ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2016; 12(7):861-871. doi:10.7150/ijbs.14213 This issue Cite
Research Paper
Asiatic Acid Protects against Cardiac Hypertrophy through Activating AMPKα Signalling Pathway
Zhen-Guo Ma1,2, Jia Dai1,2, Wen-Ying Wei1,2, Wen-Bin Zhang1,2, Si-Chi Xu1,2, Hai-Han Liao1,2, Zheng Yang1,2, Qi-Zhu Tang1,2 ![]()
1. Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, China;
2. Cardiovascular Research Institute of Wuhan University, Wuhan, China.
Abstract

Background: AMPactivated protein kinase α (AMPKα) is closely involved in the process of cardiac hypertrophy. Asiatic acid (AA), a pentacyclic triterpene, was found to activate AMPKα in our preliminary experiment. However, its effects on the development of cardiac hypertrophy remain unclear. The present study was to determine whether AA could protect against cardiac hypertrophy.
Methods: Mice subjected to aortic banding were orally given AA (10 or 30mg/kg) for 7 weeks. In the inhibitory experiment, Compound C was intraperitoneally injected for 3 weeks after surgery.
Results: Our results showed that AA markedly inhibited hypertrophic responses induced by pressure overload or angiotensin II. AA also suppressed cardiac fibrosis in vivo and accumulation of collagen in vitro. The protective effects of AA were mediated by activation of AMPKα and inhibition of the mammalian target of rapamycin (mTOR) pathway and extracellular signal-regulated kinase (ERK) in vivo and in vitro. However, AA lost the protective effects after AMPKα inhibition or gene deficiency.
Conclusions: AA protects against cardiac hypertrophy by activating AMPKα, and has the potential to be used for the treatment of heart failure.
Keywords: AMPKα, Asiatic acid, Cardiac fibrosis, Cardiac hypertrophy.
1. Introduction
Cardiac hypertrophy is characterized by an increase in size of cardiac myocyte and protein synthesis, abnormal expression of fetal gene, and excessive deposition of extracellular matrix [1-2]. Cardiac hypertrophy, which can result in heart failure and ventricular arrhythmias, is a risk factor for cardiovascular events and death [3]. The precise mechanisms controlling the process of cardiac hypertrophy are incompletely understood, but emerging data implicate the key roles for the mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK) [4-5].
Activated mTOR promotes the activation of ribosomal protein S6 kinase (P70S6K), directly regulating protein synthesis and contributing to the process of cardiac hypertrophy [6]. Conversely, inhibition of mTOR signaling with rapamycin attenuates established cardiac hypertrophy [4]. ERK regulates development of hypertrophy through the induction of the expression of hypertrophy-related genes [5]. It is reported that mTOR and ERK can be regulated by AMPactivated protein kinase α (AMPKα), which is an energy sensor of heart [6-7]. AMPKα is also closely involved in the process of cardiac hypertrophy. Activation of AMPKα by pharmacological compounds have been shown to inhibit cardiac hypertrophy [8-9]. Therefore, drugs that selectively activate AMPKα and inhibit mTOR and ERK could be of great therapeutic interest for the treatment of cardiac hypertrophy.
Asiatic acid (AA), a chemically characterized pentacyclic triterpene, naturally exists in many edible vegetables and fruits including brown mustard, basil, daylily, hawthorn fruit and spinach [10-11]. Previous study found that dietary intake of AA increased their bioavailability in the heart [10]. Accumulating evidence indicated that AA possessed a number of pharmacological activities. AA has been reported to reduce islet fibrosis, suppress mitochondria-mediated inflammasome activation, ameliorate hepatic lipid accumulation and insulin resistance, and attenuate infarct volume after focal cerebral ischemia [12-15]. AA also alleviated cardiovascular remodeling in hypertensive rat and protected against diabetic cardiomyopathy [16-17]. In our preliminary experiment, we found AA activated AMPKα in the heart. However, the effects of AA on cardiac hypertrophy and the precise mechanisms still remain unclear. Therefore, this study was to determine whether AA could alleviate hypertrophy of neonatal rat cardiac myocytes and cardiac hypertrophy induced by long-time pressure overload, as well as to reveal the precise mechanisms underlying the putative effects.
2. Materials and methods
2.1 Materials
AA was purchased from Shanghai Winherb Medical Co. (Shanghai, China), who specified above 98.5% purity confirmed by HPLC. Angiotensin II (Ang II, A9525) and Compound C (CpC, P5499) were purchased from Sigma-Aldrich. Anti-GAPDH (sc-25778) was obtained from Santa Cruz Biotechnology. Anti-AMPKα (#2603P), anti-phospho-AMPKα (#2535), anti-phospho-mTOR (#2971), anti-mTOR (#2983), anti-P70S6K (#2708), anti-phospho-P70S6K (#9234P), anti-ribosomal protein S6 (S6, #2217), anti-phospho-S6 (#5364P), anti-acetyl-CoA carboxylase (ACC, #3676), anti-phospho-ACC (#3661), anti-ERK (#4695), and anti-phospho-ERK(#4370) were obtained from Cell Signalling Technology. Anti-α-actin was purchased from Millipore. The secondary antibody was purchased from LI-COR Biosciences (at 1:10,000 dilution). All other chemicals were of analytical grade.
