin accumulation in the nucleus due to ubiquitination and degradation signaled by GSK3 phosphorylation of β-catenin. Indeed, when we examined the abundance of phosphorylated GSK3β, which has been well characterized in HCC [27], PMZ treatment reduced phospho-GSK3β (Fig 4B).
In HepG2 cells, β-catenin abundance is not controlled by phosphorylation by GSK3 due to the truncation mutation, and thus may have adapted alternative mechanisms to regulate β-catenin abundance. PMZ reduces total phospho-β-catenin Ser675 protein levels in HepG2, indicating possible inhibition of the PKA signaling cascade, leading to the observed reduction of total β-catenin abundance by ubiquitination and proteosomal destruction. One report indicates that phosphorylation of β-catenin at Ser675 by PKA may modulate signaling through a non-canonical pathway, and that this phosphorylation does not affect the phosphorylation of β-catenin by GSK3, nor does it affect the stability or intracellular location of full-length β-catenin [28]. These results could explain why β-catenin and phospho-β-catenin in HepG2 is affected by PMZ treatment, which is not under GSK3 control or regulation, whereas in Hep3B, GSK3 is the main point of control of β-catenin accumulation, thus why the abundance of phospho-β-catenin is not affected following treatment.
It has been reported that β-catenin protein can act as a co-transcription factor for the transcription of its own promoter [29]. Therefore, the decrease in β-catenin protein seen in both HCC cell lines, potentially caused by PKA signaling inhibition, could directly lead to inhibition of β-catenin gene expression as seen above. Additional studies will be required to confirm if PMZ inhibits the PKA signaling cascade and to elucidate the mechanism by which PMZ affects β-catenin protein expression.
The results of this study have clearly demonstrated that treatment with PMZ is effective in reducing cancer cell viability, possibly through down-regulation of EpCAM expression. As EpCAM is critical for HCC tumorigenesis and maintenance of the “stemness” of cancer cells, which may have increased resistance to chemotherapeutic intervention [12, 19, 30], PMZ provides a useful strategy to target cancer stem cell-like HCC, possibly through inhibition of the PKA signaling cascade. Further investigation will be required to elucidate the exact mechanism by which PMZ down-regulates wnt/β-catenin signaling. Additional off-target effects in which PMZ induces anti-cancer activity should also be examined in order to more fully characterize the effect that PMZ elicits in malignant cells, such as the growth inhibitory effect seen in EpCAM(-) Sk-Hep-1 cells. For example, PMZ has gained attention as an anti-cancer agent by acting as a STAT5 inhibitor in chronic myelogenous leukemia cells [31], as well as an inhibitor of USP/UAF1 deubiquitinase complex in non-small cell lung cancer cells [32]. These mechanisms may have importance in HCC cells or other cancers, therefore, alternative mechanisms may also play an important role in the anti-cancer effect that PMZ imparts on HCC cells, and are worth investigating in future studies.
