Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(8):1032-1040. doi:10.7150/ijbs.15624 This issue Cite
Research Paper
Rapamycin Reduced Ischemic Brain Damage in Diabetic Animals Is Associated with Suppressions of mTOR and ERK1/2 Signaling
Ping Liu1,2,*, Xiao Yang3,2,*, Changchun Hei2,4, Yvonne Meli 2, Jianguo Niu 4, Tao Sun4, P. Andy Li2, ![]()
1. Department of Endocrinology, General Hospital of Ningxia Medical University, Yinchuan 750004, China
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute Biotechnology Enterprise (BRITE), North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, USA
3. Neuroscience Center, General Hospital of Ningcia Medical University, and Key Laboratory of Craniocerebral Diseases of Ningxia Hui Autonomous Region, Yinchuan 750004, China
4. Department of Human Anatomy, Histology and Embryology, Ningxia Medical University, Yinchuan 75004, China
* These two authors contributed equally to this work.
Received 2016-3-23; Accepted 2016-5-28; Published 2016-7-18
Abstract

The objectives of the present study are to investigate the activation of mTOR and ERK1/2 signaling after cerebral ischemia in diabetic rats and to examine the neuroprotective effects of rapamycin. Ten minutes transient global cerebral ischemia was induced in straptozotocin-induced diabetic hyperglycemic rats and non-diabetic, euglycemic rats. Brain samples were harvested after 16 h of reperfusion. Rapamycin or vehicle was injected 1 month prior to the induction of ischemia. The results showed that diabetes increased ischemic neuronal cell death and associated with elevations of p-P70S6K and Ras/ERK1/2 and suppression of p-AMPKα. Rapamycin ameliorated diabetes-enhanced ischemic brain damage and suppressed phosphorylation of P70S6K and ERK1/2. It is concluded that diabetes activates mTOR and ERK1/2 signaling pathways in rats subjected to transient cerebral ischemia and inhibition of mTOR by rapamycin reduces ischemic brain damage and suppresses the mTOR and ERK1/2 signaling in diabetic settings.
Keywords: Cerebral ischemia, Diabetes, ERK1/2, mTOR, Rapamycin
Introduction
According to the newly release epidemiological report stroke is the first leading cause of death in China and fifth in the United States [1,2]. It is also the leading cause of acquired disability [1,2]. About 800,000 people experience a new or recurrent stroke each year and one dies of a stroke every 4 minutes on average in the United States [2,3]. With increased lifespan and a worldwide rise in diabetes and obesity, a drastic increase in the societal burden of stroke is inevitable [4]. Diabetes is a well-established risk factor for ischemic stroke. Diabetes significantly exacerbates ischemic brain damage by accelerating the damage maturation latency, recruiting ischemic resistant structures such as hippocampal CA3 and dentate gyrus, thalamus and cingulated cortex into damage, transforming an ischemic infarct into a hemorrhagic one, and inducing post-ischemic seizures [5]. Yet, there has been no neuroprotective drug in clinic proven to be efficacious in ameliorating diabetes-enhanced ischemic brain damage. Intensive insulin therapy with tight control of blood glucose level has not yield a beneficial outcome in three clinical trials including ACCORD, ADVANCE and VADT [6-8]. Investigating the relation between diabetes mellitus and cerebral stroke may guide us to find a better clinical management strategy down the road.
Though many drugs have been shown to be neuroprotective in various experimental stroke models, virtually none of them have been approved to be efficacious in clinical trials [9]. Recently, attention has been drawn to the macrolide antibiotic drug rapamycin, an inhibitor of mammalian target of rapamycin (mTOR). The mTOR, a typical Serine/Thronine protein kinase, is under the regulation of PI3K/Akt, Ras-MAPK, and AMPK signaling pathways. Once activated, mTOR induces phosphorylations of its downstream proteins including P70S6K, S6 and elF4E [10]. Normally, mTOR functions as a sensor of extracellular signals including stimulations from energy levels and stress. It regulates angiogenesis, cell growth, apoptosis and autophagy, and other metabolic processes [11]. Alteration of mTOR signaling is associated with cancer, cardiac diseases, neurological diseases, diabetes, and certain inflammatory diseases [12]. For example, excessive activation of mTOR causes neurodegenerative diseases and inhibition of mTOR by rapamycin ameliorates the neuronal damage in mouse model of Alzheimer's Disease [13]. Further, inhibition of mTOR also offers neuronal protection in models of Parkinson's disease (PD) and epilepsy [14-16]. We and others have demonstrated that under euglycemic condition, inhibition of mTOR by rapamycin significantly reduced brain damage in rats subjected to a transient forebrain ischemia and reperfusion [17]. Recent studies have also shown that inhibition of mTOR signaling ameliorates pathological progression of diabetic nephropathy and diabetic cardiomyopathy [18,19]. However, there has been no report to demonstrate whether inhibition of mTOR is detrimental or beneficial to the outcomes of cerebral ischemia and reperfusion damage in diabetic animals. The objectives of this study are to examine whether diabetes activates or inhibits mTOR and whether inhibition of mTOR signaling by rapamycin reduces diabetes-enhanced ischemic brain damage.