2.2 Animals and treatments
All animal experimental procedures were approved by the Guidelines for the Care and Use of Laboratory Animals of the Chinese Animal Welfare Committee and the guidelines of our hospital. All surgeries and subsequent analyses were performed in a blind fashion for all groups. C57/B6 mice (8-10 week-old; male body weight: 25.5±2g) were subjected to aortic banding (AB) according to the previous article [18]. The source of AMPKα2 knockout mice was described in our previous article [19]. Doppler analysis was performed to ensure the appropriate constriction of the aorta had been induced. AA was dissolved in 0.1% DMSO for in vitro experiments and in 1% CMCNa for in vivo experiments. One week after being subjected to AB, mice were given AA (10 mg/kg or 30mg/kg) for 7 weeks, and animals in the control group were given equal volumes of vehicle. To inhibit AMPKα, CpC (20mg/kg, every other day) was injected for 3 weeks beginning from 1 week after surgery to 4 weeks after surgery. In the inhibition experiment, the AB surgery lasts for 4 weeks. Mice were subjected to AA (30mg/kg) for 3 weeks from 1 week after surgery to 4 weeks after surgery. AMPKα2 knockout mice were subjected to AA (30mg/kg) treatment for 3 weeks from 1 week after surgery to 4 weeks after surgery. At the endpoint of study, heart weight (HW)/body weight (BW) and HW/tibial length (TL) were calculated after mice were euthanized.
2.3 Echocardiography
Mice were anesthetized using 1.5% isoflurane, and then echocardiography was performed to assess left ventricle (LV) wall and chamber dimensions, and systolic and diastolic function using a Mylab 30CV (Esaote S. P.A, Genoa, Italy) equipped with a 10-MHz linear array ultrasound transducer. M-mode was obtained under anesthesia while heart rate and body temperature were maintained.
2.4 Histological examination
The arrested hearts were fixed with 10% formalin and then sectioned transversely. After rehydration, the sections (5μm) were stained with haematoxylin-eosin (HE) or picrosirius red (PSR). The sections were observed under light microscopy, the cross-sectional areas of the myocytes were determined by a digital analysis system (Image-Pro Plus 6.0). The sections were examined blind. In each group, more than 200 myocytes were outlined. Collagen content was determined by quantification of PSR-positive area per field of view using Image-Pro Plus 6.0. Six mice each group (6 fields every animal) were used to quantify PSR staining.
2.5 Western blot and real-time PCR analysis
The hearts and iced cell lysates were lysed using a RIPA buffer. The protein concentrations were determined using the BCA Protein Assay Kit. After that, the protein extracts were loaded into SDS-PAGE gels and subsequently transferred to a PVDF membrane (Millipore, IPVH00010). After incubation with primary antibodies and secondary antibodies, the membrane was visualized with an Odyssey Imaging System. cDNA was synthesized from the total RNA extracted from the frozen heart tissues using the Transcriptor First Strand cDNA Synthesis Kit (Roche, 04896866001). Quantitative RT-PCR analysis was performed using the LightCycler 480 SYBR Green 1 Master Mix (Roche, 04707516001). All primer details were provided in Table S1. The protein levels were normalized to GAPDH. The mRNA levels were analyzed with the 2-△△Ct method and normalized to GAPDH.
2.6 Cell culture and infection with recombinant adenoviral vectors
Replication-defective adenoviral vectors under the control of the U6 promoter were used in our study. Three rat shAMPKα2 constructs were obtained from Sigma-Genosys and three Ad-shAMPKα2 adenoviruses were generated by Vigene Bioscience. We selected the one that resulted in the greatest decrease of AMPKα2 expression for further experiments. Ad-shRNA, whose sequence was GCCTGCCGTCCAAAGTTGTAA, was the non-targeting control. The cultures of neonatal rat cardiac myocytes and cardiac fibroblasts were prepared respectively as described in the previous studies [20-21]. Cells were seeded in Dulbecco's modified Eagle medium (DMEM) (GIBCO, C11995) supplemented with 10% fetal bovine serum (GIBCO, 10099), penicillin (100 U/ml) and streptomycin (100 mg/ml) (GIBCO, 15140). Bromodeoxyuridine (0.1 mM) was used to prevent fibroblast contamination in neonatal rat cardiac myocytes. Purity of the cardiac fibroblast was assessed by morphologic recognition and by positive and negative staining with antibodies against Vimentin and α-actin. The purity of cultured cardiac fibroblast in our study was greater than 98%. Cardiac fibroblasts in the second and third passages were used in our studies. After 48h, the cells were infected with Ad-shAMPKα2 or Ad-shRNA for 4h at a MOI of 10. Then, these cells were thoroughly washed and synchronized using serum-free medium for 12 hours. Hypertrophy of cardiac myocytes were induced by Ang II (1μM, 24h), and cardiac fibroblasts were also subjected to Ang II (1μM, 24h) to detect the accumulation of collagen.
