Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Experimental Procedures
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(9):1052-1062. doi:10.7150/ijbs.15811 This issue Cite
Research Paper
Alcohol Increases Liver Progenitor Populations and Induces Disease Phenotypes in Human IPSC-Derived Mature Stage Hepatic Cells
Lipeng Tian1, Abhijeet Deshmukh1, Neha Prasad1, Yoon-Young Jang1,2 ![]()
1 Department of Oncology, The Sidney Kimmel Comprehensive Cancer Center
2 Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Received 2016-4-11; Accepted 2016-6-23; Published 2016-7-18
Abstract

Alcohol consumption has long been a global problem affecting human health, and has been found to influence both fetal and adult liver functions. However, how alcohol affects human liver development and liver progenitor cells remains largely unknown. Here, we used human induced pluripotent stem cells (iPSCs) as a model to examine the effects of alcohol, on multi-stage hepatic cells including hepatic progenitors, early and mature hepatocyte-like cells derived from human iPSCs. While alcohol has little effect on endoderm development from iPSCs, it reduces formation of hepatic progenitor cells during early hepatic specification. The proliferative activities of early and mature hepatocyte-like cells are significantly decreased after alcohol exposure. Importantly, at a mature stage of hepatocyte-like cells, alcohol treatment increases two liver progenitor subsets, causes oxidative mitochondrial injury and results in liver disease phenotypes (i.e., steatosis and hepatocellular carcinoma associated markers) in a dose dependent manner. Some of the phenotypes were significantly improved by antioxidant treatment. This report suggests that fetal alcohol exposure may impair generation of hepatic progenitors at early stage of hepatic specification and decrease proliferation of fetal hepatocytes; meanwhile alcohol injury in post-natal or mature stage human liver may contribute to disease phenotypes. This human iPSC model of alcohol-induced liver injury can be highly valuable for investigating alcoholic injury in the fetus as well as understanding the pathogenesis and ultimately developing effective treatment for alcoholic liver disease in adults.
Keywords: Induced pluripotent stem cells, hepatic progenitor cells, alcoholic liver disease, acquired liver disease, fetal alcohol injury, liver cancer.
Introduction
The detrimental effects of excessive alcohol consumption on the human body, particularly in the liver, have been well established (1-5). Alcohol can induce a spectrum of liver damage, including fatty liver, fibrosis and hepatocellular carcinoma, collectively referred to as alcoholic liver disease (ALD) (6). ALD remains one of the leading causes of liver related deaths worldwide, especially in USA (3, 4). More than 90% of patients with heavy alcohol intake have some degree of steatosis and in 20 to 40% of patients, continued heavy alcohol consumption leads to liver fibrosis (3). The severe cases of liver cirrhosis confer a high risk of liver failure and short-term mortality (7). Alcohol exposure during pregnancy also leads to multiple physiological alterations in the developing fetus. The birth defects of neurons, renal, heart and liver systems after exposure to alcohol in uteri are denoted as fetal alcohol spectrum disorders (FASD), which have been reported widely (8-10). Liver biopsy from a child with FASD has demonstrated parenchymal fat with portal and peri-sinusoidal fibrosis, which resembles the pathology of adult ALD (10).
Despite the profound economic burden of ALD on society and its severe effects on fetal and adult health, few advances have been made in the prevention and treatment of this disease. And the pathogenesis of ALD in humans is not fully understood (1). This is partly due to the lack of an ideal experimental model of human ALD. Currently, most of the ALD studies heavily rely on animal models, which may not fully recapitulate the human condition, and are hence suboptimal for studying human ALD pathogenesis (11).
The advent of induced pluripotent stem cells (iPSCs) has made it feasible to study the biology and pathophysiology of human hepatic cells. Human iPSCs have been generated from diverse human somatic cells (12-17), which can then be differentiated into hepatocytes that are functional in vitro and in vivo (17-19). We have previously established a step wise hepatic differentiation protocol to induce human iPSCs into definitive endoderm, hepatic progenitor cells, early/immature- and mature- hepatocyte-like cell types under defined conditions, which recapitulates human liver development (15-19). Using some of the early stage cells, which mimic embryonic liver, and a physiological dose of alcohol, we previously observed that alcohol might negatively influence differentiation of endoderm cells into hepatic stem/progenitor cells (20).
In this study, we compared effects of pathological concentrations of alcohol on human liver cells at different developmental stages, with a focus on mature stage hepatocyte-like cells that mimic postnatal or adult liver. While alcohol exposure on early (i.e. fetal) hepatocyte stage cells significantly reduced cell proliferation, markers of fetal hepatocytes were not altered. In contrast, at a mature stage of human iPSC-derived hepatocytes, alcohol increased two distinct liver progenitor populations, oxidative mitochondrial DNA damage, and markers associated with steatosis and hepatocellular carcinoma (HCC) in a dose dependent manner. Some of these disease phenotypes were significantly improved by antioxidant treatment. Together, these results provide novel insights into the pathophysiology of alcohol injury in human liver at different developmental stages, and serve as a basis for establishing a human-relevant model of alcoholic liver injury.
Experimental Procedures
Cell culture and hepatic differentiation of human iPSCs and ethanol exposure
All human iPSCs were cultured in a feeder free condition on Matrigel (BD) using mTeSR, (Stem Cell Technologies) as we described previously (15-17). This study was done in accordance with Johns Hopkins ISCRO regulations and following a protocol approved by the Johns Hopkins IRB. Hepatic differentiation was performed by a step-wise protocol described previously (15-19, 21) and for a longer time in some cases to obtain more mature hepatocytes. Briefly, definitive endoderm (DE) stage cells were derived from five human iPSC lines after treatment with 100ng/ml Activin A for 5 days. This was followed by treatment with 10ng/ml HGF/FGF4 for 5 days to obtain hepatic progenitor (HP) stage cells. Treatment was continued with 10ng/ml HGF/FGF4/OSM and 0.1µM Dexamethasone (Dex) for 10 more days to obtain early hepatocyte-like cells and for 15 more days to obtain mature hepatocyte-like cells. Alcohol exposure was achieved by directly adding ethanol into culture media at final concentrations of 100 mM and 200 mM, which are comparable to the pathophysiological blood concentrations of a heavy drinker (22, 23). Constant alcohol level was maintained in the culture wells by using a microclimate chamber to prevent alcohol evaporation.