Materials and methods
Animals
Pathogen free male Wistar rats (weighing 250-300 g) were purchased from vendor (Charles River) and kept in a controlled environment (12:12 h light/dark cycle, 50-70% humidity, 24°C) and provided with free access to water and food. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, Revised 1996) and approved by the Institutional Animal Care and Use Committee (IACUC) at the North Carolina Central University. All efforts were made to reduce the number of animal use and to minimize animal suffering.
Induction of diabetes and cerebral ischemia
Diabetes was induced in overnight fasted rats by subcutaneous injection of freshly prepared streptozotocin (STZ, 50 mg/kg body weight, Sigma, USA), dissolved in a 0.01 M citrate buffer (pH 4.5). Blood glucose levels were measured in each rat using an OneTouch Glucometer after 72 h and one week of STZ injection. Animals with blood glucose levels exceeding 16.7 mmol/L in both readouts were included in the present study. Transient forebrain ischemia was induced in diabetic animals 2 months after the STZ injection. Animals were anesthetized with 3-4% isoflurane (Abbott Laboratories) in N2O/O2 (70:30) for induction and 1-2% isoflurane for maintenance during the surgery. A tail artery catheter was inserted to monitor the mean arterial blood pressure (MABP). Cerebral ischemia was induced by bilateral clamping of the common carotid arteries for 10 min using two aneurysm clips and combined with hypotension to 40-50 mmHg of blood pressure achieved by withdrawal of blood through a central vein catheter inserted in to the right jugular vein. Reperfusion was achieved by releasing the clamps on the common carotid arteries and re-infusing the shed blood [20]. Body temperature was maintained at 36.5 - 37.5 °C throughout the surgical procedure by an automated heating pad and a lamp. Sham-operated rats were performed with the same surgical procedures except that the arteries were not occluded. Sixteen hours of reperfusion was selected as the experimental end point based on our previous finding demonstrating that hyperglycemic animals die of post-ischemic seizures at 16-24 hours of reperfusion after being subjected to transient cerebral ischemia [21,22]. After 16 hours reperfusion, rats were re-anesthetized, their brains were briefly perfused with PBS and then carefully removed. One brain hemisphere was kept frozen in liquid nitrogen and then stored at -80°C for biochemical analyses. The other hemisphere was fixed in 10% formaldehyde solution and embedded in paraffin for histological and immunohistochemical studies. Sixteen hours of reperfusion was selected as the experimental endpoint
Experimental design and drug administration
Forty-two rats were divided into 6 groups with 6-8 rats in each subgroup: Non-diabetic sham control (NG); Non-diabetic ischemia and reperfusion (NIR); Vehicle-treated Diabetic Sham Control (DC+V); Vehicle-treated diabetic ischemia and reperfusion (DIR+V); Rapamycin-treated diabetic sham control (DC+R); and rapamycin-treated diabetic ischemia and reperfusion (DIR+R). Rapamycin (LC Laboratories, USA) was initially dissolved in 100% ethanol to make a stock solution of 20 mg/ml, which was stored in -20°C. Immediately before injection, the stock solution was diluted in 5% Tween 80 and 5% polyethyleneglycol 400 to final concentrations of 4% ethanol and 1 mg/ml rapamycin [23]. Rapamycin (1 mg/kg) was injected intraperetoneally once a day for one month prior to the induction of cerebral ischemia.
Histological and TUNEL stainings
The formalin-fixed brains were dehydrated, embedded in paraffin, and sectioned coronally at 5 μm thickness. The sections were stained with hematoxylin and eosin (HE) and the histological outcomes were evaluated under a light microscope at 400x magnification. Bright red-stained acidophilic neurons with shrunken, triangular dense purple nuclei were considered as dead neurons. The numbers of dead neurons in the cortex were counted from a minimum of four non-repeated microscopic fields located on the frontal-parietal cortex and presented as percent of cell death.
TUNEL staining was performed according to the manufacturer's instructions (The DeadEnd Fluorometric TUNEL system, Promega Corporation, USA). In brief, the sections were deparaffinized and rehydrated. Proteinase K solution was added and incubated for 8 min at room temperature. Equilibration buffer (100 µl) was added and equilibrated at room temperature for 5-10 minutes. TdT reaction mix (50 µl) was added to the sections and incubated for 60 minutes at 37°C in a humidified chamber. Slides were immersed in 2X SSC for 15 minutes, washed and mounted with Vectashield mounting medium containing PI. The results were viewed under a confocal fluorescence microscope (Nikon Eclipse E600 FN, USA) and TUNEL positive neurons were labeled in green.