2.7 Immunofluorescence staining
Immunofluorescence staining was used to outline cell surface. Neonatal rat cardiomyocytes were cultured on cover slips, infected with Ad-shAMPKα2 or Ad-shRNA for 24h, followed by stimulation with 1 µM Ang II for 24h. In this study, we used α-actin to stain the cells. Cells, fixed with 4% formaldehyde, were permeabilized in 0.1% Triton X-100, and stained with anti-α-actin (1:100 dilution). Alexa Fluor 488-goat anti-mouse (Invitrogen, A11017) was the second antibody used. The cross-sectional areas were calculated blind by Image-Pro Plus 6.0. In each group, more than 100 myocytes were outlined.
2.8 Statistical analysis
The data in our study are expressed as mean ± SEM. Differences among groups were compared by one-way ANOVA, followed by post hoc Tukey test. Comparison between two groups was assessed by an unpaired Student's t-test. All in vivo, in vitro and imaging studies were performed blind. P<0.05 was considered as statistical significance.
3. Results
3.1 AA protected against cardiac hypertrophy in vivo
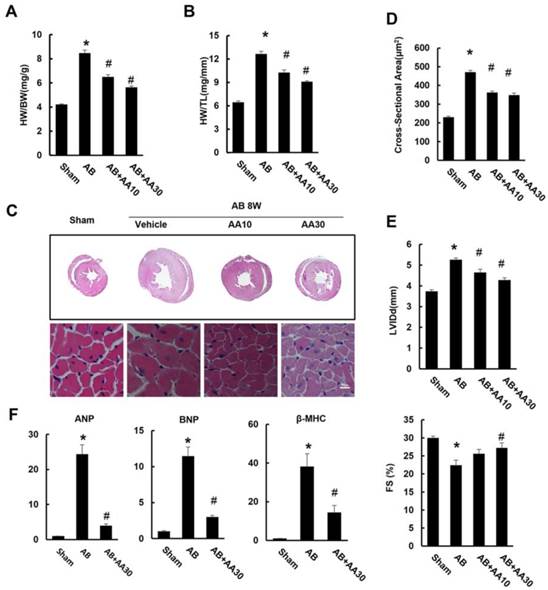
As shown by Figure 1, mice subjected to AB developed a remarkable hypertrophy, as indicated by increased ratios of HW/BW and HW/TL (Figure 1A-B). Histological examination also revealed increased cross-sectional areas of cardiomyocytes in mice with AB surgery (Figure 1C-D). Mice subjected to pressure load also developed a decline in heart function with a decrease in fractional shortening (FS), and had a significant increase in LV internal diameter at end-diastole (LVIDd) (Figure 1E). Though mice without surgery subjected to AA treatment for 7 weeks had no cardiac morphological alterations (Figure S1), mice given AA after AB surgery exhibited reduced cardiac hypertrophy (Figure 1A-E). The hypertrophic markers induced by AB, including atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC), were strikingly blunted in AA-treated mice (Figure 1F). Taken together, these data suggested that AA treatment could attenuate cardiac hypertrophy induced by chronic pressure overload.
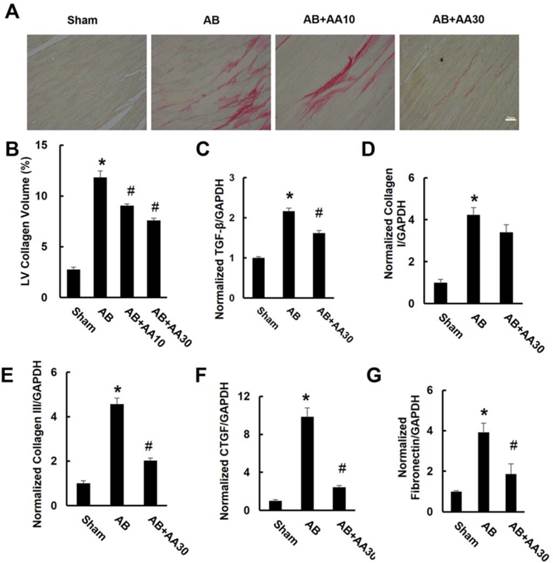
3.2 AA attenuated cardiac fibrosis induced by AB in vivo
After pressure overload, mice displayed obvious interstitial fibrosis, as indicated by increased LV collagen volume and fibrotic markers. However, the increased collagen volume was blunted by AA treatment (Figure 2A-B). This result was corroborated by subsequent analysis of mRNA levels of transforming growth factor-β (TGF-β), collagen I, collagen III, connective tissue growth factor (CTGF) and fibronectin (Figure 2C-G).
Asiatic acid (AA) prevented cardiac hypertrophy and dysfunction. A-B, Statistical results of heart weight (HW)/ body weight (BW), HW/ tibial length (TL) of the indicated groups (n=15). C, Histological results of hypertrophic heart with or without AA. D, The cross-sectional areas of myocytes (n=6). E, Echocardiographic parameters of the mice after 8 weeks of AB with or without AA treatment (n=10). F, mRNA levels of the hypertrophic markers (n=6). Compared with sham, *P < 0.05. Compared with AB, #P < 0.05.