In recent years, there has been an increased interest in repositioning current FDA-approved drugs, with main indications that are not related to oncology, as anti-cancer agents [33]. Such strategies have the advantage of rapid discovery of interesting molecular entities while decreasing time-to-approval [13]. Clinically, PMZ has been used in an oral preparation as an antipsychotic drug, and has indications for the treatment of Tourette syndrome, schizophrenia, and psychosis, among other psychiatric disorders, by acting as an antagonist of dopamine receptors. PMZ is extensively metabolized by the liver, with no reports of hepatotoxicity or acute liver injury side-effects in humans (http://livertox.nih.gov/Pimozide.htm), and our results demonstrated that growth in HHT4 immortalized primary hepatocytes was not affected by PMZ treatment. It should be noted however, that PMZ has presented with a number of serious side-effects that are extrapyramidal in nature, including akathisia and tardive dyskinesia, as well as cardiac side-effects [14]. A study of the pharmacokinetics of PMZ showed that a single 24 mg oral dose could reach a peak plasma concentration of 19 ng/mL, corresponding to a concentration of ~0.04 µM, two orders of magnitude lower than the concentration examined in this study [34]. As such, PMZ in its current formulation is not likely suitable for direct use as an anti-cancer therapy for HCC treatment, but several strategies could be employed to translate PMZ into oncologic clinical use. For example, peak plasma concentration could be increased by creating intravenous formulations of PMZ. A high intra-tumor drug concentration could also be achieved by incorporating PMZ into transcatheter arterial chemoembolization, a commonly used treatment for HCC in which a high dose of chemotherapeutic is injected directly into the tumor via the hepatic artery [35]. In order to lower the therapeutic window, and thus the concentration of PMZ required for efficacy as an anti-cancer therapy, combination therapy with other available chemotherapeutics could be considered. Additionally, it would be useful to examine PMZ analogs, with the possibility of discovering an analog that exhibits anti-cancer effects, but with less potent dopamine receptor antagonist activity. Thus, continued examination of the effect of PMZ in HCC cancer cells warrants further investigation.
Conclusion
This study demonstrated that PMZ decreases the viability of HCC cell lines by down-regulating wnt/β-catenin signaling and EpCAM gene and protein expression. Further investigation of PMZ as a potential anti-cancer therapeutic for translation into clinical medicine for targeting aggressive stem cell-like HCC should be considered.
Methods and Materials
Cell lines
For all experiments, Hep3B, HepG2 and Sk-Hep-1 cells were cultured in Minimum Essential Medium (Life Technologies) supplemented with 10% fetal bovine serum, penicillin/streptomycin, glutamine, non-essential amino acids and sodium pyruvate. HHT4 cells were cultured in HBM basal medium (Lonza) supplemented with HCM SingleQuot (Lonza), 20% knockout serum replacement (Invitrogen), penicillin/streptomycin and glutamine. Unless otherwise stated, all cell lines were seeded for experiments and were incubated at 37 °C overnight prior to experimentation. All cell lines used in this study have been authenticated by short tandem repeat (STR) analysis at the Frederick National Laboratory for Cancer Research.
Cellular proliferation
Cells were treated with various concentrations of PMZ (Sigma) in DMSO (0-40 µM) in triplicate for 48 hours. Cellular proliferation was measured using Calcein AM (Invitrogen) solution in PBS (2 ng/mL final concentration). Following 30-minute incubation at 37 °C, fluorescence was measured using a FLUOstar Omega plate reader (Ex/Em 485/520 nm).
Real-Time cell viability
DMSO or 10 µM PMZ in 100 µL media was added to each well of E-Plate VIEW xCELLigence plates (ACEA Biosciences) and incubated at 37 °C for 1 hour. Background measurements were taken and then 100 µL of cells were added to each well of the E-Plate and placed in the xCELLigence instrument. Cell index was measured in quadruplicate every 15 minutes for 48 hours.
Apoptosis
Cells were treated with 10 µM PMZ or DMSO control in triplicate for 48 hours. Cells were collected and counted to ensure equal cell number in each measurement. Apoptosis was measured using the Apo-ONE Homogenous Caspase-3/7 Assay kit (Promega), according to the manufacturer's instructions. Following addition of the Caspase 3/7 reagent to each measurement well, the measurement plate was incubated at room temperature for 4-6 hours prior to fluorescence measurement (Ex/Em: 499/521) using a FLUOstar Omega plate reader.
Colony Formation Assay
Cells were seeded in 6-well plates and treated with DMSO or 10 µM PMZ in triplicate for 10 days. Colonies were fixed with MeOH, stained with crystal violet and counted manually.