Western blotting
The brain tissues collected from the cortices were homogenized and subcellular fractions were separated through a series of centrifugation [24]. Protein concentration was measured using BCA method according to the instruction provided by the manufacturer (Millipore). An equal amount of (25 μg) protein of the cytolic fraction was loaded in each lane and separated by electrophoresis on 10%-12% SDS-polyacryl-amide gels (Introvgen), transferred to the PVDF membranes (Millipore, USA), and blocked with blocking buffer (Li-COR Biosciences, Lincoln, NE, USA) at room temperature for 1h. The membranes were incubated overnight at 4ºC with specific primary antibodies against phospho-P70S6K (1:1000 Cell Signaling Technology USA), phospho-S6, phospho-AMPK (1:500, Cell Signaling Technology USA), LC3I/II (1:1000, Abcam, USA), phospho-ERK1/2 (1:1000, Cell Signaling Technology USA), and Ras (1:1000, Abcam USA). The membranes were washed and incubated for 1h at room temperature with the anti-rabbit secondary antibody (1:10000 Odyssey USA). Immunodetection was performed using Odyssey Infrared Imaging System (Li-COR). Densitometry scanning of the bands was carried out using IRDye 800 (Li-COR Biosciences, Lincoln, NE, USA) and the ratio of the target band density to internal loading control beta-actin were calculated and presented as relative density.
Statistical Analysis
Data were analyzed using the SPSS 13.0 statistical software package. The statistical significance of was determined with one-way analysis of variance (ANOVA) and followed by LSD post-hoc test. All data are expressed as the mean ± standard deviation (SD). A value of P<0.05 was considered statistically significant.
Results
Blood glucose levels
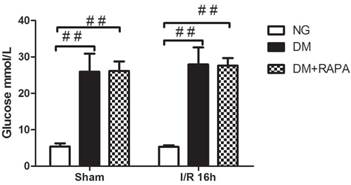
In addition to confirming hyperglycemic status after 3 and 7 days of STZ injection, blood glucose levels were also measured prior to the induction of cerebral ischemia. Figure 1 presents the mean blood glucose levels in each group. As expected, the average blood glucose contents were 26.5 mM in diabetic animals and 5 mM in non-diabetics (p<0.01). There was no difference between the vehicle and rapamycin treated rats.
Preischemic blood glucose levels in the rats. Blood glucose levels were significantly elevated in diabetic groups compared with non-diabetics. Data are mean ± SD, ## p < 0.01 vs. non-diabetic groups.
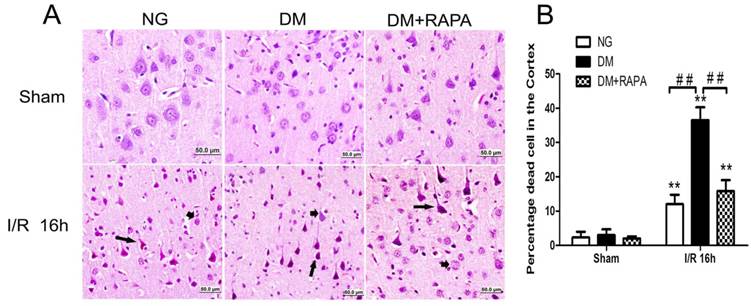
Rapamycin ameliorated ischemic neuronal death in the cortex of the diabetic rats
The histopathological outcome in the cortex was evaluated at 16h after reperfusion. There was no observed damage in any brain regions in the non-diabetic sham-operated rats. A few scattered dead neurons were observed in the cortex of the diabetic sham-operated rats. The number of dead neurons was significantly increased after 16h of ischemic/reperfusion in both non-diabetic and diabetic rats. About 12% of the cortical neurons were dead in non-diabetic animals and the percent increased to 36% in diabetic animals (p<0.01). Compared with vehicle-treated diabetic rats, rapamycin treatment dramatically reduced the damage in the cortex in the diabetic rats subjected to ischemia and reperfusion (Figure 2). The protective effects of rapamycin were also observed in the hippocampus. Diabetic ischemia caused close to 100% neuronal death in the CA1 and about 60% in the CA3 and dentate gyrus subregions. Rapamycin treatment reduced the CA1 damage to 50%, CA3 damage to 30% and almost completely spared the dentate gyrus (data not shown).
Histopathological outcomes in normoglycemic (NG), vehicle treated-diabetic (DM+V) and rapamycin-treated diabetic rats subjected to 10 min cerebral ischemia. A, Photomicrographs of the cortex; and B, summarized neuronal death in the cortex. HE staining. Arrows indicate dead neurons and arrowheads denote the survival neurons. Bar = 50 μm. Data are mean ± SD, **p <0.01 vs. sham-operated group and ## p < 0.01 vs. diabetic 16h I/R.