Asiatic acid (AA) suppressed cardiac fibrosis in vivo. A, PSR staining of hypertrophic heart with or without AA (n=6). B, Collagen volume analysis of the indicated groups (n=6). C-G, mRNA levels of the fibrotic markers (n=6). Compared with sham, *P < 0.05. Compared with AB, #P < 0.05.
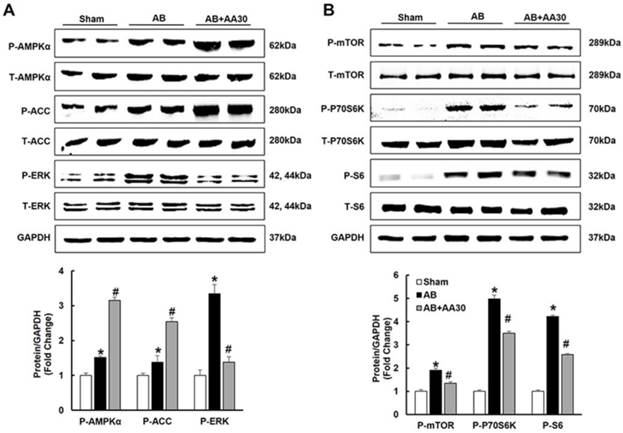
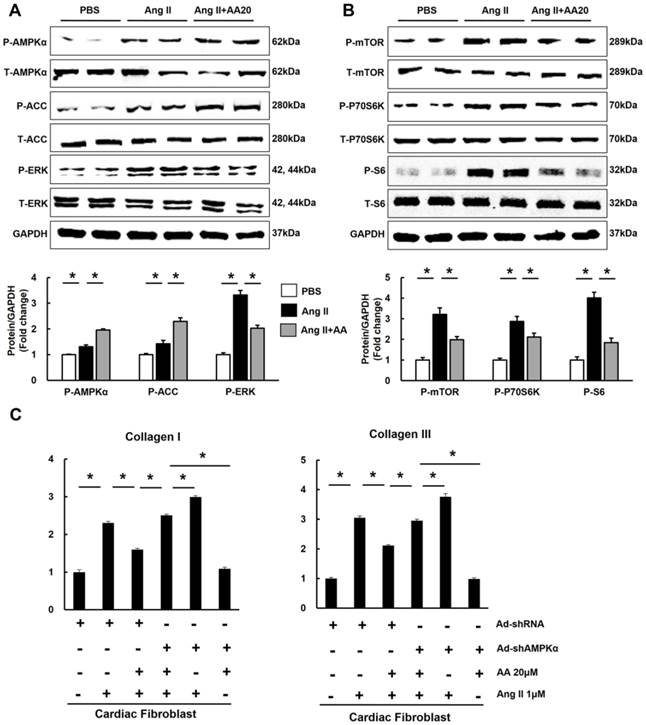
The effects of asiatic acid (AA) on AMPKα, mTOR and ERK signalling pathway. A-B, The protein levels of phosphorylated AMPKα and related targets in indicated groups (n=8). Compared with sham, *P < 0.05. Compared with AB, #P < 0.05.
3.3 AA promoted the phosphorylation of AMPKα and attenuated mTOR and ERK activation
Consistent with our preliminary finding that AA could activate AMPKα at baseline (Figure S2), we also found that AA (30mg/kg) further promoted the phosphorylation of AMPKα in the hypertrophic hearts (Figure 3A). Subsequently, the phosphorylation of ACC, which is the direct target of AMPKα and could reflect the activity of AMPKα [22], was detected. As expected, AA treatment also further promoted the phosphorylation of ACC (Figure 3A). Just as illustrated by Figure 3A-B, compared with the sham group, AB-induced phosphorylation of mTOR, P70S6K, S6 and ERK were reduced by AA treatment.