RNA extraction and Real-time PCR
Total RNA was extracted using TRIZOL reagent (Invitrogen). Reverse transcription from mRNA to cDNA was performed with high capacity cDNA reverse transcription kit (Applied Biosystems). The resulting cDNA was used as template for Real-time PCR. Quantitative polymerase chain reaction (Q-PCR) was carried out with StepOnePlus Real-Time PCR System (Applied Biosystems), using TaqMan primer-probes. PCRs were performed with 20 ng of complementary DNA, TaqMan Fast Advanced Master Mix, and TaqMan gene expression assay reagent for: AFP (Hs00173490_m1), ALB (Hs00910225_m1), CK7 (Hs00559840_m1), IL-6 (Hs00985639_m1), CK18 (Hs02827483_g1), CK19 (Hs00761767_s1), FASN (Hs01005622_m1), SOX17 (Hs00751752_s1), GPC3 (Hs01018936_m1), FLNB (Hs00181698_m1), CYP2E1 (Hs00559368_m1), CD133 (Hs01009250_m1), EpCAM (Hs00901885_m1), CYP3A4 (Hs00604506_m1), TP53(Hs01034249_m1), Neil1(Hs00908563_m1) and 18S rRNA (Hs03003631_g1) in a total volume of 20 ul.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (Sigma) for 20 minutes at room temperature, followed by addition of primary antibodies. Primary antibodies against SOX17 (1:200, R&D Systems), Ki67 (1:500, Cell Signaling), AFP (1:200, Dako), ALB (1:200, Dako), CD133 (1:100, Biolegend), and 8-OHdG (1:200, Abcam) were diluted in PBS with 0.3% BSA and 0.1% Triton X-100. Cells were incubated with appropriate primary antibodies at 4oC overnight, and then incubated with Alexa Flour 594 or 488 secondary antibodies (all of the Alexa Fluor Series from Invitrogen) at room temperature for 30 minutes. Finally, cells were counterstained with DAPI. Images were taken using the motorized Nikon Ti-E microscope and NIS-Elements software. Cell counting was performed with ImageJ software (http://imagej.nih.gov/ij/).
Flow cytometric analysis
DE stage cells were resuspended in cold PBS and then incubated with 488-SSEA3/4 (eBioscience, 1:100), PE-CXCR4 (1:100) (BD Biosciences) for 30 minutes at 4oC. HP stage cells were incubated with PE-CD133 antibody (1:100, Biolegend) for 30 minutes at room temperature. Cells were washed twice and then resuspended in PBS. Flow cytometric analysis was done with Guava easyCyte Flow Cytometer (Millipore). Cells apoptosis assay was performed using Guava Flow Cytometer and Annexin V Apoptosis Detection Kit (eBioscience).
Oil Red O staining and quantification
The cells were fixed with 4% PFA at room temperature for 30 minutes. After washing the cells with ddH2O, they were incubated in 60% isopropanol for 5 minutes, followed by 60% Oil Red O solution for 5 minutes. Finally, the cells were washed with ddH2O until excess stain was no longer apparent. Lipid droplets appear red under the microscope. After staining the cells with Oil Red O, they were washed twice with 60% isopropanol. Oil Red O was then extracted by treating the cells with 100% isopropanol for 5 minutes. 100% isopropanol was used as background control when reading absorbance at 492nm wavelength.
Alcohol metabolism assays
Activities of alcohol metabolic enzymes and alcohol metabolite acetaldehyde in ethanol treated human iPSC-derived mature hepatocytes were measured using ADH, ALDH, and aldehyde specific commercial assay kits. For ADH and ALDH, the cells were homogenized in ice-cold 0.1% Triton X-100, and centrifuged to remove insoluble material. Cell lysates were incubated with isopropanol, which results in conversion of NAD+ to NADH to generate a colorimetric (450nm) product proportional to ADH activity (Sigma, MAK053). ALDH activity was determined by mixing the cell lysates with acetaldehyde to catalyze into acetic acid, along with conversion of NAD+ to NADH which reacts with fluorometric developer. Then ALDH activity was analyzed by fluorescence microplate reader at Ex/Em=540/590 (Cayman, 700800). Aldehyde measurement was performed using a kit (Abcam, ab138882) by incubating aldehyde blue indicator with cell supernatants for 15 minutes at room temperature. After adding the reaction buffer, fluorescence was measured at Ex/Em=365/435 nm. ADH/ALDH activities and aldehyde concentration were calculated using each of the standard curves.
Statistics
Data are expressed as mean ± standard error of the mean (SEM). For pairwise comparisons, a Student's t test or Mann-Whitney test (95% confidence interval) was used. For all tests, P<0.05 was considered significant.
Results
Alcohol has little effect on endoderm differentiation from human iPSCs, but reduces hepatic progenitor formation during early hepatic specification
To evaluate the effects of alcohol on definitive endoderm (DE) differentiation, human iPSCs were treated with ethanol at multiple pathophysiological doses (22, 23) from differentiation day 0 to 4 and analyzed at day 5 (Fig. S1). The expression level of DE markers such as SOX17 and CXCR4 was not significantly affected (Fig. S1B-D). A consistent increase in the Annexin V-positive apoptotic cells was observed in cells treated with ethanol (Fig. S1E). No significant change was detected in the quantity of Ki67 (a cellular marker for proliferation) positive cells after ethanol treatment (Fig. S1F). These data demonstrated that alcohol exposure even at a high concentration (200 mM) has little effect on differentiation of human iPSCs into endoderm stage cells, suggesting that alcohol might not significantly alter endoderm formation during human embryogenesis.