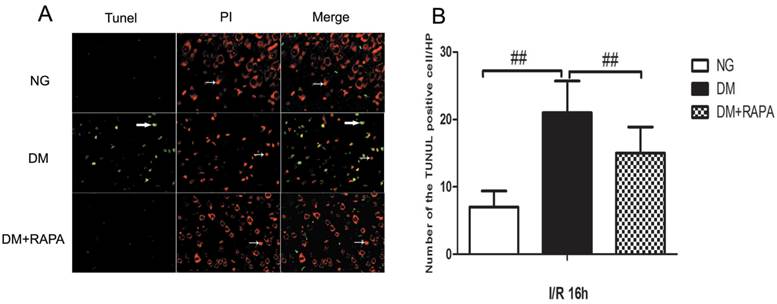
Rapamycin reduced DNA fragmentation in the cerebral cortex of the diabetic ischemic animals
TUNEL staining is a standard immunofluoroscent method for detecting DNA fragmentation. We used a commercial available TUNEL kit to detect DNA fragmentation. The brain sections were stained according the manufacturer's instruction and counter stained with PI. The results showed that transient cerebral ischemia induced a few scattered TUNEL positively stained cells in the cortex at 16 h after ischemia and reperfusion in non-diabetic animals (Figure 3, Upper panel). Diabetes caused significant increase in TUNEL positive cells after ischemia and reperfusion compared with non-diabetic ischemic animals (Figure 3, middle panel). Treatment with rapamycin drastically reduced the DNA fragmentation in the diabetic animals subjected to ischemia and reperfusion (Figure 3, lower panel). It is noticeable that not all TUNEL positive neurons were PI positive, suggesting a relative specificity of the TUNEL kit we have used to detect aopototic cell death.
Double-labeling of TUNEL and propidium iodide in the cerebral cortex after 16 h of reperfusion. A, TUNEL positive cells were labeled in green and cell nuclei were labeled in red. Necrotic cells are shown as condensed nuclear staining indicated by arrows in red channel. Most TUNEL positively stained cells are not PI positive, suggesting high specificity of TUNEL kit in detecting apoptotic cell death in this experiment. B, Bar graph shows the number of TUNEL positive cells. ## p < 0.01 vs. diabetic rats (Figure 3).
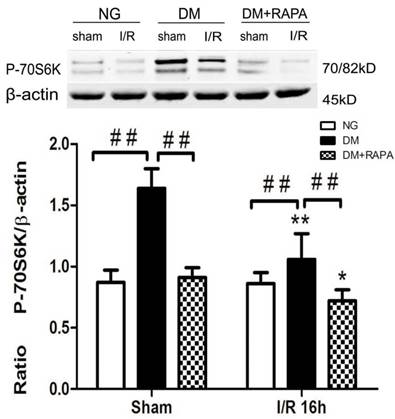
Diabetes activated and rapamycin inhibited the mTOR signaling
Phosphorylation of P70S6K has been used widely as a marker of mTOR activation. As shown in Figure 4, cerebral ischemia in non-diabetic animals did not cause activation of mTOR after 16 h of reperfusion in the cortex. In contrast, diabetes significantly increased the phosphorylation of P70S6K both prior to and after cerebral ischemia and reperfusion. Treatment with rapamycin completely prevented the diabetes-induced mTOR activation (Figure 4).
The levels of p-P70S6K in control and ischemic animals at 16 h of reperfusion in the cortex. Upper panel, representative Western blots showing protein levels of p-P70S6K in the cortex; and Lower panel, Bar graphs showing the relative band density of p-P70S6K. *p <0.05, **p <0.01 vs. each sham-operated group and ## p < 0.01 vs. diabetic rats.
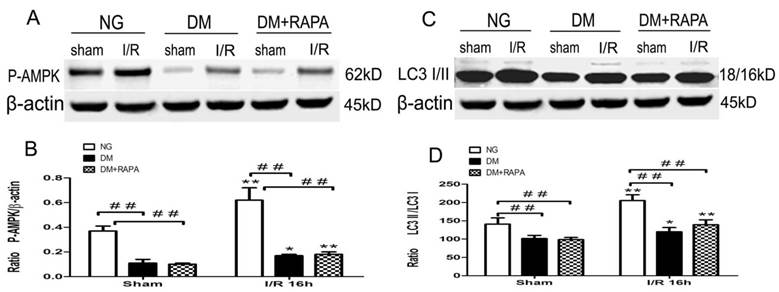
Diabetes suppressed phosphorylation of AMPKα
Changes in the levels of p-AMPKα and LC-3II after cerebral ischemic/reperfusion was evaluated by the Western blot. The p-AMPKα increased moderately in non-diabetic animals after ischemia and reperfusion compared with sham-operated control (p<0.01). Diabetes significantly reduced the levels of p-AMPKα at both baseline level and after ischemia and reperfusion comparing to non-diabetic counterparts (p<0.01), albeit an elevation of p-AMPKα after reperfusion in diabetic rats. Rapamycin increased the post-ischemic p-AMPKα comparing to diabetic ischemic rats, but did not reach to the level observed in sham-operated non-diabetic rats (Figure 5A,B). Ischemia and reperfusion caused a pronounce elevation of LC3-II in both non-diabetic and diabetic rats; however, the levels in diabetic rats were significantly lower than those of non-diabetic sham and ischemia/reperfusion groups. Rapamycin did not alter the LC3-II contents in diabetic animals (Figure 5 C,D).