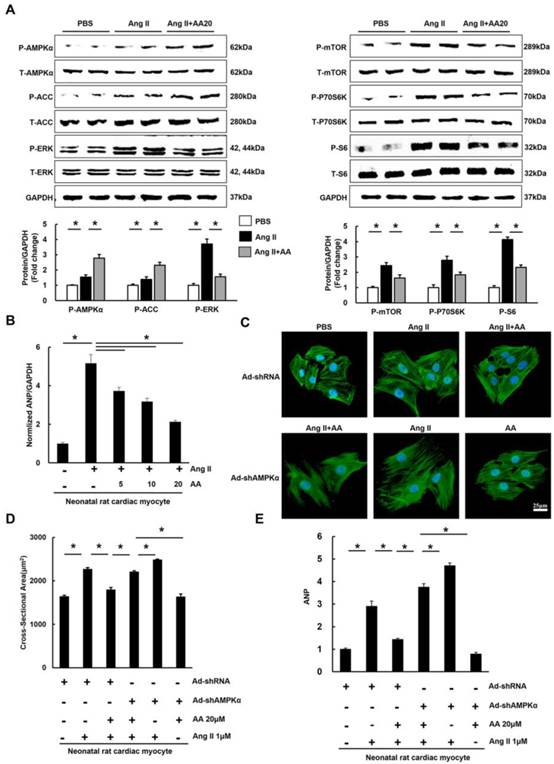
3.4 AA attenuated hypertrophy of myocytes in an AMPKα-dependent manner in vitro
To decipher the effects of AA, neonatal rat cardiac myocytes were subjected to the administration of Ang II (1μM) to induce the hypertrophic response in vitro. We first examined phosphorylated AMPKα and found that AA (20μM) further activated AMPKα, which was also reflected by the phosphorylation of ACC (Figure 4A). Consistent with the results in vivo, the Ang II-mediated increases in P-mTOR, P-P70S6K, P-S6, and P-ERK were blocked in AA-treated group (Figure 4A). Moreover, AA treatment significantly inhibited cardiomyocyte hypertrophy, as indicated by decreased surface area (Figure 4C-D). The level of ANP induced by Ang II was dose-dependently decreased after AA treatment (Figure 4B). To clearly illustrate the function of AMPKα, we knocked down AMPKα2 in cardiac myocytes. Though no significant differences in surface area and ANP were observed after knock down AMPKα2, depletion of AMPKα could completely offset the protective effects of AA in myocytes (Figure S3, Figure S4A, Figure 4C-E).
3.5 AA inhibited the accumulation of collagen in an AMPKα-dependent manner in vitro
Taken the fact that AA treatment activated AMPKα in the heart and cardiomyocytes into account, we detected the activation of AMPKα in cardiac fibroblasts. Expectedly, AA also activated AMPKα and suppressed mTOR and ERK pathways in cardiac fibroblasts (Figure 5A-B). Cardiac fibroblast directly transformed into myofibroblast, leading to the accumulation of collagen [23]. This prompted us to investigate the effects of AA on production of collagen. Treatment with AA profoundly attenuated accumulation of collagen I and collagen III, and conversely shAMPKα totally blocked the protective effects (Figure S4B, Figure 5C).
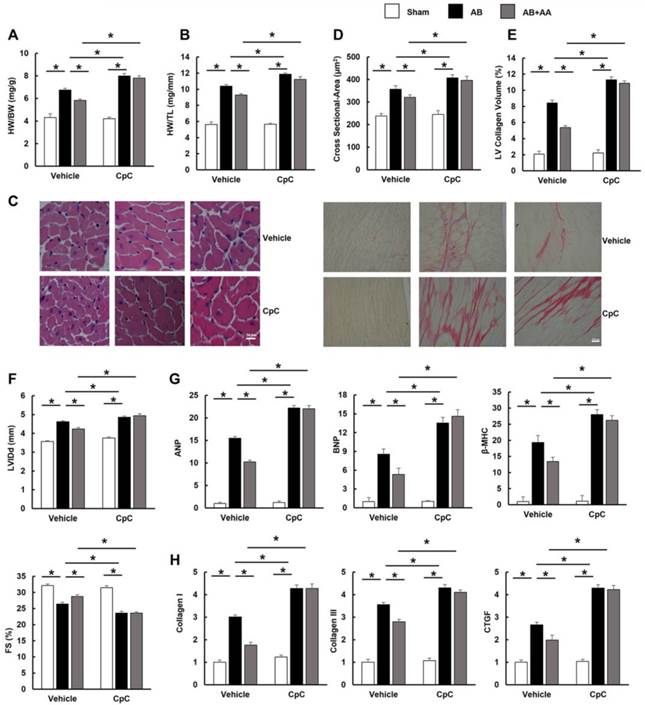
3.6 AA had no protective effects on hypertrophy and fibrosis in mice subjected to CpC
To further investigate whether AA exerted protective effects through AMPKα in vivo, mice were subjected to CpC, a common AMPKα inhibitor [24, 25]. Mice with AB surgery manifested obvious hypertrophy and fibrosis. However, AA treatment did not prevent the development of cardiac hypertrophy and fibrosis in mice with AMPKα inhibition, which is reflected by morphological changes, echocardiographic examinations and molecular markers (Figure 6A-H). Collectively, these results suggested that the protective effects of AA were dependent upon the activation of AMPKα.