Spheroid Assay
Cells were seeded into 6-well AlgiMatrix 3D cell culture system plates (Invitrogen), with 1x106 cells/well and 10% AlgiMatrix firming buffer (Invitrogen), according to the manufacturer's instructions. At the time of inoculation onto the AlgiMatrix sponges, DMSO or 10 µM PMZ was added to the culture media. Cells were incubated for 11 days, with media changed on day 4 and day 8 of culture. Spheroid number was counted by dissolving AlgiMatrix sponges with AlgiMatrix dissolving buffer (Invitrogen), collection of spheres and seeding onto a 6-well plate. Spheres were counted manually using a microscope, with 5 determinations for each condition.
Luciferase Assay
Cells were co-transfected using Lipofectamine 2000 (Life Technologies) with 500 ng M50 Super 8× TOPFlash or M51 Super 8× FOPFlash (Addgene), and 50 ng pRL-null Renilla luciferase to measure β-catenin transcriptional activity. For EpCAM transcriptional activity, cells were co-transfected with 500 ng pGL3-EpCAM2.2 and 50 ng pRL-null Renilla luciferase. Cells were then treated with DMSO or 10 µM PMZ in triplicate for 24 hours. The Dual-Luciferase Reporter Assay System (Promega) was used to determine firefly and renilla luciferase activity according to the manufacturer's instructions.
qRT-PCR
Cells were treated with DMSO or 10 µM PMZ for 6 hours. mRNA was isolated using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription of mRNA to cDNA was performed with 2 µg total RNA in 10 µL of nuclease-free water using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer's instructions. qRT-PCR was performed using TaqMan Gene Expression assays (Life Technologies, EpCAM: Hs00158980_m1; CTNNB1: Hs00170025_m1; CCND1: Hs00765553_m1) with EagleTaq Universal MMX master mix (Roche). 18S (Applied Biosystems) was used as the endogenous control.
Western Blot
For examination of total protein lysate, cells were treated with DMSO or 10 µM PMZ for 24 hours and total lysate was isolated using RIPA buffer (Cell Signaling) according to the manufacturer's instructions. For nuclear/cytoplasmic fractionation, cells were treated with DMSO or 10 µM PMZ for 6 hours and cytoplasmic and nuclear fractions were collected using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo) according to the manufacturer's instructions. Lysate was separated using NuPAGE 4-12% Bis-Tris (Novex) and transferred to a nitrocellulose membrane using the iBlot system (Life Technologies). Protein detection was performed using the following antibodies: monoclonal anti-EpCAM (R&D Systems), monoclonal anti-β-catenin (BD Transduction), monoclonal anti-phospho-β-catenin (S675) (Cell Signaling), monoclonal anti-phospho-GSK3β (S9) (Cell Signaling), monoclonal anti-β-actin (Sigma-Aldrich), monoclonal anti-α-Tubulin (Sigma-Aldrich), and monoclonal anti-Histone H3 (Sigma-Aldrich).
Statistical Analysis
GraphPad Prism software was used for all statistical calculations. Un-paired two-tailed student's t-tests with a significance level of p<0.05 were used for all statistical analyses. F-test was performed for each analysis and Welch's correction was applied when variances were significantly different.