The protein levels of p-AMPK and LC3-II in the cortex at 16h after reperfusion. A and C, Representative Western blots for p-AMPKα and LC-3II; and B and D, relative optical band density of p-AMPKα and LC-3II. **p <0.01 vs. each sham-operated group and ## p < 0.01 vs. non-diabetic rats.
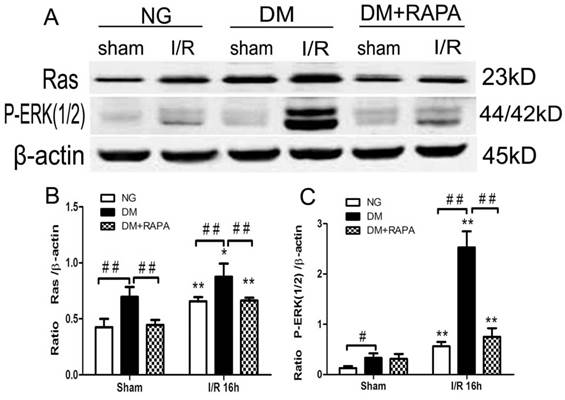
Diabetes activated and rapamycin inhibited ischemia-induced Ras-ERK1/2 pathway
MAPK is both an upstream of and downstream of mTOR. ERK1/2 has been shown to be phosphorylated after ischemia in diabetic rats [20,25]. In this study, Ras-ERK1/2 pathway was monitored using specific antibodies using the ischemic cortical tissues. The results showed that cerebral ischemia increased both Ras and p-ERK1/2 at 16 h of reperfusion in non-diabetic rats. Comparing to non-diabetic rats, diabetes resulted in a much pronounced increased in Ras and p-ERK1/2. Rapamycin intervention blocked the increases of both proteins in diabetic animals after ischemia and reperfusion (Figure 6).
Diabetes activated and rapamycin inhibited the Ras-ERK1/2 pathway. A, Western blots showing representative bands of Ras and p-ERK1/2; B and C, Bar graphs summarize average relative densitometry of Ras and ERK1/2, respectively. **p <0.01 vs. sham-operated group and ## p < 0.01 vs. diabetic rats.
Discussion
Both basic and clinical studies have shown that preischemic hyperglycemia exacerbates neuronal damage induced by cerebral ischemia [5]. In this study, we induced a transient cerebral ischemia in STZ-induced 2 month diabetic rats treated with mTOR inhibitor rapamycin or vehicle. The pathological outcome revealed that after only 16 h of reperfusion following a 10 min forebrain ischemia, the neuronal death, both necrotic and apoptotic, were significantly enhanced by diabetes, comparing to normoglycemic ischemic animals. Such increased damage was associated with activation of mTOR signaling pathway, phosporylation Ras-ERK1/2, and suppression of AMPKα. Rapamycin treatment markedly reduced ischemic brain damage in the cortex and hippocampus of the diabetic rats and its protective effect was linked to deactivation of mTOR and Ras-ERK1/2 pathways.
The mTOR gene was first described in yeast as the pharmacological targets of the microbicide rapamycin [26], and subsequently described in other invertebrate and vertebrate organisms. The mTOR contains two functionally distinct complexes, mTORC1 and mTORC2, which play central roles in translation initiation, transcription, cytoskeleton organization, cell growth, and proliferation as well as cell survival [27,28]. Though alterations of mTOR activity have been identified in several neurological diseases [29], the role of mTOR activation in cerebral ischemia remains controversial. In one hand, middle cerebral artery occlusion decreased the phosphorylation of mTOR and its downstream proteins and enhancement of mTOR phosphorylation using ferulic acid ameliorated ischemic brain damage [30, 31]. Further, decrease of phospho-P70S6K has been shown to increase neuronal cell death after cerebral focal ischemia and that ischemic postconditioning activates the mTOR [32,33]. On the other hand, activation of mTOR through inhibition of the mTOR upstream regulator PTEN increased cerebral infarction [32,33]. In a parallel study conducted in normoglycemic rats, we have observed that mTOR pathway was transiently activated after 3 h of reperfusion following forebrain ischemia and returned back to baseline level after 16 h and 7 days of reperfusion. Inhibition of mTOR using rapamycin suppressed the activation of mTOR pathway and significantly reduced ischemic brain damage in both the cortex and hippocampus [17].
The role of mTOR in mediating diabetes related complications has not been well documented. One report indicated that type-2 diabetes increased phosphorlation of mTOR myocardium of the mice and rapamycin, as well as metformine, were capable of preventing diabetes-induced cardiac malfunction through inhibiting mTOR signaling [19,34]. Activation of mTOR may also play an important role in the development of diabetic nephropathy because increased mTOR activity was observed in podocytes of the glomeruli and inhibition of mTOR prevented podocyte injury and hindered the progression of diabetic nephropathy [35,36]. Up-to-date there has been no report to document the change of mTOR in diabetic ischemic animals. Our results showed that p-P70S6K increased significantly in both diabetic sham-operated controls and ischemic rats and that rapmycin drastically inhibited the phosporylation of P70S6K caused by diabetes. These data suggest that diabetes predispose the brain tissues to be vulnerable to ischemic injury by activating the mTOR signaling and that inhibiting the activity of the mTOR is capable of reducing diabetes-exacerbated ischemic brain damage. We pre-treated the ischemic rats with rapmaycin based on the consideration that diabetic animals develop neuronal death and post-ischemic seizures after only 3 h of reperfusion. This approach limits the clinical relevance of the study.