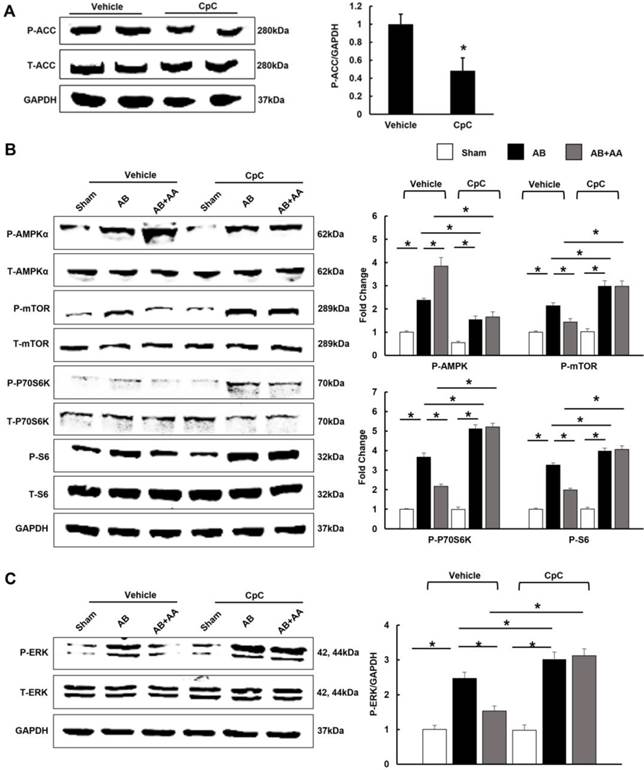
3.7 AA lost its inhibitory effects on mTOR and ERK in mice treated with CpC
After CpC injection, AMPKα was no longer activated in mice with AA treatment (Figure 7A-B). The phosphorylation levels of mTOR, P70S6K, S6 and ERK were not of statistical difference between AB+CpC and AB+AA+CpC group (Figure 7B-C).
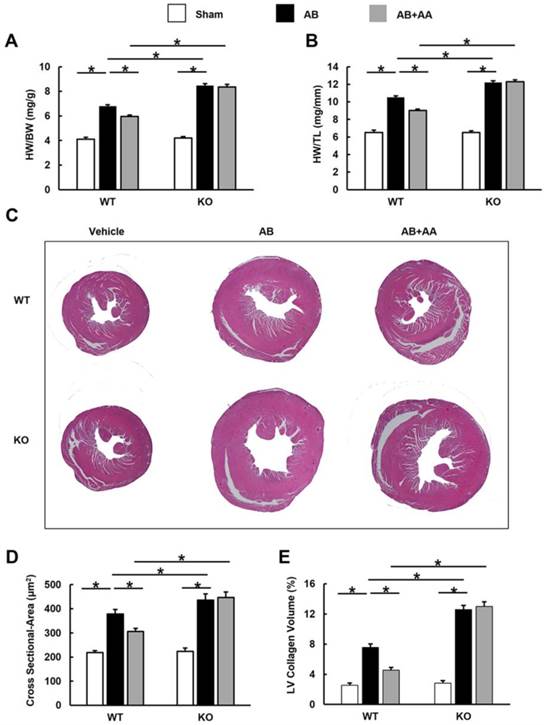
3.8 AA exhibited no protective effects in AMPKα2 knockout mice
In view of the fact that CpC had unspecific effects [26], AMPKα2 knockout mice were used. AA lost the protective effects in AMPKα2 knockout mice, as evidenced by the indistinguishable HW/BW, HW/TL, cross-sectional area and LV collagen volume.
The effects of asiatic acid (AA) on hypertrophy of neonatal rat cardiac myocytes. A, The protein levels of phosphorylated AMPKα and related targets in cardiac myocytes (n=8). B, AA inhibited increased mRNA level of ANP induced by angiotensin II (Ang II) (n=6). C-D, The cross-sectional areas of myocytes (n=6). E, AMPKα deficiency offset the inhibitory effect of AA on ANP (n=6). *P < 0.05.
The effects of asiatic acid (AA) on accumulation of collagen in cardiac fibroblasts. A-B, The protein levels of phosphorylated AMPKα and related targets in cardiac myocytes (n=8). C, AMPKα deficiency offset the inhibitory effect of AA on collagen I and collagen III (n=6). *P < 0.05.
4. Discussion
In the present study, we provided the evidence that AA attenuated pressure-overload induced cardiac hypertrophy in vivo and Ang II-induced hypertrophy of myocytes in vitro. AA also reduced cardiac fibrosis and accumulation of collagen. The cardio-protective effects of AA were medicated by activation of AMPKα, thus suppressing mTOR/P70S6K/S6 and ERK, which resulted in restricted hypertrophy and fibrosis, ultimately improving cardiac performance. Moreover, these protective effects of AA were abolished after AMPKα inhibition or deficiency.
The precise mechanisms by which AA exerts cardio-protective effects still remain elusive. AMPKα has been most extensively recognized as a sensor of cellular energy status, regulating process of energy metabolism during both physiological and pathological conditions [27]. Mounting reports have associated AMPKα with the hypertrophic response. It is reported that the loss of AMPKα activity is pro-hypertrophic and the activations of AMPKα by metformin and AICAR can suppress protein synthesis and hypertrophic growth in the cardiac myocyte [9, 28]. Previous report indicated that AA alleviated cardiovascular remodeling in rats with hypertension via restoration of eNOS expression, which has been proven a direct target of AMPKα [16, 29]. AA blocked TGF-β and MAPK signalling pathways in myocytes, which were also regulated by AMPKα [30-32]. Therefore, the inhibitory mechanism of AA on cardiac hypertrophy was examined for its effect on AMPKα activation. Our data clearly demonstrated that AA further promoted AMPKα activation induced by hypertrophic stimuli. Using adenoviral-mediated gene transfer or the inhibitor of AMPKα, we further demonstrated that AA lost cardio-protective effects in vitro and in vivo, confirming a key role of AMPKα in the restricted cardiac hypertrophy triggered by AA.