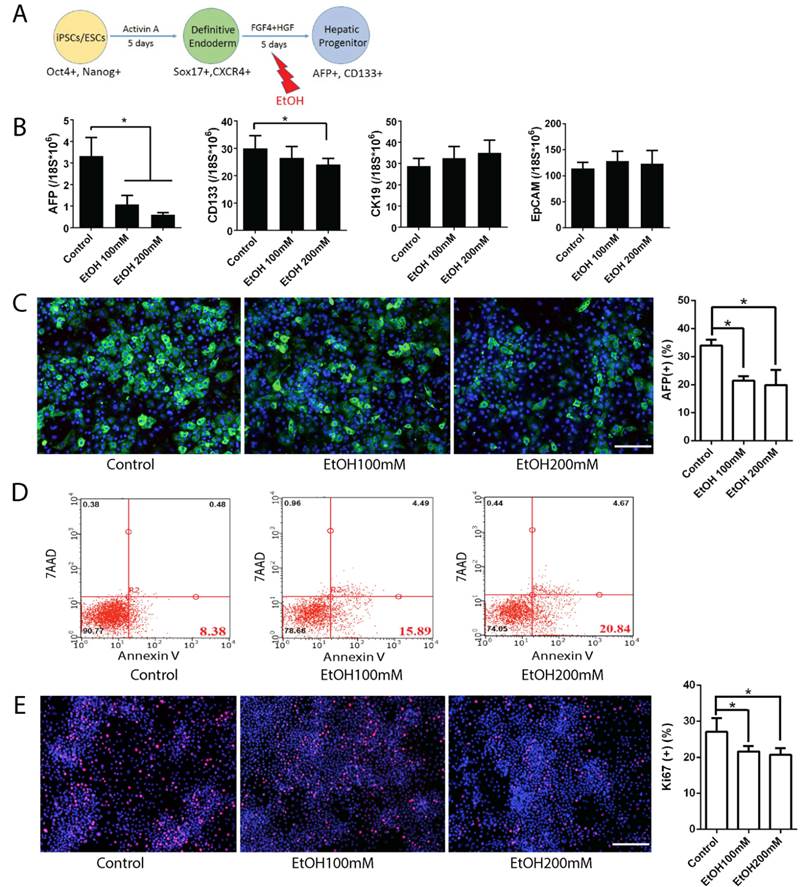
To examine the effects of alcohol on hepatic progenitor (HP) cell formation, DE cells were treated with ethanol from differentiation day 5 to 9 and analyzed at day 10 (Fig. 1). Although the cell morphology at this stage did not change significantly with alcohol treatment (Fig. S2), real time PCR results at day 10 showed decreased expression of hepatic progenitor markers including AFP and CD133 in alcohol treated group, compared to control (Fig. 1B). The inhibition effects of alcohol on AFP and CD133 were also observed at the protein level (Fig. 1C, and Fig. S2C). The amount of Annexin V-positive apoptotic cells was significantly increased after 100 mM ethanol treatment at this stage of hepatic differentiation (Fig. 1D and Fig. S2). The number of Ki67 positive proliferating cells were slightly reduced in alcohol treated cells (Fig. 1E). To further determine whether the alcohol treated HP cells maintain their progenitor potential, we purified CD133 positive HP cells from alcohol exposed day 10 plates and cultured them under normal hepatic differentiation condition. Compared to the untreated control, alcohol treated HP cells showed less mature characteristics at the end of hepatic differentiation (Fig. S2D). Taken together, ethanol exposure at a physiological concentration (100 mM) reduced expression of hepatic progenitor markers, induced apoptotic cell death, and inhibited cell proliferation during early hepatic specification. In addition, alcohol treatment, while transient, might have sustained negative effect on hepatic maturation of HP cells.
Effects of alcohol on human iPSC-derived hepatic progenitor formation. (A) Diagram of hepatic progenitor differentiation from human iPSCs, and alcohol treatment (day 5-10). (B) Expression levels of hepatic progenitor makers such as AFP, CD133, CK19, and EpCAM were examined by Real-time PCR at day 10 after 5 days of alcohol treatment. (C) Immunostaining of AFP at day 10 in control and alcohol treated groups and the percentage of AFP positive cells. (D) Annexin V positive apoptotic cells were analyzed by flow cytometry at day 10 after alcohol treatment. (E) Representative images of Ki67 staining of hepatic progenitor stage cells derived from human iPSCs and the percent of Ki67 positive cells. *:p<0.05, Scale Bar,100µm.