Autophagy is an intracellular catabolic process that responsible for recycling of degenerated proteins and subcellular organelles [37]. Autophagy could be activated by fasting, glucose depravation, exercise and other stresses. AMPK, which is known to suppress mTOR, is a major inducer of autophagy [38,39]. Rapamycin also activates autophagy by suppressing mTOR [40]. Basal level autophagy plays a house-keeping role to maintain cellular homeostasis that is important for cell survival [41]. Inhibition of this process leads to accumulation of denatured proteins in cells and causes diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease [42,43]. However, over activation of autophagy may also cause cell to die [44]. Therefore, autophagy is often described as Type II programmed cell death (PCD), to distinguish it from apoptosis, the Type I PCD [45]. Activation of autophagy after cerebral ischemia has been reported for many years [46,47]. However, it remains unclear whether activation of autophagy is beneficial or detrimental to the ischemic tissue. While several studies reported a protective effect of autophagy in experimental stroke [48,49], others observed opposite outcomes when autophagy is activated [50,51]. In the present study we observed increased levels of p-AMPKα and LC3-II to LC3-I ratio, a marker for autophagy, in non-diabetic animals after ischemia and reperfusion of 16 h in the cortex. This finding is consistent with the reports in literature [52]. The activation of p-AMPKα and autophagy is probably caused by reduced energy level during ischemic period due glucose depravation. In diabetic sham-operated rats, however, the p-AMPKα level was significantly lower than that of the non-diabetic rats, suggesting that high glucose status suppresses AMPK signaling. This is consistent with previous reports showing that hyperglycemia suppresses autophagy [53,54]. Though diabetes per se suppressed the p-AMPKα, ischemia still induced an elevation of p-AMPKα. Correspondingly, LC3-II was activated after ischemia in diabetic animals, suggesting ischemia is capable of activating autophagy even under diabetic condition. Interestingly, rapamycin treatment did not alter diabetes-induced changes in p-AMPKα and LC3-II. Although it is likely that the protective effects of rapamycin is not associated with autophagy based on LC3-II protein blot, we could not draw a conclusion regarding to the role of autophagy in mediating diabetes-enhanced ischemic brain damage and rapamycin protection because other biomarkers and indicators of autophagy were not measured in current study.
ERK1/2 plays a key role in cell proliferation and growth. Cerebral ischemia is known to increase the phosphorylation of ERK1/2. However, it is not known whether such an increase represents an defensive response or actually mediates cell [55]. Inhibition of ERK1/2 by PD98059 increased and activation of ERK1/2 decreased ischemic brain damage both in vitro and in vivo [56,57]. Whereas, emerging evidence suggested that the activation of ERK1/2 may be associated with neuronal death and the inhibition of MEK/ERK pathway can provide neuroprotection [58], especially in diabetic ischemia. Here, we found that Ras and phosphorylated ERK1/2 were increased in the cerebral cortex after ischemic injury, which is consistent to our previous reports [20]. Diabetes further enhanced the Ras and p-ERk1/2 protein levels after ischemia comparing to non-diabetic ischemic animals. Treatment with rapamycin significantly prevented the diabetes-induced elevations of Ras and P-ERK1/2, suggesting that suppression of p-ERK1/2 is associated with neuroprotection. These findings are consistent with previous reports showing that ERK1/2 is highly expressed in diabetes-recruited damage brain structures and inhibition of ERK1/2 pathway reduces ischemic brain damage in hyperglycemic animals [20,59,60]. It is not known how rapamycin suppresses Ras-ERK1/2 pathway since it is generally believed that Ras-ERK1/2 is an upstream pathway of mTOR expression. One possibility is that ERK1/2 is both an upstream regulator and a downstream target of mTOR. Another possibility is that increased reactive oxygen species (ROS) activated the ERK1/2 signaling. It has been well documented that hyperglycemia increases ROS production after ischemia [61]. ROS is capable of activating multiple signal transduction pathways, including the phosphorylation cascades leading to the activation of mitogen-activated protein kinases (MAPKs) [62,63]. We speculate that hyperglycemia induced ROS production activated ERK1/2 and rapamycin prevented the ERK1/2 activation through inhibition of ROS production. Indeed, rapamycin has been shown to protect the mitochondria against oxidative stress in rat model of Parkinson's disease [64], to improve endothelial function [65], to strengthen vascular contractility [66], and to prevent diabetic myocardia [67] by reducing oxidative stress.
In summary, our results have demonstrated that diabetes enhances ischemic brain damage and activates mTOR and Ras-ERK1/2 signaling pathways after transient cerebral ischemia and reperfusion. Inhibition of mTOR with rapamycin significantly reduces ischemic brain damage and the protective effect is associated with suppression of Ras-ERK1/2 pathways, without affecting plasma glucose levels and autophagy.