Our study also found that AA also markedly blocked activation of mTOR in the hypertrophic hearts. Activated mTOR increases phosphorylation of P70S6K, which acts on S6, exacerbating the process of hypertrophy [33]. However, the inhibitory effects of AA on mTOR/P70S6K/S6 pathway and cardiac hypertrophy were abrogated after AMPKα depletion, which were also consistent with several previous reports. MicroRNA-451 blocked cardiac hypertrophy via activating AMPKα and inhibiting phosphorylation of mTOR [34]. Metformin, a known activator of AMPKα, reduced phosphorylation of mTOR and its downstream target P-P70S6K and protected against systolic overload-induced heart failure [35]. ERK, a key regulator activated in cardiac myocytes in response to hypertrophic stimuli [36], was also found to mediate the cardiac protection of AA, as indicated by the fact that AA inhibited the activation of ERK and lost inhibitory effects on ERK after AMPKα inhibition or knock down. Collectively, these results indicate that AMPKα activation by AA inhibits cardiac hypertrophy partly through suppressing mTOR and ERK.
The effects of asiatic acid (AA) were blocked by Compound (CpC) in vivo. A-B, Statistical results of heart weight (HW)/ body weight (BW), HW/ tibial length (TL) of the indicated groups (n=12). C, HE staining and PSR staining (n=6). D, The cross-sectional areas of myocytes (n=6). E, Collagen volume (n=6). F, Echocardiographic parameters (n=8). G, mRNA levels of the hypertrophic markers (n=6). H, mRNA levels of the fibrotic markers (n=6). *P < 0.05.
The effects of asiatic acid (AA) and Compound (CpC) on AMPKα, mTOR and ERK signalling pathway. A-C, The protein levels of phosphorylated AMPKα and related targets in indicated groups (n=6). *P < 0.05.
Cardiac fibrosis, a common feature of heart diseases, is characterized by accumulation of collagen and increase of the extracellular matrix. In this study, we found that AA reduced cardiac fibrosis induced by pressure overload. Furthermore, anti-fibrotic effects of AA were evaluated in vitro using cardiac fibroblasts, and we observed that AA attenuated Ang II-induced collagen synthesis. To further elucidate the underlying mechanism, we detected effects of AA on AMPKα in fibroblast, which could suppress fibrotic actions[37], and found that AA promoted phosphorylation of AMPKα. In contrast, depletion of AMPKα by genetic approach or a specific inhibitor disrupted the protective effects of AA in vitro and in vivo, indicating the restrained cardiac fibrosis by AA may be attributed to activation of AMPKα. mTOR/P70S6K and ERK, the downstream targets of AMPKα, have been demonstrated to regulate the process of fibrosis[38, 39]. Limited fibrosis by AA may partly be due to the attenuation of mTOR and ERK.
In conclusion, AA protects against cardiac hypertrophy and fibrosis via AMPKα. We also show that depletion of AMPKα results in the loss of the protective effects of AA against hypertrophy and fibrosis. Our study provide an experimental evidence for the application of AA in the treatment of cardiac hypertrophy and heart failure. Future study aiming at elucidating the potential clinical use of AA will be of great interest.
Asiatic acid (AA) lost the inhibitory effects on cardiac hypertrophy and fibrosis in AMPKα knockout (KO) mice. A-B, Statistical results of heart weight (HW)/ body weight (BW), HW/ tibial length (TL) of the indicated groups (n=8). C, Histological results of hypertrophic heart with or without AA. D, The cross-sectional areas of myocytes (n=5). E, PSR staining of hypertrophic heart with or without AA (n=5). WT: wild type. *P < 0.05.
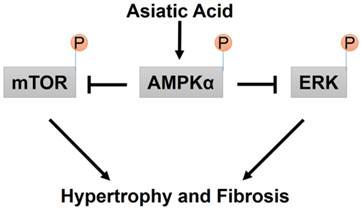
A proposed mechanism for the effects of AA on cardiac hypertrophy and fibrosis.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81270303, 81470516, 81470402), the Fundamental Research Funds for the Central Universities of China (No. 2014302020202), and the Hubei Province's Outstanding Medical Academic Leader program.
Conflict of Interest
None.