Alcohol reduces proliferation of human iPSC-derived early stage hepatocyte-like cells
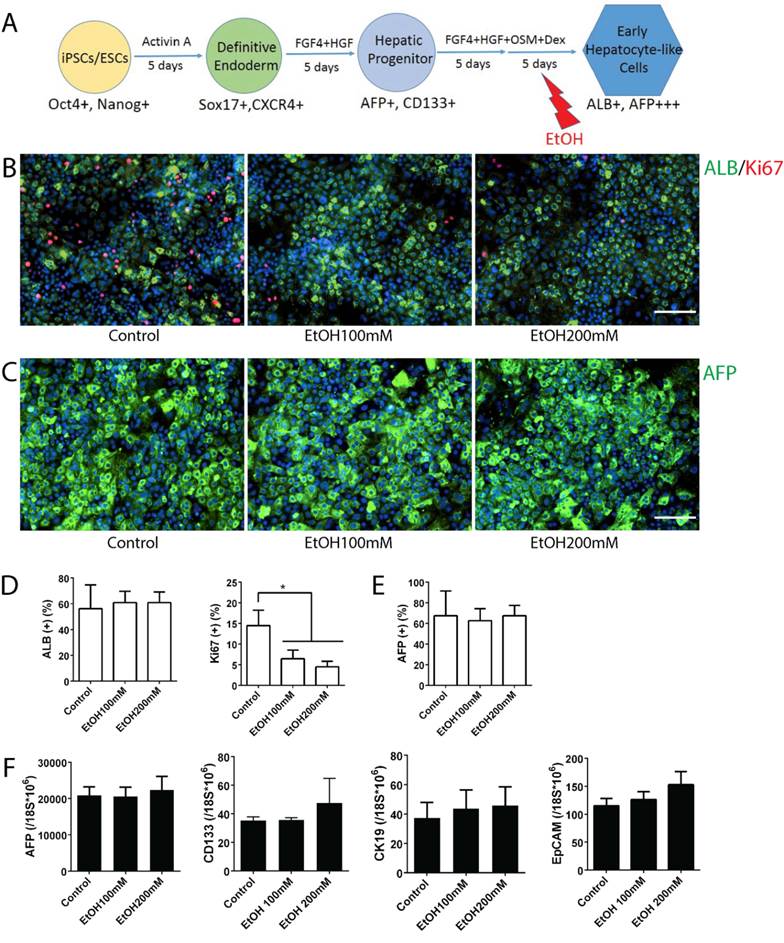
AFP expression is high in fetal hepatocytes and reduces sharply as they mature to an adult hepatocyte phenotype (24-26). Similarly, human iPSC-derived early hepatocyte-like cells (~day 20) express AFP at the highest level, and the levels decrease sharply with further maturation (~day 25) at mature stage hepatocyte-like cells (Fig. S3A). In contrast, a mature hepatocyte marker ALB increases significantly during early to mature hepatocyte transition (Fig. S3B). Thus, the early hepatocyte-like cells generated in this in vitro system represent fetal hepatocytes, and the mature stage hepatocyte-like cells represent postnatal or adult hepatocytes. To examine the effects of alcohol on early stage hepatocytes, we utilized early hepatocyte-like cells differentiated from iPSCs, which express high AFP and low ALB (Fig. S3). After these cells were treated with ethanol for 5 days, more uniform small sized early hepatocyte-like cells were observed in alcohol treated groups (Fig. 2B). The quantity of AFP or ALB positive cells did not increase after alcohol treatment; however, the number of Ki67 positive proliferating cells was significantly reduced after 100 mM or 200 mM ethanol exposure (Fig. 2B-E). Expressions of hepatic progenitor markers such as AFP, CK19, CD133 and EpCAM (27-32) were not altered after alcohol treatment even at a high concentration (200 mM) (Fig. 2F). These results indicate that alcohol at a physiological concentration (100 mM) negatively influences proliferation of early stage liver (i.e. fetal liver) rather than affecting hepatic differentiation.
Effects of alcohol on human iPSC-derived early stage hepatocytes. (A) Diagram of early stage hepatocyte-like cell differentiation and alcohol treatment (day 15-20). (B) ALB-positive cells (green) and Ki67 (red) positive cells were shown at this stage with or without alcohol treatment. (C) Immunostaining of AFP (green) at day 20 in alcohol treated and untreated groups. (D, E) The percentages of ALB, AFP, or Ki67 positive cells are expressed as the mean of three independent experiments. (F) Markers for hepatic progenitors are not changed at early hepatocyte stage cells by ethanol treatment. AFP, CD133, CK19 and EpCAM expression levels were examined by Real-time PCR at day 20. *:p<0.05, Scale Bar, 100µm.
Alcohol increases two hepatic progenitor populations in mature stage hepatocyte-like cells derived from human iPSCs
To examine the effects of alcohol on mature stage hepatocytes, we treated hepatocyte-like cells differentiated from human iPSCs with ethanol for 5 days at the mature hepatocyte stages (Fig. 3A). Strikingly, treatment with a physiological concentration of ethanol (100 mM) upregulated a hepatic progenitor marker AFP, while a high concentration (200 mM) upregulated both AFP and another progenitor marker, CD133 (Fig. 3C, D, E). Other hepatic progenitor markers such as CK19 and EpCAM were not altered significantly (Fig. 3E). Mature hepatocyte markers including ALB, CDP3A4 and CK18 were not significantly changed after alcohol treatment even at a high concentration (200 mM) (Fig. 3B, D and Fig. S4). As expected, activities of major alcohol metabolic enzymes (ADH and ALDH, CYP2E1) and its metabolite, acetaldehyde, were increased after ethanol treatment (Fig. 3F). To determine whether AFP and CD133, two progenitor markers that were most significantly affected by alcohol treatment, mark the same hepatic progenitor population, we co-stained these markers in mature stage hepatic cells (Fig. S5). Interestingly, these two markers are rarely co-expressed in the cells. In addition, a majority of CD133 positive cells did not co-stain with either hepatocytic (ALB) or non-hepatocytic (SMA, or CK7) liver cell markers.
Effects of alcohol on human iPSC-derived mature stage hepatocytes. (A) Diagram of mature stage hepatocyte-like cell differentiation and alcohol treatment (day 20-25). (B, C) Immunostaining of ALB and Ki67 (B), and AFP and Ki67 (C) at day 25 mature hepatic cells with or without alcohol treatment. (D) The percentages of ALB, AFP or Ki67 positive cells expressed as the mean of three independent experiments. Expressions of hepatic progenitor markers, AFP, CD133, CK19 and EpCAM examined by Real-time PCR (E) and Alcohol metabolic activities measured (F) at day 25 mature stage hepatocytes. *:p<0.05, Scale Bar, 100µm.