Acknowledgements
This study is supported by American Epileptic Society to PAL, National Natural Science Foundation of China (81560226) and Natural Science Foundation of Ningxia Hui Autonomous (NZ14281) to XY, Chinese National Science Foundation 973-Preproject (2014CB560711) to JN. The Biomanufacturing Research Institute and Technology Enterprise (BRITE) is partially funded by the Golden Leaf Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank Mr. Brent Caligan for proofreading.
Author Contributions
PL, PAL, JN, ST conceived and designed the experiments. PL, XY, CH, YM performed the experiments. PL, XY, CH, PAL analyzed the data. PAL, JN, TS contributed reagents, materials, and analysis tools. PL, PAL wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kochanek KD, Xu JQ, Murphy SL, Arias E. Mortality in the United States, 2013. NCHS Data Brief, No. 178. Hyattsville, MD: National Center for Health Statistics, Centers for Disease Control and Prevention, US Dept. of Health and Human Services. 2014
2. Mozaffarian D, Benjamin EJ, Go A. et al. Executive summary: Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:447-454
3. Murphy SL, Xu JQ, Kochanek KD. Deaths: final data for 2010. National Vital Statistics Report. 2013;6:1-117
4. Livingston-Thomas JM, Tasker RA. Animal models of post-ischemic forced use rehabilitation: methods, considerations, and limitations. Exp Transl Stroke Med. 2013;5:2
5. Li PA, He QP. Mechanisms of hyperglycemia-enhanced ischemic brain damage. Transl Med Res. 2013;3:1-11
6. Gerstein HC, Miller ME, Byington RP. et al. Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545-2559
7. Patel A, MacMahon S, Chalmers J. et al. ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560-2572
8. Duckworth W, Abraira C, Moritz T. et al. VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360(2):129-139
9. Chauhan A, Sharma Uma, Jagannathan NR. et al. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res. 2011;225(11):603-609
10. Wilfred L, Jerrold SL. Mammalian target of rapamycin and the kidney. I. The signaling pathway. Am J Physiol Renal Physiol. 2012;303:1-10
11. Chen HJ, Xiong T, Qu Y. et al. mTOR activate shypoxia-inducible factor-1 and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Neurosci Lett. 2012;507:118-123
12. Dazert Eva, Hall MN. mTOR signaling in disease. Curr Opinion Cell Biol. 2011;23:744-755
13. Spilman P, Podlutskaya N, Hart MJ. et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979
14. Malagelada C, Jin ZH, Jackson-Lewis V. et al. Rapamycin protects against neuron death in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166-1175
15. Liu K, Liu C, Shen L. et al. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Int. 2013;38:201-207
16. Talos DM, Sun H, Zhou X. et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS ONE. 2012;7:35885-35901
17. Yang X, He C.C, Liu P, Song Y. et al. Inhibition of mTOR pathway by rapamycin reduces brain damage in rats subjected to transient forebrain ischemia. Int J Biol Sci. 2015;11(12):1424-1435
18. Mori H, Inoki K, Masutani K. et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun. 2009;384:471-475
19. Das A, Durrant D, Koka S. et al. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice. J Biol Chem. 2014;289:4145-4160
20. Li PA, He QP, Ouyang YB. et al. Phosphorylation of extracellular signal-regulated kinase after transient cerebral ischemia in hyperglycemic rats. Neurobiol Dis. 2001;8:127-135
21. Li PA, Shamloo M, Smith M-L, Katsura K, Siesjö BK. The influence of plasma glucose concentration on ischemic brain damage is a threshold function. Neurosci Lett. 1994;177:63-65
22. Li PA, Shamloo M, Katsura K, Smith M-L, Siesjö BK. Critical values for plasma glucose in aggravating ischemic brain damage: correlation to extracellular pH. Neurobiol Dis. 1995;2:97-108
23. Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J neurosci. 2011;31(6):2337-47
24. Midori A. Yenari, Satoshi Iwayama, Danye Cheng, Guo Hua Sun,Miki Fujimura, et al. Mild hypothermia attenuates cytochrome c release but does not alter Bcl-2 expression or caspase activation after experimental stroke. J Cereb Blood Flow Metab. 2002;22(1):29-38
25. Zhang F, Signore A.P, Zhou Z. et al. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: potential signaling mechanisms. J Neurosci Res. 2006;83:1241-1251
26. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immuno- suppressant rapamycin in yeast. Science. 1991;253:905-909
27. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-293
28. Chong ZZ, Shang YC, Wang S. et al. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog Neurobiol. 2012;99(2):128-148
29. Michael GG, Brian KK. TOR on the brain. Exp Gerontol. 2011;46:155-163
30. Koh PO. Ferulic acid attenuates focal cerebral ischemia-induced decreases in p70S6 kinase and S6 phosphorylation. Neurosci Lett. 2013;555:7-11
31. Xie R, Wang P, Cheng M. et al. Mammalian target of rapamycin cell signaling pathway contributes to the protective effects of ischemic postconditioning against stroke. Stroke. 2014;45:2769-2776