Acknowledgements
This work was supported by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research and Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Abbreviations
CK1: casein kinase 1; EpCAM: epithelial cell adhesion marker; GSK3: glycogen synthase kinase 3; HCC: hepatocellular carcinoma; PKA: protein kinase A; PMZ: pimozide; TCF/LEF: T-cell factor/lymphoid enhancement factor
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer JClin. 2013;63:11-30
2. Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23:2892-9
3. Raza A, Sood GK. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World J Gastroenterol. 2014;20:4115-27
4. Ferenci P, Fried M, Labrecque D, Bruix J, Sherman M, Omata M. et al. Hepatocellular carcinoma (HCC): a global perspective. Journal of clinical gastroenterology. 2010;44:239-45
5. Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? Journal of hepatology. 2012;56:686-95
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. NEnglJ Med. 2008;359:378-90
7. Sun H, Zhu MS, Wu WR, Shi XD, Xu LB. Role of anti-angiogenesis therapy in the management of hepatocellular carcinoma: The jury is still out. World J Hepatol. 2014;6:830-5
8. Wang XW, Thorrgeirsson SS. The Biological and clinical challenge of liver cancer heterogeneity. Hepatic Oncology. 2014;1:5
9. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989-5005
10. Dan YY, Riehle KJ, Lazaro C, Teoh N, Haque J, Campbell JS. et al. Isolation of multipotent progenitor cells from human fetal liver capable of differentiating into liver and mesenchymal lineages. ProcNatlAcadSci USA. 2006;103:9912-7
11. Schmelzer E, Wauthier E, Reid LM. The Phenotypes of Pluripotent Human Hepatic Progenitors. Stem Cells. 2006;24:1852-8
12. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-β-catenin signaling in hepatocellular carcinoma. Cancer research. 2007;67:10831-9
13. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673-83
14. Pinder RM, Brogden RN, Swayer R, Speight TM, Spencer R, Avery GS. Pimozide: a review of its pharmacological properties and therapeutic uses in psychiatry. Drugs. 1976;12:1-40
15. Henrich CJ, Budhu A, Yu Z, Evans JR, Goncharova EI, Ransom TT. et al. High-throughput screening for identification of inhibitors of EpCAM-dependent growth of hepatocellular carcinoma cells. ChemBiolDrug Des. 2013;82:131-9
16. Gao C, Xiao G, Hu J. Regulation of Wnt/beta-catenin signaling by posttranslational modifications. Cell Biosci. 2014;4:13
17. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O. et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. ProcNatlAcadSci USA. 1998;95:8847-51
18. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
19. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-24
20. Takai A, Fako V, Dang H, Forgues M, Yu Z, Budhu A. et al. Three-dimensional Organotypic Culture Models of Human Hepatocellular Carcinoma. Scientific reports. 2016;6:21174
21. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-80
22. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R. et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5522-7
23. Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Molecular and cellular biology. 2005;25:9063-72
24. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL. et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136-47
25. Fanti M, Singh S, Ledda-Columbano GM, Columbano A, Monga SP. Tri-iodothyronine induces hepatocyte proliferation by protein kinase A-dependent beta-catenin activation in rodents. Hepatology. 2014;59:2309-20
26. Fang X, Yu SX, Lu Y, Bast RC Jr, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11960-5
27. Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866-84
28. Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971-6
29. Bandapalli OR, Dihlmann S, Helwa R, Macher-Goeppinger S, Weitz J, Schirmacher P. et al. Transcriptional activation of the beta-catenin gene at the invasion front of colorectal liver metastases. J Pathol. 2009;218:370-9
30. Dolle L, Theise ND, Schmelzer E, Boulter L, Gires O, van Grunsven LA. EpCAM and the biology of hepatic stem/progenitor cells. American journal of physiology Gastrointestinal and liver physiology. 2015;308:G233-G50
31. Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB. et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421-9
32. Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J. et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390-400
33. Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. International journal of biological sciences. 2014;10:654-63
34. McCreadie RG, Heykants JJ, Chalmers A, Anderson AM. Plasma pimozide profiles in chronic schizophrenics. Br J Clin Pharmacol. 1979;7:533-4
35. Nishikawa H, Kita R, Kimura T, Osaki Y. Transcatheter arterial embolic therapies for hepatocellular carcinoma: a literature review. Anticancer Res. 2014;34:6877-86
Author contact
![]() Corresponding author: Anuradha Budhu or Xin Wei Wang, National Cancer Institute, 37 Convent Drive, Building 37, Room 3044A, Bethesda, Maryland 20892; Email: xw3ugov; Phone: 301-496-2099.
Corresponding author: Anuradha Budhu or Xin Wei Wang, National Cancer Institute, 37 Convent Drive, Building 37, Room 3044A, Bethesda, Maryland 20892; Email: xw3ugov; Phone: 301-496-2099.