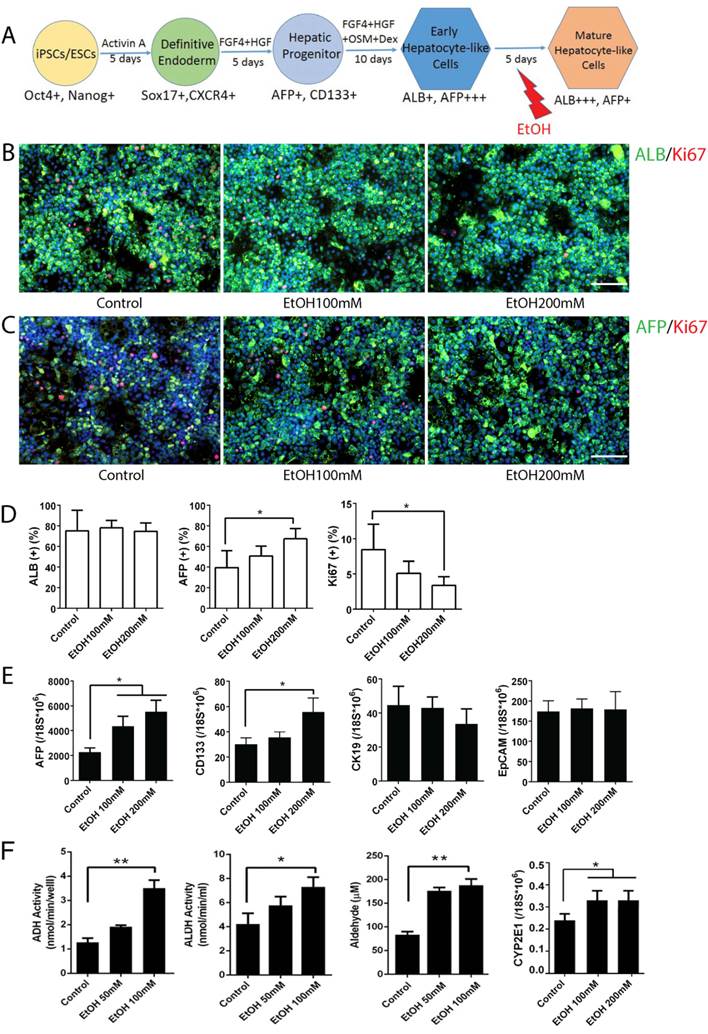
These results collectively suggest that a physiological concentration of ethanol (100 mM) induces AFP+ liver progenitor subset, while a high concentration (200 mM) induces both AFP+ progenitors and CD133+ progenitors in mature stage hepatocytes, but has little effect on other subsets of liver progenitors (CD19+ or EpCAM+ cells).
Alcohol significantly increases disease phenotypes in mature stage hepatic cells derived from human iPSCs
To detect the effects of alcohol on ALD pathogenesis, we analyzed multiple ALD phenotypes in the alcohol treated mature stage (day 25) hepatocyte-like cells derived from human iPSCs (Fig. 4). By using Oil red O staining, we observed intracellular lipid droplet accumulation in the iPSC-derived mature hepatocyte-like cells after alcohol treatment at both 100 and 200 mM concentrations (Fig. 4A). An increase in number as well as size of the lipid droplets was observed in the ethanol treated mature stage hepatic cells compared to untreated controls in a dose dependent manner (Fig. 4A, B). Analysis of fatty acid synthase (FASN) by real time PCR showed increased FASN transcription in alcohol treated cells when compared to the untreated control (Fig. 4C).
Alcohol increases disease phenotypes including lipid accumulation and hepatocellular carcinoma markers, and oxidative mitochondrial injury in human iPSC-derived mature stage hepatocytes. (A) Lipid droplets were stained with Oil Red O in control groups and ethanol treated groups. (B) After staining, Oil Red O was extracted by isopropanol and the absorbance at 492nm was measured. (C) Expression levels of fatty acid synthase (FASN), hepatocellular carcinoma markers glypican3 (GPC3) and filamin B (FLNB), and tumor suppressor, TP53 were measured by Real-time PCR. (D) Immunostaining of 8-OHdG, a marker for oxidative mitochondrial DNA damage, in alcohol treated day 25 mature stage hepatocytes derived from human iPSCs. (E) Expression of IL-6 and Neil 1 were examined by Real-time PCR in the day 25 hepatocytes exposed to alcohol 0 to 200 mM. (F) NAC treatment restored expression of Neil-1, FASN, and GPC3 in the alcohol treated mature hepatocytes. The treatment period and cell stage for Figure 4 is the same as those used for Figure 3. *:p<0.05, Scale Bar, 100µm.
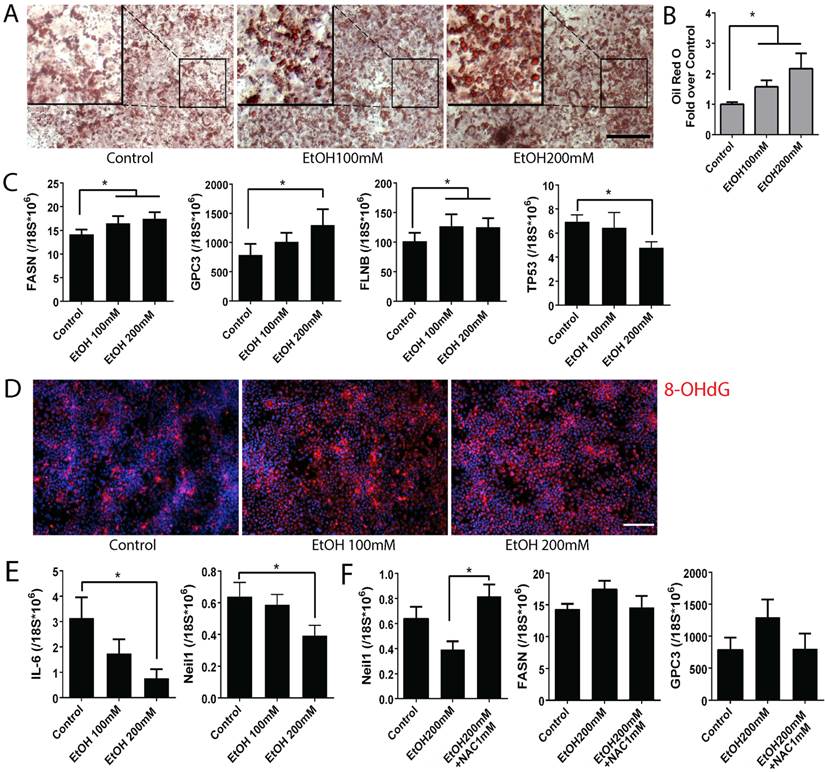
Glypican3 (GPC3) and Filamin B (FLNB) are widely accepted as tumor biomarkers for hepatocellular carcinoma, in addition to AFP (33, 34). Similar to the effect of alcohol on increasing AFP at the mature stage hepatocytes (Fig. 3), GPC3 and FLNB were also considerably induced after alcohol exposure (Fig. 4C). A well-known tumor suppressor TP53 was significantly reduced in the 200 mM ethanol treated group (Fig. 4C). These results together indicate that alcohol exposure results in ALD associated phenotypes (i.e, fat accumulation and tumorigenic changes) in human iPSC-derived mature stage hepatocyte-like cells in vitro.