32. Shi GD, OuYang YP, Shi JG. et al. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun. 2011;404:941-945
33. Zhao ZC, Yan CS, Wang SH. et al. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog Neurobiol. 2012;99:128-148
34. Xie ZL, Lau K, Eby B. et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770-1778
35. Gödel M, Hartleben B, Herbach N. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121(6):2197-2209
36. Inoki K, Mori H, Wang J. et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181-2196
37. Lee YJ, Hong YK, Lee SR. et al. Autophagy contributes to retardation of cardiac growth in diabetic rats. Lab Anim Res. 2012;28(2):99-107
38. Egan DF, Shackelford DB, Mihaylova MM. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456-46
39. Kim J, Kundu M, Viollet B. et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-141
40. Urbanek T, Kuczmik W, Basta-Kaim A. et al. Rapamycin induces of protective autophagy in vascular endothelial cells exposed to oxygen-glucose deprivation. Brain Res. 2014;1553:1-11
41. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:252-261
42. Alirezaei M, Kemball CC, Lindsay WJ. Autophagy, inflammation and neurodegenerative disease. Eur J Neurosci. 2011;33:197-204
43. Bozena G, Alicja K, Daniela K. Neuronal autophagy in cerebral ischemia -a potential target for neuroprotective strategies? Pharmacol Reports. 2012;64:1-15
44. Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70-77
45. Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther. 2011;16:239-243
46. Xu F, Gu JH, Qin ZH. Neuronal autophagy in cerebral ischemia. Neurosci Bull. 2012;28(5):658-666
47. Loos B, Genade S, Ellis B. et al. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp Cell Res. 2011;317:1437-1453
48. Tanabe F, Yone K, Kawabata N. et al. Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: Functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy. 2011;7:1462-1471
49. Guo Y, Wang J, Wang Z. et al. Melatonin protects N2a against ischemia/reperfusion injury through autophagy enhancement. J Huazhong Univ Sci Technolog Med Sci. 2010;30:1-7
50. Wang JY, Xia Q, Chu KT. et al. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011;70:314-322
51. Zheng C, Han J, Xia W. et al. NAD(+) administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett. 2012;512:67-71
52. Liu C, Gao Y, Barrett J. et al. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68-78
53. Sciarretta S, Zhai P, Shao D. et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134-46
54. Xu XM, Kobayash S, Chen K. et al. Diminished Autophagy Limits Cardiac Injury in Mouse Models of Type 1 Diabetes. J Biol Chem. 2013;25:18077-18092
55. Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflamm. 2010;7:14-26
56. Zhang F, Wang SP, Signore AP. et al. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38:2329-2336
57. Cao Q, Qian M, Wang XF. et al. Negative feedback regulation of Raf/MEK/ERK cascade after sublethal cerebral ischemia in the rat hippocampus. Neurochem Res. 2011;36:153-162
58. Li XZ, Yan HX, Zhou HR. et al. Hypoxic Preconditioning attenuates neuronal cell death by preventing MEK/ERK signaling pathway activation after transient global cerebral ischemia in adult rats. Mol Neurobiol. 2013;48:109-119
59. Zhang JZ, Jing L, Ma AL. et al. Hyperglycemia increased brain ischemia injury through extracellular signal-regulated protein Kinas. Pathol Res Practice. 2006;202:31-36
60. Zhang JZ, Jing L, Guo FY. et al. Inhibitory effect of ketamine on phosphorylation of the extracellular signal-regulated kinase1/2 following brain ischemia and reperfusion in rats with hyperglycemia. Exp Toxicol Pathol. 2007;59:227-235
61. Tsuruta R, Fujita M, Ono T. et al. Hyperglycemia enhances excessive superoxide anion radical generation, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Brain Res. 2010;1309:155-163
62. Akhiani AA, Werlenius O, Aurelius J. et al. Role of the ERK pathway for oxidant-induced parthanatos in human lymphocytes. PLoS One. 2014;9:e89646
63. Son Y, Cheong YK, Kim NH. et al. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639
64. Jiang J, Jiang J, Zuo Y, Gu Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson's disease. Int J Mol Med. 2013;31:825-832
65. Rajapakse AG, Yepuri G, Carvas JM, Stein S, Matter CM, Scerri I, Ruffieux J Montani, JP Ming, XF Yang, Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS ONE. 2011;6:e19237
66. Gao G, Li JJ, Li Y. et al. Rapamycin inhibits hydrogen peroxide-induced loss of vascular contractility. Am J Physiol Heart Circ Physiol. 2011;300:1583-1594
67. Das A, Durrant D, Koka S. et al. mTOR inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem. 2014;289(7):4145-60
Author contact
![]() Corresponding author: P. Andy Li, M.D., Ph.D. Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707. Fax: 919-530-6600; E-mail: pliedu
Corresponding author: P. Andy Li, M.D., Ph.D. Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707. Fax: 919-530-6600; E-mail: pliedu