References
1. Hill JA, Olson E N. Cardiac plasticity. N Engl J Med. 2008;358:1370-1380
2. Chien KR. Stress pathways and heart failure. Cell. 1999;98:555-558
3. de Simone G, Gottdiener JS, Chinali M, Maurer MS. Left ventricular mass predicts heart failure not related to previous myocardial infarction: The Cardiovascular Health Study. Eur Heart J. 2008;29:741-747
4. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T. et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050-3055
5. Liang F, Lu S, Gardner DG. Endothelin-dependent and-independent components of strain-activated brain natriuretic peptide gene transcription require extracellular signal regulated kinase and p38 mitogen-activated protein kinase. Hypertension. 2000;35:188-192
6. Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307-318
7. Meng R, Pei Z, Zhang A, Zhou Y, Cai X, Chen B. et al. AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch Biochem Biophys. 2011;511:1-7
8. Ma ZG, Dai J, Zhang WB, Yuan Y, Liao HH, Zhang N. et al. Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor / AMPKα signalling pathway. Br J Pharmacol. 2016;173:1502-1516
9. Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771-32779
10. Yin MC, Lin MC, Mong MC, Lin CY. Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J Agric Food Chem. 2012;60:7697-7701
11. Hashim P, Sidek H, Helan MH, Sabery A, Palanisamy UD, Ilham M. Triterpene composition and bioactivities of Centella asiatica. Molecules. 2011;16:1310-1322
12. Wang X, Lu Q, Yu DS, Chen YP, Shang J, Zhang LY. et al. Asiatic acid mitigates hyperglycemia and reduces islet fibrosis in Goto-Kakizaki rat, a spontaneous type 2 diabetic animal model. Chin J Nat Med. 2015;13:529-534
13. Guo W, Liu W, Jin B, Geng J, Li J, Ding H. et al. Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int Immunopharmacol. 2015;24:232-238
14. Yan SL, Yang HT, Lee YJ, Lin CC, Chang MH, Yin MC. Asiatic acid ameliorates hepatic lipid accumulation and insulin resistance in mice consuming a high-fat diet. J Agric Food Chem. 2014;62:4625-4631
15. Lee KY, Bae ON, Serfozo K, Hejabian S, Moussa A, Reeves M. et al. Asiatic acid attenuates infarct volume, mitochondrial dysfunction, and matrix metalloproteinase-9 induction after focal cerebral ischemia. Stroke. 2012;43:1632-1638
16. Bunbupha S, Prachaney P, Kukongviriyapan U, Kukongviriyapan V, Welbat JU, Pakdeechote P. Asiatic acid alleviates cardiovascular remodeling in rats with L-NAME-induced hypertension. Clin Exp Pharmacol Physiol. 2015;42:1189-1197
17. Hung YC, Yang HT, Yin MC. Asiatic acid and maslinic acid protected heart via anti-glycative and anti-coagulatory activities in diabetic mice. Food Funct. 2015;6:2967-2974
18. Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K. et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun. 2014;5:3303
19. Deng W, Zong J, Bian ZY, Zhou H, Yuan Y, Zhang R. et al. Indole-3-carbinol protects against pressure overload induced cardiac remodeling via activating AMPK-alpha. Mol Nutr Food Res. 2013;57:1680-1687
20. Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413-423
21. Simpson P, McGrath A, Savion S. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ Res. 1982;51:787-801
22. Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:299-304
23. Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116:1269-1276
24. Dai Y L, Huang S L, Leng Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms. Int J Biol Sci. 2015;11:1272-1280
25. Noyan-Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li RK. et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127:74-85
26. Ljubica V, Maja M, Kristina J, Vilimanovich U, Sudar E, Isenovic E. et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40-50
27. Hardie DG. AMPK-sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939-952
28. Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B. et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918-924
29. Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y. et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res. 2009;104:496-505
30. Si L, Xu J, Yi C, Xu X, Wang F, Gu W. et al. Asiatic acid attenuates cardiac hypertrophy by blocking transforming growth factor-beta1-mediated hypertrophic signaling in vitro and in vivo. Int J Mol Med. 2014;34:499-506
31. Chan CY, Mong MC, Liu WH, Huang CY, Yin MC. Three pentacyclic triterpenes protect H9c2 cardiomyoblast cells against high-glucose-induced injury. Free Radic Res. 2014;48:402-411
32. Thakur S, Viswanadhapalli S, Kopp JB, Shi Q, Barnes JL, Block K. et al. Activation of AMP-activated protein kinase prevents TGF-β1-induced epithelial-mesenchymal transition and myofibroblast activation. Am J Pathol. 2015;185(8):2168-80
33. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549-564
34. Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S. et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. 2015;116:279-288
35. Xu X, Lu Z, Fassett J, Zhang P, Hu X, Liu X. et al. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase alpha2. Hypertension. 2014;63:723-728
36. Kehat I, Molkentin JD. Extracellular Signal-Regulated Kinase 1/2 (Erk1/2) Signaling In Cardiac Hypertrophy. Ann N Y Acad Sci. 2010;1188:96-102
37. Ma X, Fu Y, Xiao H, Song Y, Chen R, Shen J. et al. Cardiac Fibrosis Alleviated by Exercise Training Is AMPK-Dependent. PLoS One. 2015;10:e129971
38. Yu SY, Liu L, Li P, Li J. Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac Cardiovasc Surg. 2013;61:223-228
39. Du J, Guan T, Hui Z, Xia Y, Liu F, Zhang Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem Biophys Res Commun. 2008;368:402-407
Author contact
![]() Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University; Cardiovascular Research Institute, Wuhan University at Jiefang Road 238, Wuhan 430060, China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: qztangedu.cn.
Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University; Cardiovascular Research Institute, Wuhan University at Jiefang Road 238, Wuhan 430060, China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: qztangedu.cn.
Received 2015-10-24
Accepted 2016-3-23
Published 2016-5-25