Alcohol induces oxidative mitochondrial damage in mature stage hepatic cells derived from human iPSCs
Since ALD phenotypes were induced in the ethanol treated mature stage hepatic cells, we tested if these phenotypic changes are also associated with a known ALD pathogenesis, oxidative stress in liver. We first measured a marker for mitochondrial oxidation, 8-OHdG. Interestingly, 8-OHdG was increased in the ethanol treated mature hepatocytes derived from human iPSCs compared to the untreated controls (Fig. 4D). It has been reported that IL-6 promotes liver recovery from mitochondrial DNA oxidation in ALD and that reduction of IL-6 results in inactivation of mitochondrial repair enzymes such as Neil-1 and induces cell cycle arrest (35). IL-6 and Neil-1 were decreased in the ethanol treated mature hepatocyte-like cells derived from human iPSCs in a dose dependent manner (Fig. 4E) and a cell cycle check point P53 was also reduced (Fig. 4C). To further evaluate the contribution of oxidative stress to ALD phenotype, N-Acetyl-L-Cysteine (NAC) was added to the ethanol treated mature hepatocytes derived from human iPSCs. The decreased Neil-1was restored after NAC treatment (Fig. 4F). Although not as significant as seen in Neil-1, similar effects were observed in markers associated with liver cancer or steatosis including GPC3 and FASN (Fig. 4F). Collectively, these results suggest that alcohol induces damage in mature hepatocytes through oxidative mitochondrial damage and that ALD phenotypes can be reversed by antioxidant treatment.
Discussion
Significant progress (i.e. vaccine and drug) has been made for prevention and treatment of viral hepatitis during the last decades. However there have been few strategies that can effectively prevent or treat another major liver disease, ALD. To date, how alcohol affects liver development and hepatocytes in human remains largely unknown. Human iPSCs and their hepatic derivatives have been proven to be a great resource for modeling inherited or monogenic liver diseases (16, 18, 36, 37) while their use in modeling acquired liver diseases are limited. In this study, we attempted to evaluate the effects of alcohol on human liver development and to model alcoholic liver disease, a common acquired liver disease, using human iPSC-derived multistage hepatic cells. Different stages of human iPSC-derived hepatic cells were exposed to alcohol at multiple pathologic concentrations (22, 23). The effects of alcohol on endoderm formation were not significant (Fig. S1). However, generation of AFP or CD133 expressing hepatic progenitor cell subsets was significantly reduced by alcohol treatment during the early hepatic specification stage (Fig. 1). While alcohol treatment only resulted in a reduction in proliferative activity of early stage hepatocytes (Fig. 2), alcohol damage in mature stage hepatocytes (Fig. 3, 4) induced ALD phenotypes including steatosis and HCC markers, which may be associated with increased oxidative stress and increased AFP or CD133 expressing progenitor populations (Fig. 5). Based on this pure effect of ethanol on mature stage human (iPSC-derived) hepatocytes, it is possible to postulate that mature hepatocytes may obtain progenitor or disease phenotypes independent of significant interaction with non-hepatic cells in response to alcohol injury. This study also suggests that human iPSCs can be used for modeling certain aspects of ALD (i.e., steatosis and initiation of HCC) and studying its pathogenesis. In addition, the new finding that two progenitor markers (AFP and CD133) were differentially affected by alcohol in early hepatic specification stage (Fig. 1) and the mature hepatocyte stage (Fig. 3) may provide potential candidates as disease targets for both fetal alcohol injury and adult ALD.
Direct effect of alcohol on mature stage human hepatic cells and potential pathogenesis of ALD. Based on this pure effect of ethanol on mature stage human hepatic cells, it is possible to postulate that alcohol can alter cell fate (i.e., obtain multiple liver progenitor phenotypes), and induce pathogenic changes within mature hepatocytes, without major interaction with non-hepatic cell types. Ethanol may directly increase two progenitor phenotypes (AFP and CD133) in addition to impaired proliferation and oxidative damage in postnatal or mature human hepatocytes. Damaged hepatocytes increase fat content and may directly contribute to cancer. It is likely that increased AFP+ hepatic progenitors lead to cancerous changes. It is possible that CD133+ hepatic stem/progenitors might be partly responsible for both liver fibrosis and cancer, however it needs to be determined if CD133+ progenitors activates hepatic stellate cells (HSCs) to increase fibrosis. This cell fate change and oxidative damage in mature hepatocyte may be more ALD specific, compared to liver diseases with other causes. Black line; known/likely, Red line; new/possible, and dotted line; possible/no evidence.
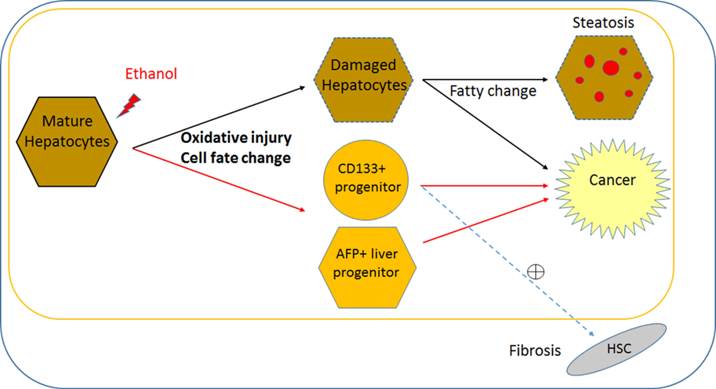
While AFP is relatively well known as a marker for fetal hepatocytes, hepatoblast/bi-potent liver progenitors, as well as a HCC bio-marker, the role of CD133 (Prominin 1) has not been clear and is rather complicated. CD133 has long been regarded as a primitive hematopoietic and neural stem cell marker (38), however evidence suggests it may also be a cancer stem cell marker in solid cancers such as brain tumors (39), renal tumors (40), liver cancer (32), and colon (41) and prostate carcinomas (42). Recent evidence suggests that CD133 is also a marker for oval cells in adult murine liver, which have the gene expression profile and function of bi-potent liver progenitors (31). However, CD133 is also suggested as a fibrosis marker, since hepatic stellate cells express CD133 and are involved in liver fibrosis (43, 44). Therefore, in human liver CD133 may serve as a liver progenitor marker with ability to activate hepatic stellate cells in a disease condition (Fig. 5), or a marker for a more multipotent stem/progenitor, which produces not only hepatocytes and ductal cells but also hepatic stellate cells. In our human iPSC-based model, CD133 and AFP increased with alcohol treatment in MH stage cells (Fig. 3). Future studies will determine whether these two distinct hepatic progenitor subsets (Fig. S5) are the main targets involved in the pathology of ALD, as well as the functional differences between CD133+ progenitors and AFP+ progenitors in ALD pathogenesis (Fig. 5).
It has long been suggested that oxidative stress is one of the main pathogenic mechanisms underlying ALD initiation and progression (1, 45, 46). Our data based on the human iPSC model support these findings by demonstrating the critical role of mitochondrial injury in mature hepatocytes in response to alcohol (Fig. 4). Interestingly, NAC treatment has not only restored the decreased mitochondrial enzyme Neil-1, but also improved markers associated with HCC and steatosis (Fig. 4F). Antioxidants such as S-Adenosylmethionine have been part of clinical studies, but those with clear benefits are yet to be identified (47). Prospective studies are needed to uncover the key pathogenic driver(s) in alcohol induced oxidative liver injury. Importantly, as we have successfully shown using a human iPSC model of a metabolic liver disease (18), this in vitro human iPSC model recapitulating certain features of ALD may also allow high-throughput screening of new antioxidant and anti-ALD drug candidates.
Alcoholic liver disease is a complex acquired human disease involving multiple cell types. While our human in vitro cellular models mimic some of the ALD features, it does not recapitulate the natural history or a whole feature of ALD. Therefore, animal ALD models would still be very important to study the complex in vivo environment where non-hepatic cells including inflammatory cells interact with liver cells. In this study, we investigated the direct effects, without presence of other complicating factors present in vivo, of alcohol on early and mature stage hepatic cells derived from human iPSCs, which mimics fetal and post-natal liver, respectively. This human iPSC based cellular model of alcohol-induced liver injury can be a very useful tool for studying FASD and ALD as well as for developing preventive or therapeutic strategies for alcoholic liver disorders.
Supplementary Material
Supplementary figures.
Abbreviations
ALD, alcoholic liver disease; iPSCs, induced pluripotent stem cells; FASD, fetal alcohol spectrum disorders; DE, definite endoderm; HP, hepatic progenitor cells; MH, mature hepatocyte-like cells; AFP, alpha-fetoprotein; CK19, cytokeratin 19; CK7, cytokeratin 7; SOX17, SRY-box 17; EpCAM, epithelial cell adhesion molecule; TP53, tumor protein p53; Neil1, nei endonuclease VIII-like 1; CXCR-4, C-X-C chemokine receptor type 4; ALB, albumin; FASN, fatty acid synthase; GPC3, glypican3; FLNB, filamin B; NAC, N-Acetyl-L-Cysteine.
Acknowledgements
This work was supported by grants from Maryland Stem Cell Research Funds (2010-MSCRFII-0101, 2013-MSCRFII-0170 and 2014-MSCRFF-0655) and by NIH (R21AA020020).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572-1585
2. Williams R. Global challenges in liver disease. Hepatology. 2006;44:521-526
3. Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491-501
4. Paula H, Asrani SK, Boetticher NC, Pedersen R, Shah VH, Kim WR. Alcoholic liver disease-related mortality in the United States: 1980-2003. Am J Gastroenterol. 2010;105:1782-1787
5. O'Shea RS, Dasarathy S, McCullough AJ, Practice Guideline Committee of the American Association for the Study of Liver D, Practice Parameters Committee of the American College of G. Alcoholic liver disease. Hepatology. 2010;51:307-328
6. Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343-368
7. European Association for the Study of L. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol. 2012;57:399-420
8. Hofer R, Burd L. Review of published studies of kidney, liver, and gastrointestinal birth defects in fetal alcohol spectrum disorders. Birth Defects Res A Clin Mol Teratol. 2009;85:179-183
9. Burd L, Klug MG, Li Q, Kerbeshian J, Martsolf JT. Diagnosis of fetal alcohol spectrum disorders: a validity study of the fetal alcohol syndrome checklist. Alcohol. 2010;44:605-614
10. Lefkowitch JH, Rushton AR, Feng-Chen KC. Hepatic fibrosis in fetal alcohol syndrome. Pathologic similarities to adult alcoholic liver disease. Gastroenterology. 1983;85:951-957
11. Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH. Rodent models of alcoholic liver disease: of mice and men. Alcohol. 2012;46:715-725
12. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-872
13. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-1920
14. Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV. et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473-5480
15. Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810-1819
16. Choi SM, Liu H, Chaudhari P, Kim Y, Cheng L, Feng J, Sharkis S. et al. Reprogramming of EBV-immortalized B-lymphocyte cell lines into induced pluripotent stem cells. Blood. 2011;118:1801-1805
17. Liu H, Kim Y, Sharkis S, Marchionni L, Jang YY. In vivo liver regeneration potential of human induced pluripotent stem cells from diverse origins. Sci Transl Med. 2011;3:82ra39
18. Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, Liu JO. et al. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology. 2013;57:2458-2468
19. Choi SM, Kim Y, Liu H, Chaudhari P, Ye Z, Jang YY. Liver engraftment potential of hepatic cells derived from patient-specific induced pluripotent stem cells. Cell Cycle. 2011;10:2423-2427
20. Tian L, Prasad N, Jang YY. In Vitro Modeling of Alcohol-Induced Liver Injury Using Human-Induced Pluripotent Stem Cells. Methods Mol Biol. 2014
21. Chaudhari P, Prasad N, Tian L, Jang YY. Determination of Functional Activity of Human iPSC-Derived Hepatocytes by Measurement of CYP Metabolism. Methods Mol Biol. 2014
22. Henzel K, Thorborg C, Hofmann M, Zimmer G, Leuschner U. Toxicity of ethanol and acetaldehyde in hepatocytes treated with ursodeoxycholic or tauroursodeoxycholic acid. Biochim Biophys Acta. 2004;1644:37-45
23. Dolganiuc A, Szabo G. In vitro and in vivo models of acute alcohol exposure. World J Gastroenterol. 2009;15:1168-1177
24. Spear BT, Jin L, Ramasamy S, Dobierzewska A. Transcriptional control in the mammalian liver: liver development, perinatal repression, and zonal gene regulation. Cell Mol Life Sci. 2006;63:2922-2938
25. Morford LA, Davis C, Jin L, Dobierzewska A, Peterson ML, Spear BT. The oncofetal gene glypican 3 is regulated in the postnatal liver by zinc fingers and homeoboxes 2 and in the regenerating liver by alpha-fetoprotein regulator 2. Hepatology. 2007;46:1541-1547
26. Xie Z, Zhang H, Tsai W, Zhang Y, Du Y, Zhong J, Szpirer C. et al. Zinc finger protein ZBTB20 is a key repressor of alpha-fetoprotein gene transcription in liver. Proc Natl Acad Sci U S A. 2008;105:10859-10864
27. Saxena R, Theise N. Canals of Hering: recent insights and current knowledge. Semin Liver Dis. 2004;24:43-48
28. Shiojiri N. Enzymo- and immunocytochemical analyses of the differentiation of liver cells in the prenatal mouse. J Embryol Exp Morphol. 1981;62:139-152
29. Hua M, Zhang W, Li W, Li X, Liu B, Lu X, Zhang H. Molecular mechanisms regulating the establishment of hepatocyte polarity during human hepatic progenitor cell differentiation into a functional hepatocyte-like phenotype. J Cell Sci. 2012;125:5800-5810
30. Wang P, Zhang H, Li W, Zhao Y, An W. Promoter-defined isolation and identification of hepatic progenitor cells from the human fetal liver. Histochem Cell Biol. 2008;130:375-385
31. Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM. A CD133-expressing murine liver oval cell population with bilineage potential. Stem Cells. 2007;25:2419-2429
32. Rountree CB, Ding W, He L, Stiles B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells. 2009;27:290-299
33. Suzuki M, Sugimoto K, Tanaka J, Tameda M, Inagaki Y, Kusagawa S, Nojiri K. et al. Up-regulation of glypican-3 in human hepatocellular carcinoma. Anticancer Res. 2010;30:5055-5061
34. Kim H, Kim K, Yu SJ, Jang ES, Yu J, Cho G, Yoon JH. et al. Development of biomarkers for screening hepatocellular carcinoma using global data mining and multiple reaction monitoring. PLoS One. 2013;8:e63468
35. Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology. 2010;52:2137-2147
36. Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y. et al. Rescue of ATP7B function in hepatocyte-like cells from Wilson's disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum Mol Genet. 2011;20:3176-3187
37. Cayo MA, Cai J, Delaforest A, Noto FK, Nagaoka M, Clark BS, Collery RF. et al. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology. 2012
38. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J. et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002-5012
39. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821-5828
40. Bruno S, Bussolati B, Grange C, Collino F, Graziano ME, Ferrando U, Camussi G. CD133+ renal progenitor cells contribute to tumor angiogenesis. Am J Pathol. 2006;169:2223-2235
41. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-110
42. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-10951
43. Kordes C, Sawitza I, Muller-Marbach A, Ale-Agha N, Keitel V, Klonowski-Stumpe H, Haussinger D. CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun. 2007;352:410-417
44. Mavila N, James D, Shivakumar P, Nguyen MV, Utley S, Mak K, Wu A. et al. Expansion of prominin-1-expressing cells in association with fibrosis of biliary atresia. Hepatology. 2014;60:941-953
45. Zhu H, Jia Z, Misra H, Li YR. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J Dig Dis. 2012;13:133-142
46. Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141-154
47. Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515-1542
Author contact
![]() Corresponding author: Yoon-Young Jang, M.D., Ph.D. 1550 Orleans Street, CRB2 Rm552, Baltimore, MD 21231. Office (410)-502-8195 Email: yjang3edu.
Corresponding author: Yoon-Young Jang, M.D., Ph.D. 1550 Orleans Street, CRB2 Rm552, Baltimore, MD 21231. Office (410)-502-8195 Email: yjang3edu.